
Vývoj disolučnej metódy pre tablety s obsahom warfarínu sodného
Development of dissolution method for warfarin sodium tablets
Warfarin is intensively discussed drug with narrow therapeutic index. In the past, its generic substitution was identified as a cause of bleeding. Altered quality of the active substance or varying drug content was discussed. The substance quality can be evaluated with adequate dissolution method. An official dissolution method with aqueous medium exists, however this method is non-discriminatory. In the first 15 minutes the whole amount of the active pharmaceutical ingredient is released from a tested dosage form, which does not allow comparison between tablets from different producers and it also makes difficult to track the changes throughout stability testing. In the literature, there is a well known method using pH 6.8 buffer, which seems to be a suitable alternative to water. The aim of this study was to prove, that this alternative medium, when two stirring speeds for dissolution (50 or 25 rpm) are used, will be suitable for calculation of similarity and difference factor and if it will be eventually discriminatory with regard to particle size and radial hardness. For this purpose we prepared tablets with 10 mg of warfarin sodium in form of crystalline clathrate with isopropanol. Tablets differed by particle size of active pharmaceutical ingredient (d50 = 4.8, or d50 = 22.5 μm respectively) and by radial hardness (30, or 100 N respectively). The content uniformity of the tablets was determined using process capability index (Cpk) and Bergum method. It was confirmed that the dissolution medium with pH of 6.8 allows comparison of dissolution profiles by similarity and difference factors but under given conditions it is not discriminatory.
Key words:
warfarin • dissolution method • particle size distribution • radial hardness • similarity factor • difference factor
:
Ivana Lukášová; Jan Muselík; Aleš Franc
:
Čes. slov. Farm., 2017; 66, 281-286
:
Original Articles
Warfarín je stále rozsiahle diskutovaným liečivom s úzkym terapeutickým indexom. Jeho generické náhrady boli v minulosti príčinou krvácavosti. Ako príčina boli diskutované kvalita substancie a miera obsahovej rovnorodosti. Kvalitu substancie je možné posúdiť vhodnou disolučnou metódou. Doteraz existuje pre tablety s obsahom warfarínu oficiálna miešadlová disolučná metóda s vodným médiom, ktorá nemá rozlišujúci charakter. V priebehu prvých 15 minút u nej dochádza k takmer úplnému uvoľneniu účinnej látky z liekovej formy, čím je znemožnené vzájomné porovnanie tabliet od rôznych výrobcov alebo sledovanie prípadných zmien v priebehu stabilitných testov. Z literatúry je známa aj metóda s médiom s pH 6,8, ktorá sa odporúča ako vhodná alternatíva. Cieľom tejto práce bolo vyskúšať, či táto metóda pri dvoch rýchlostiach otáčok (50 a 25 rpm) bude vhodná k výpočtom faktorov podobnosti a rozdielnosti a prípadne bude mať rozlišujúci charakter vzhľadom k veľkosti častíc účinnej látky a pevnosti tabliet. Za týmto účelom boli vyrobené tablety s obsahom 10 mg sodnej soli warfarínu vo forme kryštalického klathrátu s izopropanolom. Pripravené tablety sa líšili veľkosťou častíc účinnej látky (d50 = 4,8, resp. 22,5 μm) a ich radiálnou pevnosťou (30, resp. 100 N). Obsahová rovnorodosť tabliet, bola overená pomocou indexu spôsobilosti procesu (Cpk) a Bergumovej metódy. Bolo potvrdené, že disolučné médium s pH 6,8 umožňuje vzájomné porovnanie disolučných profilov pomocou výpočtov faktorov podobnosti a rozdielnosti, no za daných podmienok nemá rozlišujúci charakter.
Kľúčové slová:
warfarín • disolučná metóda • distribúcia veľkosti častíc • radiálna pevnosť • faktor podobnosti • faktor rozdielnosti
Úvod
Warfarín sodný, či už vo forme kryštálu alebo amorfu, patrí medzi liečivá s úzkym terapeutickým indexom1). Z chemického hľadiska sa jedná o soľ slabej kyseliny a silnej zásady s hodnotou pKa 5,052). Rozpustnosť tejto účinnej látky závisí od stupňa ionizácie molekuly a úzko súvisí s pH okolitého prostredia. K rýchlemu rozpusteniu sodnej soli warfarínu dochádza vo vode. K spomaleniu rozpúšťania dochádza v kyslom prostredí. V zásaditom prostredí sa rozpustnosť warfarínu zvyšuje vplyvom ionizácie molekúl. Z hľadiska biofarmaceutického klasifikačného systému (BCS) patrí warfarín do II. triedy, pre ktorú je typická nízka rozpustnosť a vysoká permeabilita3).
Skúška disolúcie je jedným zo základných testov pri zabezpečovaní kvality liekovej formy. Táto liekopisná skúška sa využíva tiež na sledovanie zmien liberácie v priebehu skladovania a u bioekvivalenčných štúdií. Pri bioekvivalenčných a stabilitných štúdiách sa obvykle testujú tablety s najvyšším obsahom účinnej látky (active pharmaceutical ingredient – API), čo v prípade warfarínu zodpovedá tabletám s obsahom 10 mg liečiva. Ak sa bioekvivalencia potvrdí na najvyššej terapeutickej sile, je výsledky často možné aplikovať aj na ostatné sily4). U stabilitných štúdií môžu byť ostatné sily redukované metódami matrixingu a bracketingu5). Toto riešenie je pre výrobcov ekonomicky a časovo výhodné.
Rôzne prípravky s rovnakou účinnou látkou, môžu mať odlišné kvalitatívne vlastnosti (napr. veľkosť častíc API), môžu obsahovať rôzne pomocné látky či môžu byť vyrobené rôznymi technologickými postupmi, čo môže ovplyvňovať rýchlosť a množstvo uvoľnenej API. Ku zmenám kvality prípravku môže dôjsť aj v dôsledku variability výroby alebo v priebehu testovania stability prípravku, ktorý má už dané zloženie a technológiu výroby. U liečivých látok s úzkym terapeutickým indexom, kde uvoľňovanie účinnej látky môže vo zvýšenej miere ovplyvniť odpoveď organizmu, je preto vhodné nájsť takú disolučnú metódu, ktorá umožní vzájomné porovnanie disolučných profilov pomocou výpočtu faktora podobnosti f2 a faktora rozdielnosti f1.
USP, resp. FDA metóda, ktoré pre tablety s obsahom sodnej soli warfarínu používajú vodné médium pri otáčkach 50 rpm (prístroj s lopatkovým miešadlom), nemá rozlišujúcu schopnosť 6–8), nie je biorelevantná a neumožňuje spravidla výpočet f1 a f2 9). Použitie média s pH 1,2, ktoré sa používa u tabliet s okamžitým uvoľňovaním, je nevhodné z dôvodu nízkej rozpustnosti warfarínu v kyslom prostredí10). Pri testoch s médiom s pH 4,5 sa zistilo nedostatočné množstvo uvoľneného warfarínu (testované na tabletách s obsahom 5 mg API). Pri tomto pH je štandardný limit pre liekové formy s okamžitým uvoľňovaním dosiahnutý len v niektorých prípadoch7, 11). Pri obsahu 10 mg warfarínu sa dá preto predpokladať ďalšie zníženie uvoľneného množstva, lebo väčšina jeho molekúl sa pri pH 4,5 nachádza v neionizovanej forme. Existujú aj biorelevantné metódy, ako je kombinácia média s pH 1,2 buď s následným zvýšením na pH 7,412) alebo s n-oktanolom metódou bifázickej disolúcie13). Uvedené metódy sú však pre rutinnú kontrolu technicky aj časovo náročné. Dá sa skonštatovať, že iba disolučné metódy s použitím neutrálnych a mierne bázických médií, v ktorých je warfarín dobre rozpustný, sú vhodné pre disolúciu tabliet s najvyšším terapeutickým obsahom 10 mg warfarínu. Ako alternatíva k liekopisnej metóde využívajúcej vodu býva preto ponúkané médium s pH 6,82, 14). Pri použití tohto média sa predpokladá veľmi dobrá rozpustnosť warfarínu, čo vyplýva z jeho chemickej povahy. Vplyvom výskytu spoločného iónu sodíka v pufri a v molekule warfarínu môže pri disolúcii však paradoxne dochádzať k spomaleniu rozpúšťania warfarínu a celkovo disolúcie15). Vďaka spomaleniu rozpúšťania by toto médium mohlo byť použité pre výpočet faktorov podobnosti a rozdielnosti a umožniť rozlíšenie medzi prípravkami s rozličnými kvalitatívnymi vlastnosťami, ktoré môžu byť ovplyvnené variabilitou výroby.
Všetky tieto spomenuté metódy používajú otáčky lopatkového miešadla nie menej ako 50 rpm, čo je doporučená rýchlosť pre lieky s okamžitým uvoľňovaním16). Niektoré súčasné práce naznačujú, že pre okamžité uvoľňovanie s dobre rozpustnou API je možné použiť aj nižšie otáčky, čo by mohlo mať rozlišujúci účinok11).
Cieľom práce bolo overiť vhodnosť média s pH 6,8 pre disolúciu warfarínu na základe výpočtov faktorov podobnosti a rozdielnosti a overenie rozlišujúcej schopnosti použitého média s ohľadom na kvalitu použitej substancie a liekovej formy. K tomuto účelu boli pripravené tablety s obsahom 10 mg warfarínu vo forme kryštalického klathrátu s izopropanolom, ktoré sa líšili veľkosťou častíc warfarínu (d50 = 4,8, resp. 22,5 μm) a ich radiálnou pevnosťou (30, resp. 100 N). Vyrobené tablety boli podrobené disolučnej skúške pri liekopisom stanovených otáčkach (50 rpm) a znížených otáčkach (25 rpm).
Pokusná časť
Príprava tabliet
Zloženie zmesí a základné fyzikálno-chemické vlastnosti jednotlivých zložiek sú uvedené v tabuľke 1. Zvolenými premennými bola veľkosť častíc warfarínu. Celkovo boli pripravené 2 šarže zmesí. Všetky suroviny okrem warfarínu boli preosiate cez sito s veľkosťou ôk 500 μm. Približne 50,0 g warfarínu bolo preosiatych sústavou dvoch sít umiestnených nad sebou (väčšie hore), ktorých veľkosť ôk bola 250 a 80 μm. Sitá boli umiestnené do sitovacieho zariadenia Retch AS 200 Basic (Nemecko). Sústava sa nechala vibrovať 10 minút pri amplitúde 60 (podľa stupnice prístroja). Frakcia zachytená na site o veľkosti ôk 250 μm bola použitá pri príprave šarže II a frakcia zachytená pod sitom o veľkosti ôk 80 μm sa použila k príprave šarže I. Zmesi boli pripravené patentovanou technológiou17). Veľkosť jednej šarže bola 500,0 g. Z pripravených zmesí (šarža I a II) bolo odobratých 10 vzoriek postupom prevzatým z experimentu18), u ktorých bol stanovený obsah. Pred lisovaním bola každá zmes podrobená testovaniu fyzikálnych vlastností podľa ČL 200919) (viď fyzikálne hodnotenie zmesí a tabliet). Zvyšná zmes bola vylisovaná výstredníkovým tabletovacím lisom Korsch EK0 (Nemecko) do tabliet s použitím plochých razidiel s priemerom 13 mm. Z každej zmesi boli pripravené tablety s obsahom 10 mg warfarínu o rôznej distribúcii veľkosti častíc warfarínu (šarža I a II), ktorých teoretická hmotnosť bola 542,0 mg. Nastavenie lisovacej sily sa v priebehu lisovania menilo tak, aby tablety dosiahli dve cieľové radiálne pevnosti ~ 30 a 100 N. V priebehu lisovania bolo v pravidelných časových intervaloch odobratých 10 vzoriek pre kontrolu obsahovej rovnorodosti a zvyšné tablety boli použité k hodnoteniu podľa ČL 2009.

Fyzikálne hodnotenie zmesí a tabliet
U zmesí bol stanovený sypný uhol, index stlačiteľnosti, Hausnerov pomer a rýchlosť vytekania otvorom. Sypná a zhustená hustota bola stanovená podľa čl. 2.9.15 a 2.9.34 s použitím zariadenia SVM 102 (Erweka, Nemecko). Rýchlosť vytekania otvorom bola stanovená podľa čl. 2.9.16 ČL s použitím násypky s uzatvoreným spodným koncom od firmy Medipo (ZT, Česká republika).
U tabliet bola hodnotená mechanická odolnosť – pevnosť, oder, výška, rozpad a hmotnostná rovnorodosť. Pevnosť, výška a hmotnosť tabliet (n = 20) bola stanovená pomocou zariadenia Pharmatest WHT-1 (Pharmatest, Nemecko). Hmotnostná rovnorodosť bola stanovená podľa čl. 2.9.5 a pevnosť tabliet podľa čl. 2.9.8. Oder bol hodnotený podľa čl. 2.9.7 pomocou friabilitátora TAR 10 (Erweka, Nemecko). Rozpad bol stanovený podľa čl. 2.9.1 s použitím ZT 4 (Erweka, Nemecko).
Stanovenie obsahu warfarínu
Obsah warfarínu v pripravených zmesiach a tabletách bol stanovený pomocou HPLC. Postup prípravy vzoriek a priebeh analytického stanovenia bol prevzatý z literatúry18).
Skúška disolúcie
Pripravené tablety (12 tabliet z každej šarže) boli hodnotené miešadlovou disolučnou metódou na prístroji Sotax (AT 7 Donau Lab, Švajčiarsko). Na základe tejto skúšky bolo stanovené množstvo uvoľnenej účinnej látky v čase. Ako disolučné médium bol použitý fosfátový pufer s pH 6,8 pripravený podľa ČL 200919). Objem pufru bol 900 ml. Rýchlosť otáčania lopatkových miešadiel bola nastavená na 50, resp. 25 rpm. V priebehu skúšky bola v systéme udržiavaná teplota 37 ± 0,5 °C. V čase 5, 10, 20, 30, 60 a 120 minút boli z disolučných nádob pomocou 2ml pipety ručne odobraté vzorky, ktoré boli analyzované metódou HPLC za rovnakých podmienok, za akých bola analyzovaná zmes a tablety pri stanovení obsahu. Pre každý odberový bod bolo vypočítané relatívne množstvo uvoľneného liečiva a smerodajná odchýlka.
Štatistické spracovanie výsledkov
Výsledky stanovenia obsahu jednotlivých vzoriek zmesí a tabliet z každej šarže (n = 10) boli prepočítané relatívne voči teoretickému obsahu warfarínu. Z týchto hodnôt bol pre každú pripravenú šaržu vypočítaný priemerný obsah, RSD a Cpk index pre limity ČL 2009 čl. 2.9.6 (85–115 %). Zmesi boli hodnotené aj podľa kritérií FDA, podľa ktorých je limit pre obsahovú rovnorodosť 90 až 110 % a RSD ˂ 5 %20). Tablety boli navyše hodnotené tiež podľa čl. 2.9.40 ČL 200919). Získané hodnoty RSD tabliet boli porovnané s tabelovanými hodnotami Bergumovej metódy.
Pre porovnanie disolučných profilov tabliet bol vypočítaný faktor podobnosti f2 a faktor rozdielnosti f1 21) a ďalej účinnosť disolúcie (D.E.) vypočítaná na základe plochy pod disolučnou krivkou22). Obidva faktory boli vypočítané z priemerného relatívneho množstva uvoľneného liečiva vo všetkých odberových bodoch (5–120 minút).
Výsledky a diskusia
V tejto experimentálnej práci bola testovaná vhodnosť použitia alternatívneho disolučného média s pH 6,8 pre výpočet faktorov podobnosti a rozdielnosti disolučných kriviek uvoľňovania warfarínu pri otáčkach 50 a 25 rpm (prístroj s lopatkovým miešadlom). Za týmto účelom boli pripravené tablety s teoretickým obsahom 10 mg warfarínu sodného (zloženie v tab. 1), ktoré sa líšili veľkosťou častíc warfarínu (šarža I s d50 = 4,8 a šarža II s d50 = 22,5 μm) a ich radiálnou pevnosťou (30 a 100 N) z dôvodu simulácie variability výroby. Výsledky hodnotenia tokových vlastností pripravených zmesí sú uvedené v tabuľke 2. Na základe indexu stlačiteľnosti a Hausnerovho pomeru bol tok zmesí vyhodnotený ako primeraný. Podľa stanoveného sypného uhla bol charakter toku výborný. Dobré tokové vlastnosti je možné pripísať plnivu Di-cafos, ktorého zastúpenie v zmesi činí 70 % a vyznačuje sa veľmi dobrými tokovými vlastnosťami23). Zmesi boli následne vylisované do tabliet.

Fyzikálno-chemické vlastnosti tabliet s teoretickým obsahom 10 mg warfarínu sú zhrnuté v tabuľke 3. Rozpad tabliet všetkých šarží vyhovuje požiadavkám liekopisu, a to bez ohľadu na veľkosť častíc či pevnosť. Čas rozpadu tabliet s radiálnou pevnosťou 100 N bol v porovnaní s tabletami s pevnosťou 30 N dlhší. Predĺženie času rozpadu so vzrastajúcou pevnosťou je v zhode s očakávaním, nakoľko čím vyššia lisovacia sila sa pri lisovaní použije, tým pevnejšie sú aj hotové výlisky a tým pomalší je rozpad. Oder tabliet bol nepatrne vyšší u tabliet s pevnosťou 30 N, čo zodpovedá všeobecným zákonitostiam. Všetky tablety vyhoveli skúške oderu. Na základe výsledkov hodnotenia tabletových zmesí a tabliet sa zistilo, že zvolený technologický postup vedie k výrobe štandardných tabliet. Tablety majú vyhovujúce liekopisné vlastnosti a medzi šaržami neboli zistené významné rozdiely, ktoré by mohli negatívne skresliť výsledky disolučnej skúšky.

Vyhodnotenie obsahovej rovnorodosti zmesí a tabliet s obsahom 10 mg warfarínu
Zmesi a tablety boli podrobené tiež skúške obsahovej rovnorodosti, nakoľko nevyhovujúca obsahová rovnorodosť by mohla negatívne ovplyvniť výsledky disolučnej skúšky. Pre zabezpečenie obsahovej rovnorodosti, s ohľadom na úzky terapeutický index warfarínu, používajú viacerí výrobcovia prísnejší limit obsahovej rovnorodosti pre tablety v porovnaní s liekopisnými limitmi. Konkrétne sa jedná o limit firmy DuPont Pharma, ktorý postupne prebrali aj ďalší generickí výrobcovia (Apotex, Taro Pharmaceuticals)24, 25). Podľa tohto limitu musí obsah desiatich testovaných jednotiek spadať do intervalu 92,5–107,5 % a RSD musí byť do 3 %26). Pre zabezpečenie, že nie len testovaná šarža, ale aj všetky nasledujúce splnia predpísané limity sa používajú rôzne štatistické nástroje. Jedná sa napr. o index spôsobilosti procesu Cpk27, 28) a Bergumovu metódu29), ktoré na danej štatistickej hladine významnosti zabezpečujú, že aj ďalšie vyrobené šarže splnia liekopisné požiadavky na obsahovú rovnorodosť. Výsledky obsahovej rovnorodosti tabletových zmesí a tabliet sú zhrnuté v tabuľke 4 a 5 a sú pre všetky testované zmesi a tablety vyhovujúce, čím je zabezpečené, že výsledky disolučnej skúšky nebudú negatívne ovplyvnené kolísaním obsahu liečiva v tabletách. Tablety spĺňajú tiež limit výrobcu DuPont.


Vyhodnotenie skúšky disolúcie s médiom s pH 6,8
Vyrobené tablety boli následne podrobené skúške disolúcie v prostredí pufru s pH 6,8. Vplyvom výskytu spoločného iónu sodíka v pufre a v molekule warfarínu sa očakávalo spomalenie uvoľňovania warfarínu oproti vode, na základe ktorého bude možné vzájomné porovnanie disolučných profilov, a prípadne tak bude možné rozlíšiť tiež kvalitu tabliet (veľkosť častíc warfarínu a pevnosť tabliet).
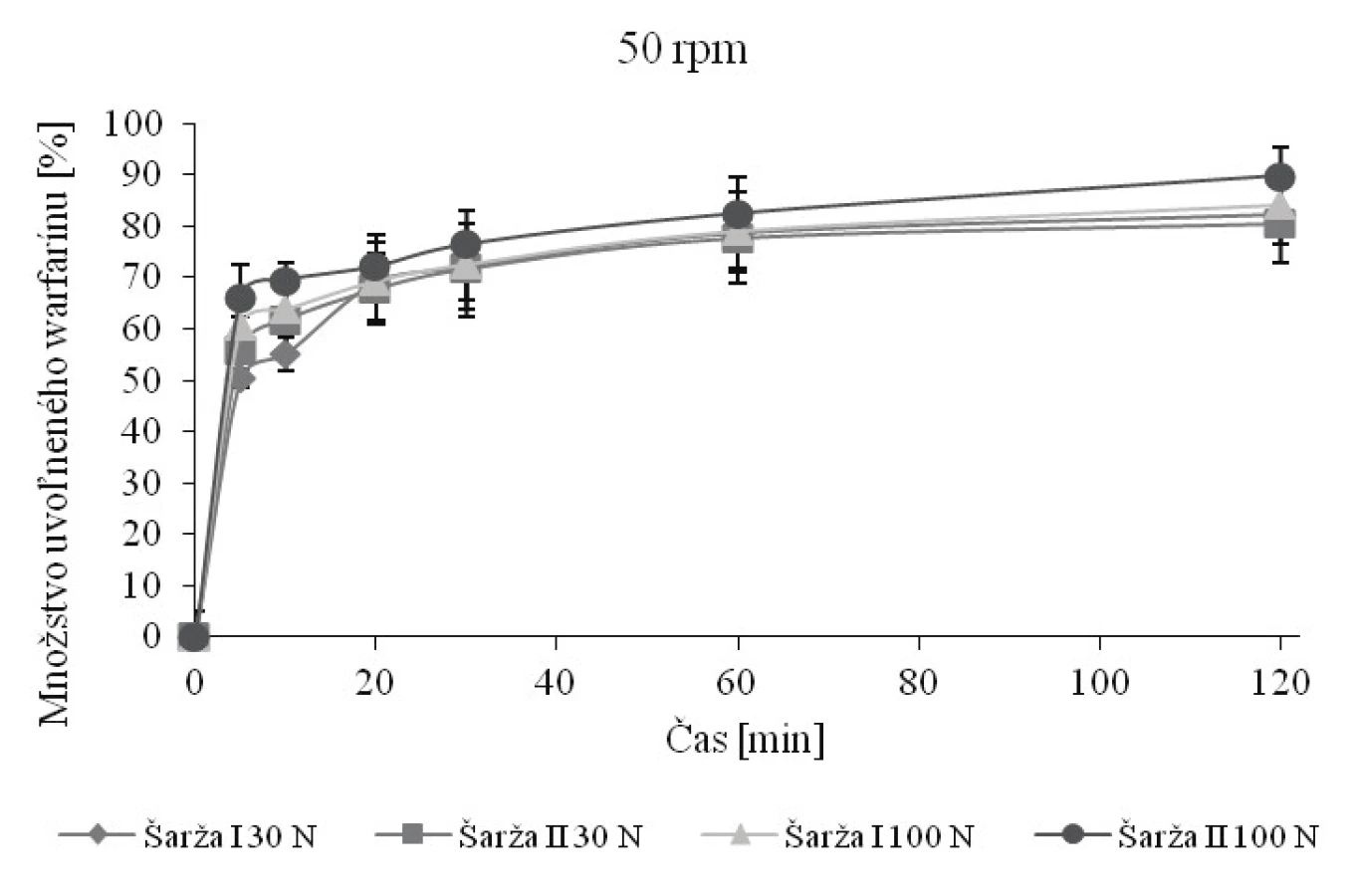
Na obrázku 1, resp. 2 sú vyobrazené disolučné krivky pri 50 rpm, resp. 25 rpm. Obe metódy umožňujú výpočet faktorov podobnosti a rozdielnosti, no nemajú tu rozlišujúci charakter (tab. 6, resp. 7). Pri otáčkach 25 rpm dochádza k zníženiu rýchlosti uvoľňovania v porovnaní s otáčkami 50 rpm a ani po 120 minútach nedôjde k uvoľneniu 80 % warfarínu. Na základe výpočtov faktorov podobnosti a rozdielnosti (tab. 7) sa zistilo, že niektoré podobné krivky podľa f2 (54,55, resp. 54,62) sú zároveň rozdielne podľa f1 (19,16, resp. 16,75). Nesúlad medzi f1 a f2 je dobre známy aj z literatúry13, 22), no pre legislatívne účely je rozhodujúci faktor podobnosti f2 30). Vzhľadom k nejednotnosti záverov na základe výpočtov f1 a f2 boli disolučné profily porovnané taktiež pomocou výpočtu D.E. (tab. 6 a 7). Literatúra uvádza, že pokiaľ je zmena D.E. medzi hodnotenými profilmi menšia ako 10, je možné profily považovať za podobné22). Pretože absolútna hodnota maximálnej zmeny D.E. medzi hodnotenými profilmi nepresiahla hodnotu 7, je možné na základe tohto parametra považovať profily za podobné. Zníženie otáčok teda nielen že nevedie k rozlišujúcemu charakteru, no nejaví sa ako vhodné ani z hľadiska nedostatočného množstva uvoľnenej látky. Je teda možné konštatovať, že disolučné médium s pH 6,8 umožní porovnanie disolučných kriviek na základe výpočtov f1 a f2, ale za daných podmienok bez rozlišujúceho charakteru. Len otáčky 50 rpm sa javia ako relevantné, pretože umožňujú splniť obvyklý liekopisný limit Q limitu (75 % + 5 %)19).



Záver
V tejto práci bolo pomocou disolučnej skúšky hodnotené uvoľňovanie sodnej soli warfarínu z tabliet s teoretickým obsahom 10 mg. Pri skúške disolúcie bolo použité médium s pH 6,8, ktoré sa používa ako alternatíva k liekopisnej metóde používajúcej vodu, ktorá nie je vhodná pre výpočet faktorov podobnosti a rozdielnosti. Zvolená miešadlová disolučná metóda pri otáčkach 50 rpm nemá rozlišujúci charakter s ohľadom na použitú veľkosť častíc warfarínu a pevnosť. Tvarom disolučnej krivky sa však umožní výpočet faktorov podobnosti a rozdielnosti a teda porovnanie kriviek, čo je dôležité v priemysle napríklad pri transfere technológií alebo v hodnotení stabilitných štúdií.
Zoznam skratiek
API - účinná látka
Cpk - index spôsobilosti procesu
ČL 2009 - Český liekopis 2009
D.E. - efektivita disolúcie
FDA - Úrad pre kontrolu potravín a liečiv
rpm - počet otáčok za minútu
RSD - relatívna smerodajná odchýlka
Stret záujmov: žiadny.
Došlo 30. října 2017
Prijato 24. listopadu 2017
PharmDr. Ivana Lukášová
J. Muselík
A. Franc
Ústav technologie léků, Farmaceutická fakulta
Veterinární a farmaceutická univerzita Brno
Palackého tř. 1946/1, 612 42 Brno
e-mail: ivana.lukasova@gmail.com
Sources
1. Benet L. Z., Goyan J. E. Bioequivalence and narrow therapeutic index drugs. Pharmacotherapy. 1995; 15, 433–440.
2. Zhang X., Wen H., Fan J., Vince B., Li T., Gao W., Kinjo M., Brown J., Sun W., Jiang W., Lionberger R. Integrating in vitro, modeling, and in vivo approaches to investigate warfarin bioequivalence. CPT Pharmacometrics Syst. Pharmacol. 2017; 6, 523–531.
3. Benet L. Z. The role of BCS (Biopharmaceutics Classification System) and BDDCS (Biopharmaceutics Drug Disposition Classification System) in drug development. J. Pharm. Sci. 2013; 102, 34–42.
4. EMEA. Guideline on the investigation of bioequivalence (2010). http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2010/01/WC500070039.pdf (25. 10. 2017).
5. EMEA. ICH Topic Q 1 D; Bracketing and matrixing designs for stability testing of drug substances and drug products (2002). http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500002652.pdf (20. 10. 2017).
6. U.S. Pharmacopeia. USP 29 Warfarin sodium tablets. http://www.pharmacopeia.cn/v29240/usp29nf24s0_m88790.html (18. 10. 2017).
7. Nguyenpho A., Ciavarella A. B., Siddiqui A., Rahman Z., Akhtar S., Hunt R., Korang-Yeboah M., Khan M. A. Evaluation of in-use stability of anticoagulant drug products: Warfarin sodium. J. Pharm. Sci. 2015; 104, 4232–4240.
8. Ali S. L., Krämer J. Pharmaceutical quality of warfarin sodium tablets: A multinational postmarket comparative study. Pharm. Ind. 1999; 61, 363–368.
9. O’Reilly R. A., Nelson E., Levy G. Physicochemical and physiologic factors affecting the absorption of warfarin in man. J. Pharm. Sci. 1966; 55, 435–437.
10. Stella V. J., Mooney K. G., Pipkin J. D. Dissolution and ionization of warfarin. J. Pharm. Sci. 1984; 73, 946–948.
11. Quereshi S. A. Choice of rotation speed (rpm) for bio-relevant drug dissolution testing using a crescent-shaped spindle. Eur. J. Pharm. Sci. 2004; 23, 271–275.
12. Wagner J. G., Welling P. G., Lee K. P., Walker J. E. In vivo and in vitro availability of commercial warfarin tablets. J. Pharm. Sci. 1971; 60, 666–677.
13. Franc A., Muselík J., Goněc R., Vetchý D. Biphasic dissolution method for quality control and assurance of drugs containing active substances in the form of weak acid salts. Acta Pharm. 2016; 66, 139–145.
14. McCormick T. J., Gibson A. B., Diana F. J. Development and validation of a dissolution method for warfarin sodium and aspirin combination tablets. J. Pharm. Biomed. Anal. 1997; 15, 1881–1891.
15. Serajuddin A. T., Sheen P. C., Augustine M. A. Common ion effect on solubility and dissolution rate of the sodium salt of an organic acid. J. Pharm. Pharmacol. 1987; 39, 587–591.
16. FDA. Guidance for Industry: Dissolution testing of immediate release solid oral dosage forms (1997). https://www.fda.gov/downloads/drugs/guidances/ucm070237.pdf (18. 10. 2017).
17. University of Veterinary and Pharmaceutical Sciences Brno, Franc, A., Muselík, J. Způsob přípravy pevné lékové formy se sodnou solí warfarínu ve formě klathrátu izopropanolu. Patent CZ 304136 Czech Republic, 2013.
18. Muselík J., Franc A., Doležel P., Goněc R., Krondlová A., Lukášová I. Influence of process parameters on content uniformity of a low dose active pharmaceutical ingredient in a tablet formulation according to GMP. Acta Pharm. 2014; 64, 355–367.
19. Český lékopis 2009. 1. vydání. Praha: Grada Publishing 2009.
20. FDA. Guidance for industry. ANDAs: Blend uniformity analysis (1999). https://www.fda.gov/ohrms/dockets/98fr/992635gd.pdf (20. 10. 2017).
21. Shah V. P., Tsong Y., Sathe P., Williams R. L. Dissolution profile comparison using similarity factor, f2. http://www.dissolutiontech.com/DTresour/899Art/DissProfile.html (18. 10. 2017).
22. Anderson N. H., Bauer M., Boussac N., Khan-Malek R., Munden P., Sardaro M. An evaluation of fit factors and dissolution efficiency for the comparison of in vitro dissolution profiles. J. Pharm. Biomed. Anal. 1998; 17, 811–822.
23. Ondrejček P., Svačinová P., Vraníková B., Holas O., Rabišková M., Škubalová Z. Compaction equation and its use in the determination of compressibility of pharmaceutical materials. Poster 2016.
24. Sawoniak A.E., Shalansky K.F., Zed P.J., Sunderji R. Formulary considerations related to warfarin interchangeability. Can. J. Hosp. Pharm. 2002; 55, 215–218. http://cjhp-online.ca/index.php/cjhp/article/viewFile/578/679
25. Interchangeability of warfarin sodium preparations. https://www.ab.bluecross.ca/dbl/pdfs/dblreportdec00update.pdf (13. 10. 2017).
26. Wittkowsky A. K. Generic warfarin: implications for patient care. Pharmacotherapy 1997; 17, 640–643.
27. Perez-Wilson M. Machine/Process Capability Study. A five stage methodology for optimizing manufacturing processes. 6th ed. Arizona: Advanced Systems Consultants 2014; s. 357.
28. Pearn W. L., Shu M. H. Manufacturing capability control for multiple power-distribution switch processes based on modified Cpk MPPAC. Microelectron. Reliab. 2003; 43, 963–975.
29. Bergum J. S., Li H. Acceptance limits for the new ICH USP 29 content-uniformity test. Pharm. Technol. 2007; 31, 90–100.
30. FDA. Guidance for industry. Immediate release solid oral dosage forms (1995). https://www.fda.gov/downloads/drugs/guidances/ucm070636.pdf (22. 10. 2017).
Labels
Addictology Clinical biochemistry Paediatric clinical oncology Paediatric psychiatry Diabetology Endocrinology Pharmacy Clinical pharmacology Intensive Care Medicine Internal medicine General practitioner for children and adolescents General practitioner for adults Psychiatry Toxicology Pharmaceutical assistantArticle was published in
Czech and Slovak Pharmacy

2017 Issue 6
- Advances in the Treatment of Myasthenia Gravis on the Horizon
- Memantine in Dementia Therapy – Current Findings and Possible Future Applications
- Memantine Eases Daily Life for Patients and Caregivers
- Possibilities of Using Metamizole in the Treatment of Acute Primary Headaches
Most read in this issue
- Gut microbiota: its development and relation to certain diseases
- Development of dissolution method for warfarin sodium tablets
- Carboxylic acids from brown algae Fucus vesiculosus and Padina pavonica
- The size-reduced Eudragit® RS microparticles prepared by solvent evaporation method – monitoring the effect of selected variables on tested parameters