
Tuberózní skleróza
Tuberous Sclerosis
Tuberous sclerosis (TSC) is a hereditary autosomal dominant multisystemic disease caused by mutation of tumor-suppressor genes with multiple hamartomas and benign tumors. There are two types of TSC caused by mutation of various tumorsuppressor genes – type 1 (TSC1) and type 2 (TSC2); these two types are not discernable based on the clinical symptomatology. The clinical symptomatology is variable and age-dependent. Typical cutaneous symptomatology of tuberous sclerosis includes hypomelanotic maculae, angiofibromas, fibrous plaques, fibromas of the nails, shagreen patch, confetti skin lesion, other manifestations include hamartomas and benign tumors of the central nervous system, eyes, lungs, kidneys and heart. A common finding includes mental retardation, epilepsy and autism. The prognosis is variable and depends on the clinical symptomatology and complications; the cause of death being brain or kidney tumors. The diagnosis of TSC is based on genetic analysis or clinical diagnostic criteria (11 primary and 6 secondary). The treatment of tuberous sclerosis is symptomatic, with promising results using the mTOR inhibitors. Patients should be observed in specialized centers.
Keywords:
tuberous sclerosis complex type 1, 2 – hypomelanotic macule – angiofibroma – shagreen patch – subependymal nodules – subependymal giant cell astrocytoma – rhabdomyoma – lymphangioleiomyomatosis – angiomyolipoma – epilepsy – mTOR – rapamycin – everolimus – specialized centers’ care
Authors:
D. Humhejová 1,2
Authors‘ workplace:
Dětská klinika UJEP, Masarykova nemocnice v Ústí nad Labem o. z., Krajská zdravotní a. s.
přednosta MUDr. Jaroslav Škvor, CSc.
1; Ústecká poliklinika, s. r. o., Ústí nad Labem
2
Published in:
Čes-slov Derm, 91, 2016, No. 2, p. 43-59
Category:
Reviews (Continuing Medical Education)
Overview
Tuberózní skleróza (TSC) je autozomálně dominantně dědičné multisystémové onemocnění způsobené mutací tumor supresorových genů se vznikem mnohočetných hamartomů a benigních nádorů. Rozlišují se 2 typy TSC způsobené mutací různých tumor supresorových genů – typ 1 (TSC1) a typ 2 (TSC2), podle klinického obrazu jsou tyto typy neodlišitelné. Klinický obraz TSC je variabilní, manifestace projevů je věkově vázaná. Typickými kožními projevy tuberózní sklerózy jsou hypomelanotické makuly, angiofibromy, fibrózní plaky, fibromy nehtů, šagrénová kůže, drobné skvrnité depigmentace, dále se vyskytují hamartomy a benigní nádory v centrálním nervovém systému, oku, plicích, ledvinách a srdci. Častým nálezem je mentální retardace, epilepsie a autismus. Prognóza je individuální podle klinického obrazu a komplikací, příčinou úmrtí bývají komplikace nádorů mozku a ledvin. K diagnostice TSC slouží genová analýza a klinická diagnostická kritéria (11 hlavních a 6 vedlejších). Léčba tuberózní sklerózy je symptomatická, slibné výsledky má léčba mTOR inhibitory. Pacienti by měli být sledováni ve specializovaných centrech.
Klíčová slova:
tuberózní skleróza typ 1, 2 – hypomelanotické makuly – angiofibrom – šagrénová kůže – subependymální noduly – subependymální obrovskobuněčný astrocytom – rhabdomyom – lymfangioleiomyomatóza plic – angiomyolipom – epilepsie – mTOR – rapamycin – everolimus – specializovaná centra odborné péče
ÚVOD
Tuberózní skleróza je po neurofibromatóze prvního typu druhým nejčastějším neurokutánním syndromem. Klinicky se projevuje především epilepsií, mentální retardací a kožními projevy, současně bývají postiženy další orgánové systémy. K vyjádření multisystémového charakteru onemocnění se v současné době používá název komplex tuberózní sklerózy (Tuberous Sclerosis Complex, TSC) [4, 7].
První popisy kožních projevů tuberózní sklerózy se objevují v literatuře již v 19. století. V atlasu francouzského dermatologa Pierre-Olive Françoise Rayera z roku 1835 lze nalézt ilustraci angiofibromů v obličeji mladého muže (obr. 1). Friedrich Daniel von Recklinghausen publikoval v roce 1862 popis patologického obrazu novorozence s nádory srdce („myomaty“) a ložisky sklerózy v mozku. Désiré-Magloire Bourneville v roce 1880 spojuje nález mentální retardace a epileptických záchvatů s makroskopickým popisem „tuberózních změn mozkových závitů“ [30] a jako první vyslovil název tuberózní skleróza. Na konci 19. století se věnoval tuberózní skleróze také anglický kožní lékař John James Pringle, v literatuře se proto můžeme setkat s historickými názvy tuberózní sklerózy – Bournevillova nemoc nebo Bournevillova-Pringlova nemoc či epiloia.
![Ilustrace angiofibromů v obličeji mladého muže v atlasu Pierre-Olive Françoise Rayera z roku 1835 [20]](https://www.prelekara.sk/media/cache/resolve/media_object_image_large/media/image/b70d01a34ef7b392ebba099056945e3b.jpg)
Do 80. let 20. století byla tuberózní skleróza poddiagnostikována, byla popisována incidence 1 : 100 000 až 1 : 200 000 obyvatel, méně závažné případy nebyly odhaleny [16]. Podle dnešních literárních pramenů se tuberózní skleróza vyskytuje celosvětově s frekvencí 1 : 6 000 až 10 000 obyvatel, postihuje všechny rasy a obě pohlaví stejně často [30]. V České republice žije přibližně 1 000 až 1 500 osob s TSC, na celém světě je postiženo přibližně 1,5 milionu obyvatel [31].
ETIOPATOGENEZE
Tuberózní skleróza je onemocnění s autozomálně dominantním typem dědičnosti, vznikající na podkladě mutace tumor-supresorového genu. Pozitivní rodinná anamnéza je však pouze u jedné třetiny případů, zbývajících 60–80 % případů vzniká na podkladě nově vzniklé mutace.
TSC může být způsobena mutací 2 různých genů, podle nichž se dělí na onemocnění TSC1 a TSC2. Gen TSC1 o délce 45 kb se nachází na 9. chromozomu (9q34.3), kóduje protein hamartin. Gen TSC2 je lokalizován na 16. chromozomu (16p13.3), jeho délka je rovněž 45 kb a kóduje protein tuberin. Gen TSC2 leží v sousedství PKD1 genu, zodpovědného za polycystickou chorobu ledvin [29]. Celkem bylo popsáno více než 1500 mutací genů TSC1 a TSC2 s náhodnou distribucí v zodpovědných genech. Asi 30 % mutací bylo popsánu v genu TSC1, většinou charakteru bodových mutací vedoucích ke zkrácení kódovaného proteinu. V genu TSC2 bylo identifikováno 70 % mutací, tyto mutace zahrnovaly mnoho rozsáhlých přestaveb genu, velkých delecí a záměnových mutací [29]. Onemocnění TSC2 je častější, vyskytuje se u dvou třetin nemocných tuberózní sklerózou. U sporadických případů jsou častěji nalézány mutace v genu TSC2. Minimálně 1 % případů onemocnění TSC vzniká na podkladě somatického mozaicismu v genu TSC1 nebo TSC2, v literatuře byly popsány případy germinální mozaiky v rodinách zdravých rodičů s dvěma a více dětmi s tuberózní sklerózou [15].
Hamartin a tuberin jsou bílkoviny, které spolu tvoří nitrobuněčný komplex hamartin-tuberin regulující buněčný růst, proliferaci a diferenciaci buněk především cestou kinázy mTOR (mammalian target of rapamycin), ale také ovlivněním dalších metabolických cest [18]. Porucha hamartinu nebo tuberinu zabrání účinku celého komplexu hamartin-tuberin s následným vznikem mnohočetných hamartomů a benigních nádorů.
Na základě dosavadních poznatků nelze rozlišit fenotypy TSC1 a TSC2 na základě klinického vyšetření, podle klinických studií je u TSC2 popisován závažnější klinický průběh onemocnění [18], zvýšené riziko výskytu renálních projevů TSC, ale nižší výskyt mentální retardace.
KLINICKÝ OBRAZ
Klinický obraz TSC je velmi variabilní. Extrémní variabilita exprese je částečně dána typem mutace a náhodným výskytem mutace druhé alely tumor-supresoru [27]. Rozdílná exprese je pozorována u pacientů z různých rodin, ale i mezi postiženými příbuznými téže rodiny [27, 29]. Spektrum projevů TSC se u různých pacientů pohybuje od bezpříznakových kožních projevů až po těžké poškození CNS s epilepsií, psychomotorickou retardací a mnohočetnými orgánovými projevy včetně život ohrožujících komplikací tuberózní sklerózy.
Klinický obraz se vyvíjí v závislosti na věku pacienta. U novorozence mohou být přítomny rhabdomyomy srdečního svalu a subependymální noduly, v kojeneckém a batolecím věku se často objevuje těžká, na terapii rezistentní epilepsie a subependymální obrovskobuněčný astrocytom. Postižení ledvin se objevuje v adolescenci a dospělém věku, u žen středního věku se objevuje závažné postižení plic lymfangioleiomyomatózou. Typickou manifestaci projevů TSC v závislosti na věku zobrazuje graf 1.

KOŽNÍ PROJEVY
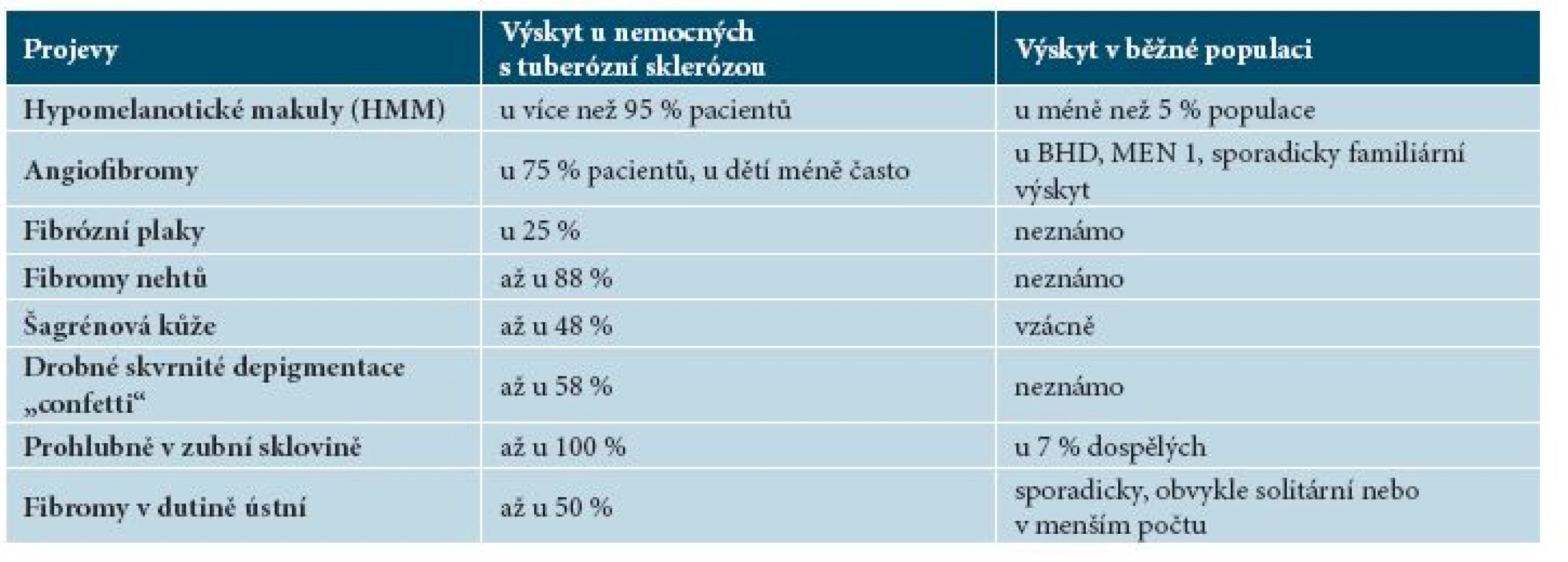
Kožní projevy TSC se nachází takřka u 100 % pacientů. Objevují se již v prvních měsících života, v dalších letech přibývají. Tabulka 1 uvádí přehled kožních projevů tuberózní sklerózy.
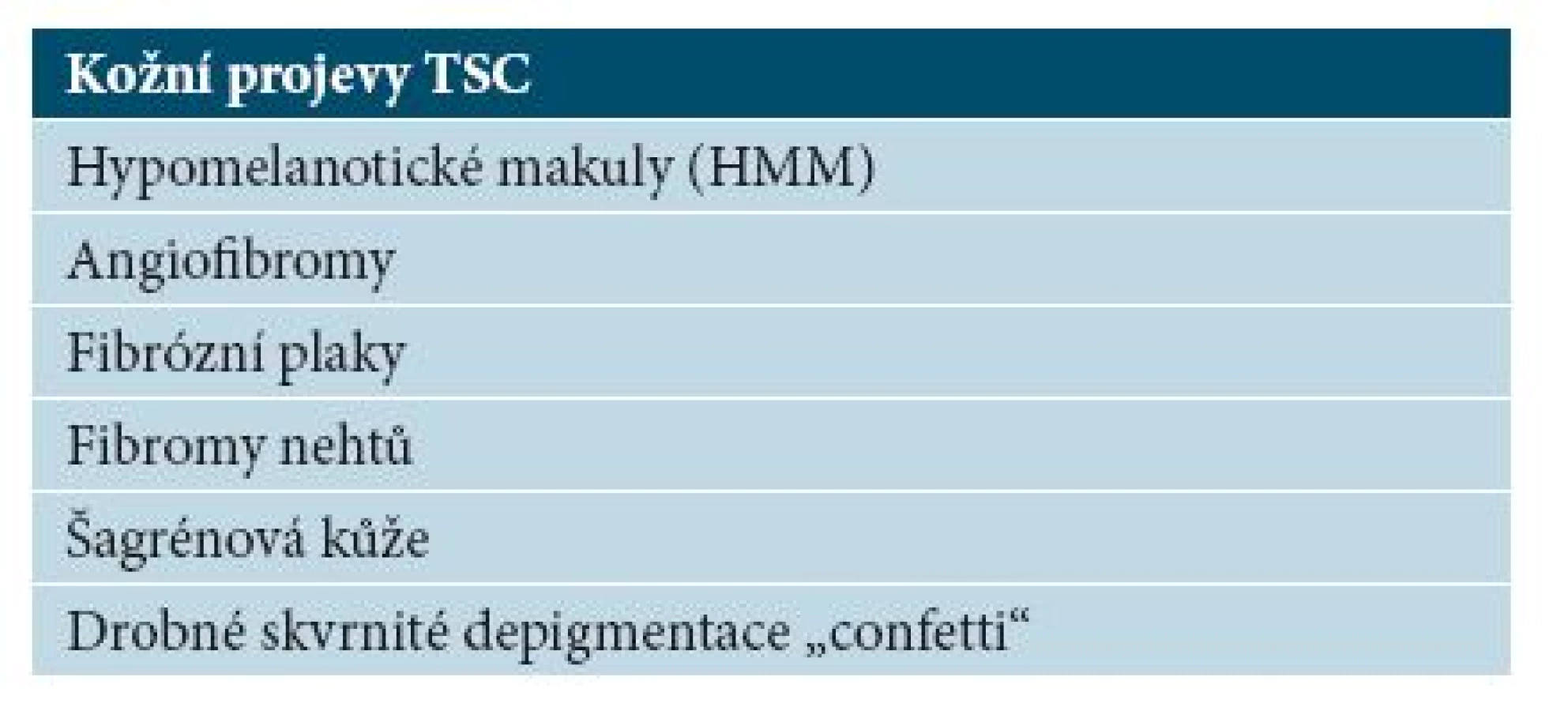
Hypomelanotické makuly (HMM)
Jsou nejčastějším kožním nálezem u tuberózní sklerózy, vyskytují se již u více než 90 % dětí mladších 5 let [9]. Jedná se o nejčasnější klinicky zjevný příznak nemoci, přítomný často již při narození nebo v kojeneckém věku. S věkem se počet makul zvyšuje, u zdravých jedinců bez TSC mohou být přítomny až dvě hypomelanotické makuly [16]. Jedná se o ostře ohraničené depigmentované makuly oválného tvaru, které mají vzhled jasanového listu (tzv. ash-leaf spot), mohou však být i polygonální či prstovitého tvaru [9]. Makuly mohou mít velikost až několik centimetrů (obr. 2), diagnosticky významné jsou makuly velikosti alespoň 5 mm [16]. V hypomelanotických makulách jsou melanocyty přítomny v obvyklém množství, ale mají méně dendritických výběžků, menší a méně četné melanozomy [6]. HMM se vyskytují nejvíce na trupu a na horních končetinách. U nemocných TSC se vyskytují také ostře ohraničená ložiska bílých vlasů ve kštici (poliosis). Ložiska poliózy se v rámci diagnostických kritérií započítávají do součtu hypomelanotických makul [16].
Angiofibromy
Angiofibromy jsou dalším častým nálezem u TSC, v minulosti byly označovány jako adenoma sebaceum. Prevalence angiofibromů stoupá s věkem, podle studií jsou přítomny až u 75 % dětí nad 9 let [9]. Časné léze se objevují ve věku 1–5 let ve formě purpurových makul uložených symetricky ve střední části obličeje, mohou připomínat pavoučkové névy. Později dochází ke zmnožení fibrózní složky a vyklenování projevů se vznikem ostře ohraničených papul barvy kůže až červenohnědé. Projevy se postupně zvětšují, přibývají a mohou splývat do rozsáhlých klinicky nápadných ložisek zejména v oblasti tváří, nazolabiální rýhy a na bradě (obr. 3), zpravidla bývá ušetřena oblast horního rtu a laterální části obličeje [6].

Histologický obraz angiofibromů je charakteristický, v dermis se nachází objemné fibroblasty s ostnitými výběžky a perifolikulární koncentrické uspořádání fibrózy připomínající vrstvení cibule [6].
Podle nejnovějších diagnostických kritérií musí být přítomny alespoň 3 angiofibromy v obličeji, neboť jeden nebo dva izolované projevy mohou být přítomny u zdravého jedince, avšak i v těchto případech je nutné na diagnózu TSC pomýšlet.
Mnohočetné angiofibromy objevující se v dětství jsou charakteristické pro TSC, splňují hlavní kritéria TSC [9]. Pokud se angiofibromy objevují až v dospělosti, jsou hodnoceny jako vedlejší kritérium.
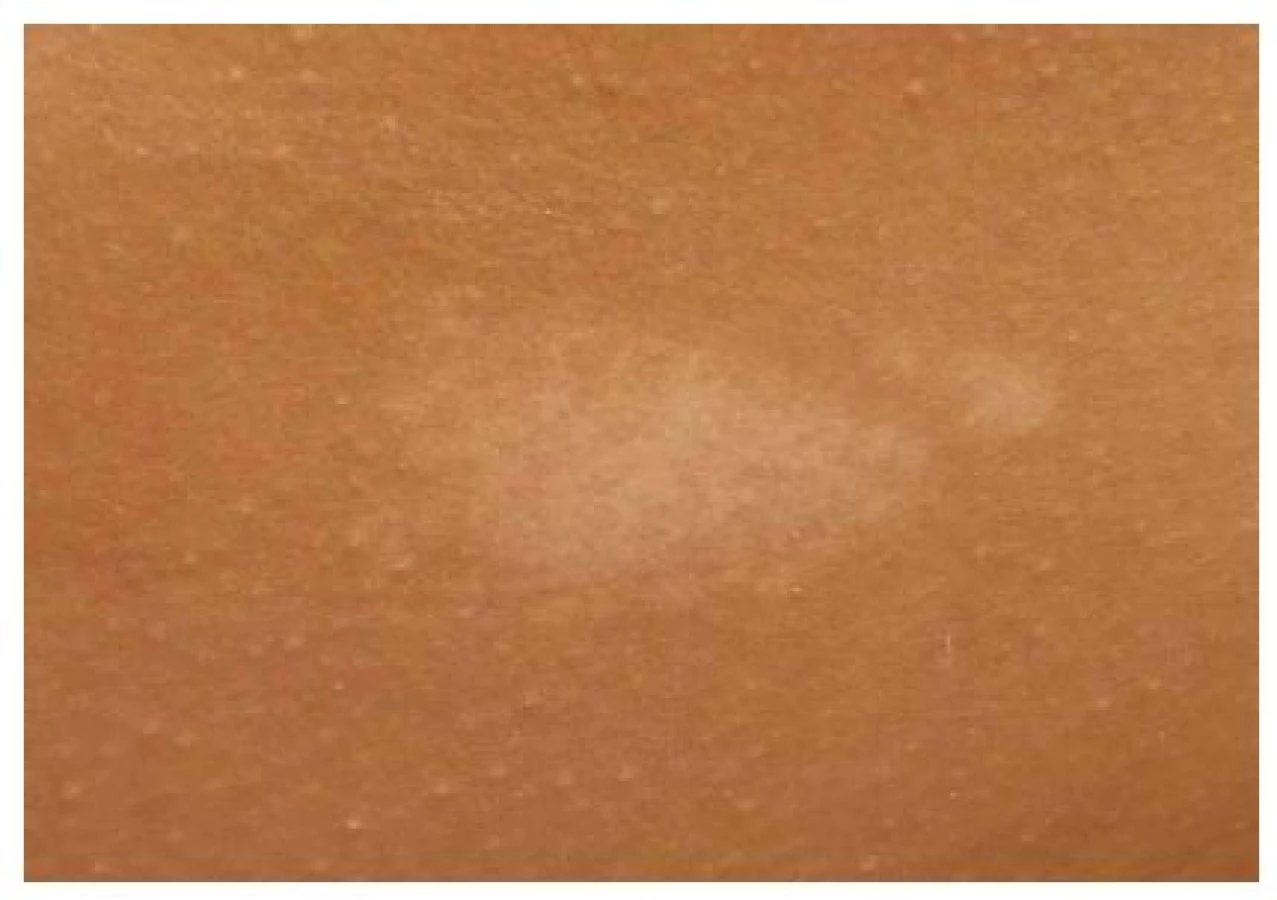
Fibrózní plaky
Jedná se o solitární či mnohočetné ostře ohraničené plaky barvy kůže nebo růžovohnědé, s jemně hrbolatým povrchem (obr. 4). Vyskytují se kdekoliv v obličeji, často jednostranně na čele, v oblasti očních víček, podél hranice vlasů, v obočí nebo ve kštici. Histologicky se jedná o plošné angiofibromy. Jsou pozorovány asi u 25 % pacientů s TSC, přítomnost jednoho fibrózního plaku splňuje hlavní kritérium TSC [9].
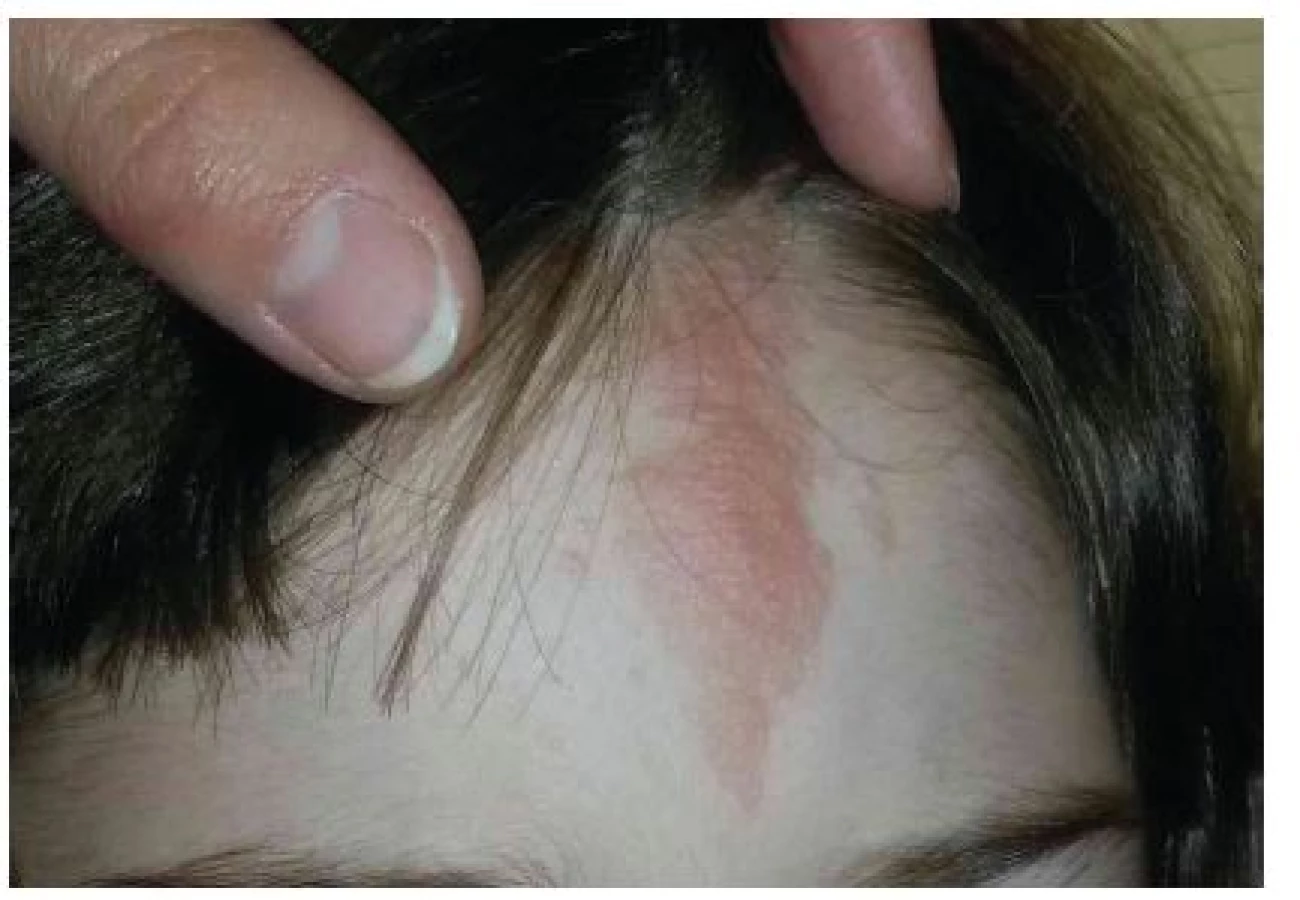
Fibromy nehtů
Fibromy nehtů, v minulosti označované jako Koenenovy tumory, se obvykle objevují v pubertě nebo v časné dospělosti, u dospělých s TSC nad 30 let věku se vyskytují až u 88 % [9]. Tyto výrůstky barvy kůže nebo žlutočervené barvy se vyskytují v bezprostředním okolí nehtu nebo pod nehtovou ploténkou, která bývá často postižena podélným rýhováním, třepením až úplnou destrukcí (obr. 5). Nejčastěji vznikají v okolí nehtů dolních končetin, kde bývají vícečetné, bolestivé a často krvácejí. V běžné populaci zpravidla vznikají fibromy nehtů v návaznosti na trauma a jsou obvykle solitární. U TSC se objevují mnohočetné fibromy i bez anamnézy předchozího traumatu [16]. Ke splnění hlavního diagnostického kritéria TSC je potřebná přítomnost 2 nebo více fibromů.
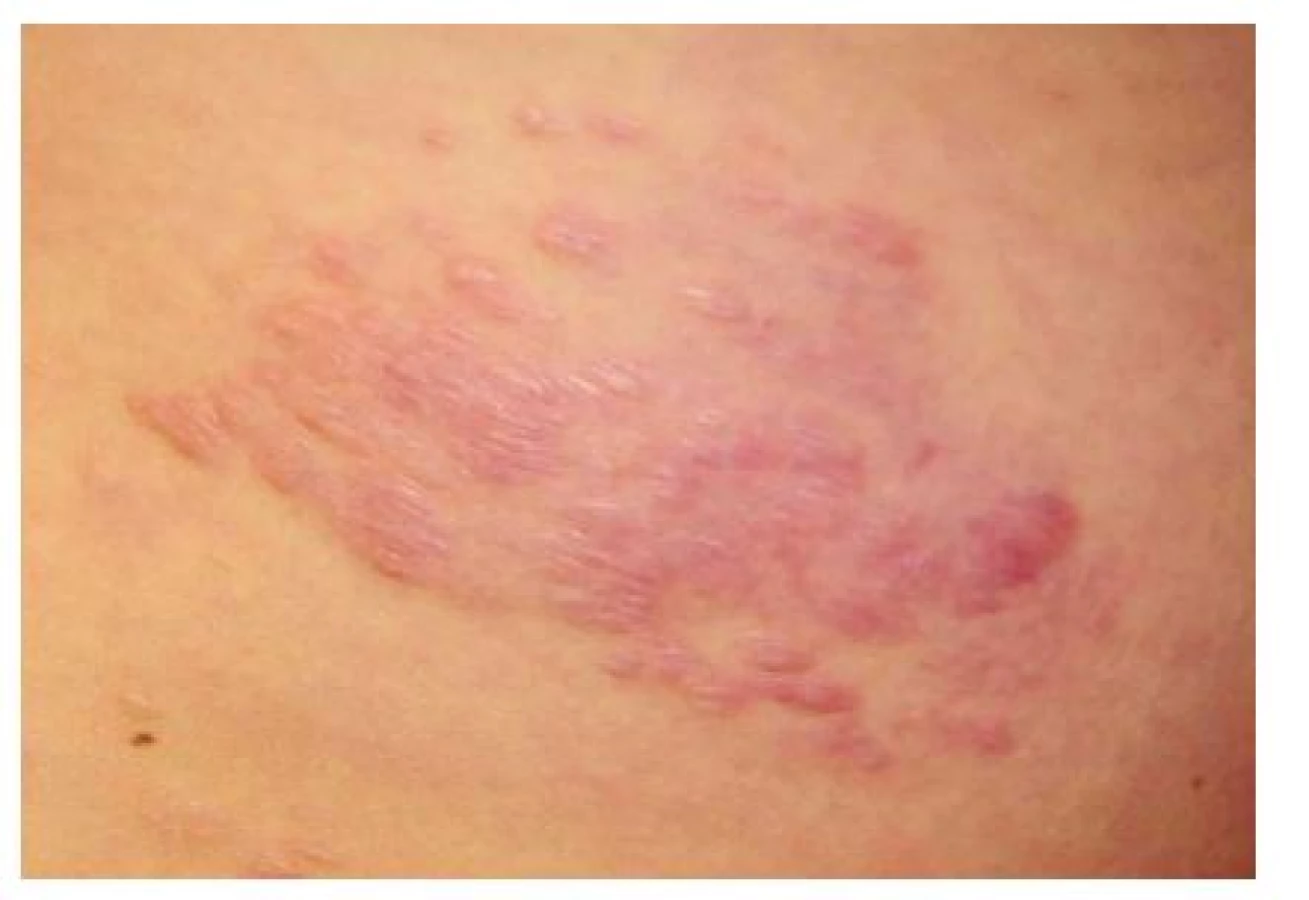
Šagrénová kůže (shagreen patch)
Vyskytuje se až u 50 % nemocných TSC, obvykle se objevuje u dětí v prvních 10 letech života [16]. Jedná se o solitární ostře ohraničené ložisko velikosti dlaně zpravidla v lumbosakrální oblasti (obr. 6). Povrch ložiska bývá nerovný, obvykle barvy kůže nebo červený, díky vkleslým folikulárním ústím připomíná vzhled pomerančové kůry. Histopatologický nález odpovídá kolagenomu [9]. Nález šagrénové kůže v lumbosakrální oblasti splňuje hlavní diagnostické kritérium.
Drobné skvrnité depigmentace
Mnohočetné ostře ohraničené drobné skvrnité depigmentace velikosti 1–3 mm bez šupin (obr. 7) jsou označovány jako „confetti“ (confetti skin lesions). Bývají lokalizované zejména na končetinách, jejich výskyt je udáván u 2,8 % dětí a až u 58 % všech nemocných tuberózní sklerózou [9, 16]. Přítomnost skvrn confetti splňuje vedlejší kritérium TSC.

Skvrny cafe au lait
Nachází se u 7–16 % nemocných TSC, tato incidence není zvýšena oproti běžné populaci [16]. Nález skvrn cafe au lait nepatří mezi diagnostická kritéria TSC.
MIMOKOŽNÍ PROJEVY TUBERÓZNÍ SKLERÓZY
U TSC je popisována široká paleta klinických příznaků postihujících řadu orgánových systémů. Tabulka 2 uvádí přehled mimokožních projevů TSC.
Projevy v dutině ústní
Prohlubně v zubní sklovině (dental enamel pits)
Jedná se o drobné prohlubně v zubní sklovině velikosti do 1 mm. Vyskytují se u nemocných TSC až ve 100 %, zatímco v běžné populaci pouze u 7 % obyvatel [9, 16]. Podle aktualizovaných doporučení musí být přítomny aspoň 3 prohlubně ke splnění vedlejšího diagnostického kritéria TSC.
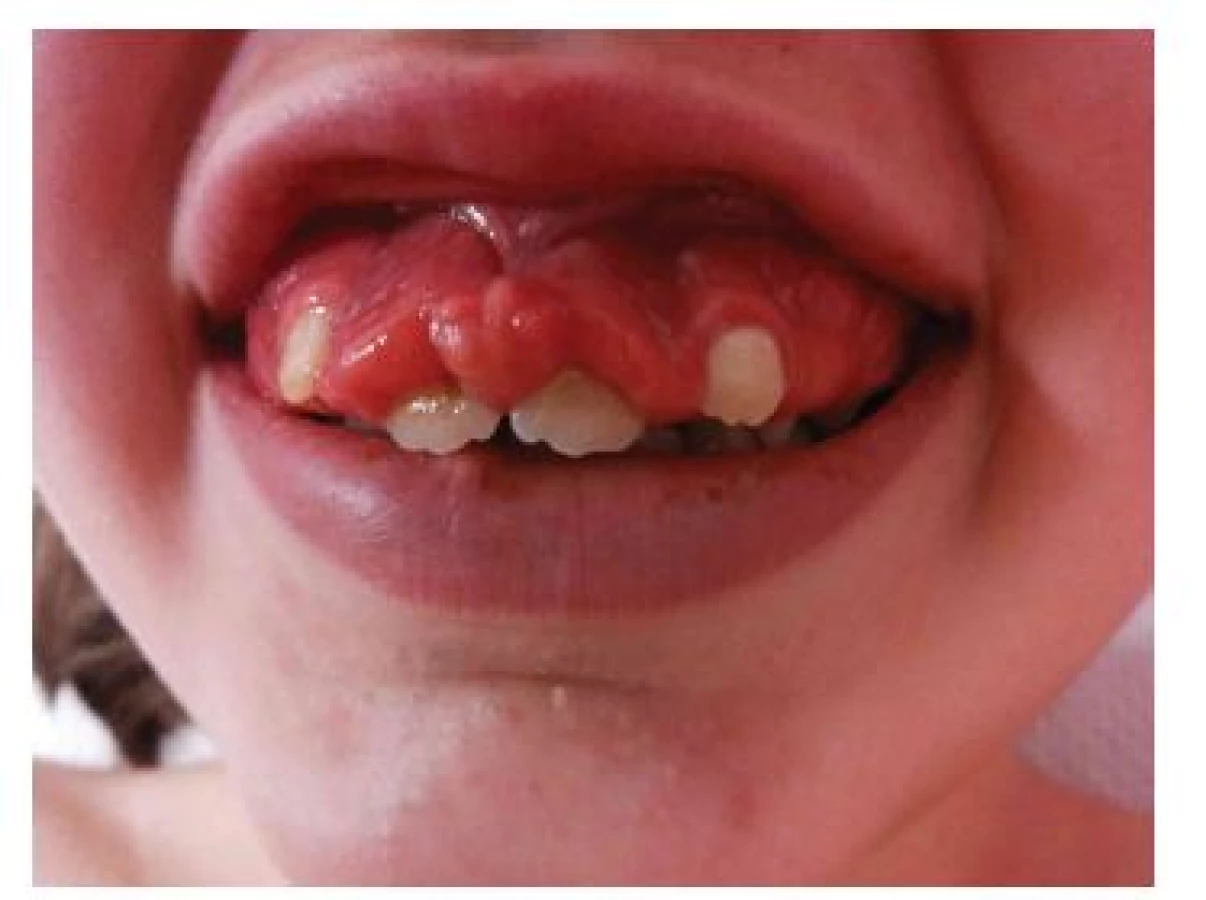
Fibromy v dutině ústní
Fibromy v dutině ústní jsou solitární nebo mnohočetné papuly až noduly barvy okolní dásně nebo načervenalé barvy (obr. 8). Fibromy dásní se vyskytují u 20 až 50 % nemocných TSC, častěji u dospělých [9, 16]. Ve starších diagnostických kritériích byly hodnoceny jako vedlejší diagnostické kritérium. Nová doporučení rozšířila toto kritérium o fibromy v kterékoliv intraorální lokalizaci, včetně bukální a labiální sliznice a jazyka. Solitární posttraumatické fibromy v dutině ústní jsou poměrně častým nálezem v běžné populaci, pro splnění diagnostického kritéria TSC je třeba přítomnost 2 nebo více fibromů [9, 16].
Oční projevy
Až u 50 % nemocných TSC se vyskytují hamartomy v oblasti sítnice, hyalinní nebo cystické noduly, přesuny pigmentu a achromatické skvrny na sítnici.
Mnohočetné retinální hamartomy (multiple retinal hamartomas)
Nachází se u 30–50 % pacientů s tuberózní sklerózou, prevalence u osob bez TSC není známa. Tyto hamartomy jsou obvykle vícečetné, mají podobný histopatologický obraz jako tubery CNS u tuberózní sklerózy. Retinální hamartomy obvykle nezpůsobují poruchy vidění, jsou časným příznakem onemocnění TSC u malých dětí, u kterých dosud nejsou přítomny další projevy tuberózní sklerózy [16]. Nález více než jednoho hamartomu sítnice je signifikantním nálezem TSC klasifikovaným jako hlavní kritérium.
Achromatické skvrny na sítnici (retinal achromic patch)
Jedná se o ložiskové hypopigmentace sítnice. Nachází se u 39 % pacientů s TSC, v celkové populaci se nachází u 1 z 20 000 obyvatel. Jsou vedlejším diagnostickým kritériem TSC [16].
Rhabdomyomy srdce (cardiac rhabdomyoma, CR)
Jedná se o benigní nádor srdečního svalu, nejčastěji je lokalizován v oblasti septa mezi srdečními komorami (obr. 9). Rhabdomyomy srdce jsou často prvním příznakem nemoci. Mohou být nalezeny již při prenatálním ultrazvukovém vyšetření srdce prováděném v rámci screeningu vrozených vývojových vad ve 20. týdnu těhotenství. Rhabdomyomy zpravidla spontánně regredují v dětském věku, někdy zcela vymizí do 2–3 let věku, byly však popsány případy opětovného zvětšení rhabdomyomů v období puberty s následnou regresí. Pouze u 4 % nemocných TSC rhabdomyomy rostly, nebo se objevily během života de novo [1].
![Objemný rhabdomyom (*) vycházející ze septa levé komory (LV) a další mnohočetné hyperechogenní rhabdomyomy ve stěně a septu pravé komory (RV) a úponu mitrální chlopně na echokardiografickém vyšetření pacienta s tuberózní sklerózou LA – levá síň, AO – aorta [18].](https://www.prelekara.sk/media/cache/resolve/media_object_image_large/media/image/f19d793bfe7c1724878bd0e9bafb2dc8.jpg)
CR jsou většinou klinicky němé, mohou ale způsobit již prenatálně arytmie, hydrops plodu nebo jeho úmrtí [14]. Závažné hemodynamické obtíže včetně obstrukce krevního průtoku srdcem a městnavé srdeční slabosti vyžadující kardiochirurgický výkon v novorozeneckém věku se vyskytují asi u 10 % dětí. Elektrofyziologické abnormality (síňové a komorové arytmie, WPW syndrom) zpravidla přetrvávají i po vymizení rhabdomyomů.
Rhabdomyomy srdce se vyskytují až u 66 % všech nemocných s TSC, v běžné populaci jsou velmi vzácné (výskyt < 0,1 %) [16]. Nález rhabdomyomů srdce je vysoce specifický pro TSC, prenatální nález solitárního CR představuje 75–80% riziko TSC [16], u nálezu mnohočetných CR je riziko TSC ještě vyšší [18].
Nález rhabdomyomu u plodu musí vést k vyšetření novorozence dětským kardiologem a klinickým genetikem. Se souhlasem rodičů má být provedena molekulární analýza, která může odhalit kauzální mutaci v genu TSC1 nebo TSC2.
Plicní nálezy
Nejčastějším plicním nádorem u nemocných TSC je lymfangioleiomyomatóza plic, mezi méně časté plicní nálezy patří multifokální mikronodulární pneumocytová hyperplazie (MMPH) a vzácný plicní nádor z jasných buněk (clear cell tumor of the lung, CCSTL) [16].
Lymfangioleiomyomatóza plic (LAM)
Vyskytuje se téměř výhradně u dospělých žen s TSC2, výskyt s věkem stoupá. Histologicky se jedná o intersticiální plicní proces s abnormální proliferací buněk hladkého svalstva, vedoucí k destrukci plicní tkáně. Patofyziologický podíl na postižení plic mají kromě mTOR signální dráhy pravděpodobně také estrogeny. U 40 % premenopauzálních žen s tuberózní sklerózou se nachází rentgenologické změny na plicích, ale jen 5 % z nich má klinicky významné příznaky [30]. LAM se projevuje obvykle ve 3.–4. dekádě progredující dušností a recidivujícími pneumothoraxy, chylothoraxy z obstrukce lymfatických cest, kritická respirační nedostatečnost v ojedinělých případech vyžaduje transplantaci plic. U mužů je LAM zpravidla asymptomatická, pouze 10–12 % z nich má klinické obtíže. Přítomnost LAM patří mezi hlavní diagnostická kritéria TSC.
Lymfangioleiomyomatóza se vyskytuje i u pacientů bez TSC, bývá označována jako sporadická LAM (S-LAM), obvykle mívá závažnější průběh než u nemocných s TSC [16].
Neurologické a neuropsychiatrické nálezy
Morfologickými projevy tuberózní sklerózy v mozku jsou kortikální dysplazie (kortiko-subkortikální tubery a radiální migrační dráhy v bílé hmotě), subependymální noduly a subependymální obrovskobuněčné astrocytomy. Klinickými projevy TSC jsou epilepsie, mentální retardace a TSC asociované neuropsychiatrické poruchy (TAND).
Kortikální dysplazie
Jedná se o kongenitální abnormality mozkové tkáně, způsobené poruchou proliferace, růstu a migrace zárodečných buněk neurální lišty v časném embryonálním období. Podle morfologie rozlišujeme kortiko-subkortikální tubery a radiální migrační dráhy.
Kortiko-subkortikální tubery jsou solitární nebo vícečetná ložiska mozkové tkáně s abnormální strukturou. Mohou být lokalizovány v kůře kterékoliv části mozkových hemisfér, nejčastěji se nachází ve frontálních lalocích, nejvýraznější ložiska bývají v parietálních oblastech (obr. 10), počet tuberů u pacienta kolísá mezi 5–50 [10]. Skleroticky změněné oblasti vykazují změny tvaru a velikosti gliových buněk i neuronů. Dysplastické neurony mají chudé větvení dendritů a abnormální tvar těla, atypické gliové buňky jsou obrovské. Kortikální tubery jsou pozorovány u 90 % pacientů s TSC, podle nich je pojmenováno onemocnění tuberózní sklerózou.
Radiální migrační dráhy v bílé hmotě vznikají obdobným patologickým procesem. Jedná se o ostrůvky heterotopních buněk v bílé hmotě, které z neurální lišty nedocestovaly do cílového místa mozkové kůry. U TSC bývají často současně přítomny tubery i radiální migrační dráhy [16].
Oba typy kortikální dysplazie u TSC jsou často asociovány s farmakorezistentní epilepsií a poruchami učení. Jedno nebo více ložisek kortikální dysplazie mohou být nalezena také bez TSC, podle diagnostických kritérií je mnohočetný nález ložiskové kortikální dysplazie pouze jedním hlavním diagnostickým kritériem, ke stanovení definitivní diagnózy TSC je nezbytná přítomnost dalších klinických příznaků [16].
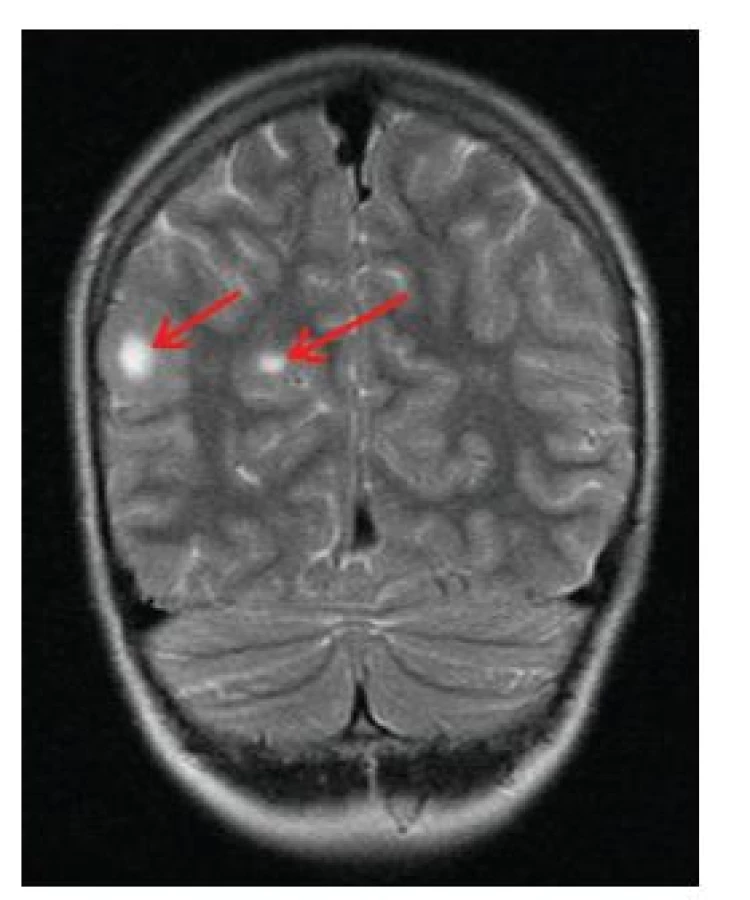
Subependymální noduly (SEN)
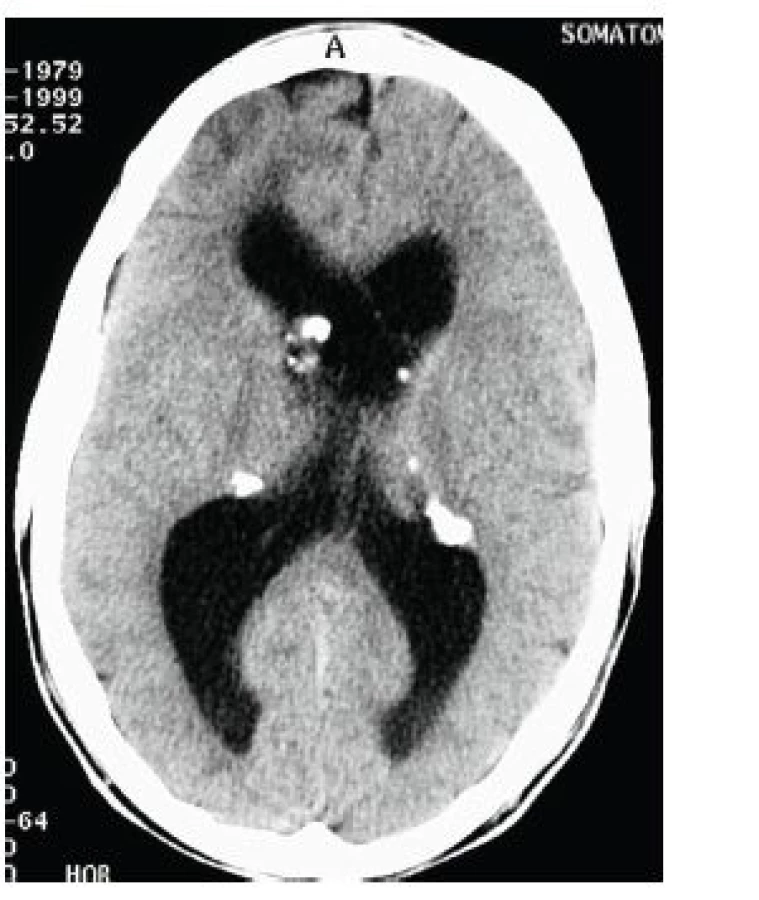
Jedná se o mnohočetné benigní drobné hamartomy nacházející se pod ependymální výstelkou mozkových komor, nejčastěji ve stěně postranních komor a ve foramen Monroi (obr. 11). Častým nálezem jsou kalcifikace subependymálních nodulů, díky nimž jsou SEN velmi dobře patrny i na CT snímku mozku (obr. 12). Jejich počet stoupá s věkem, jsou často prokazatelné již prenatálně nebo při narození, popisovány jsou až u 80 % nemocných TSC [16].
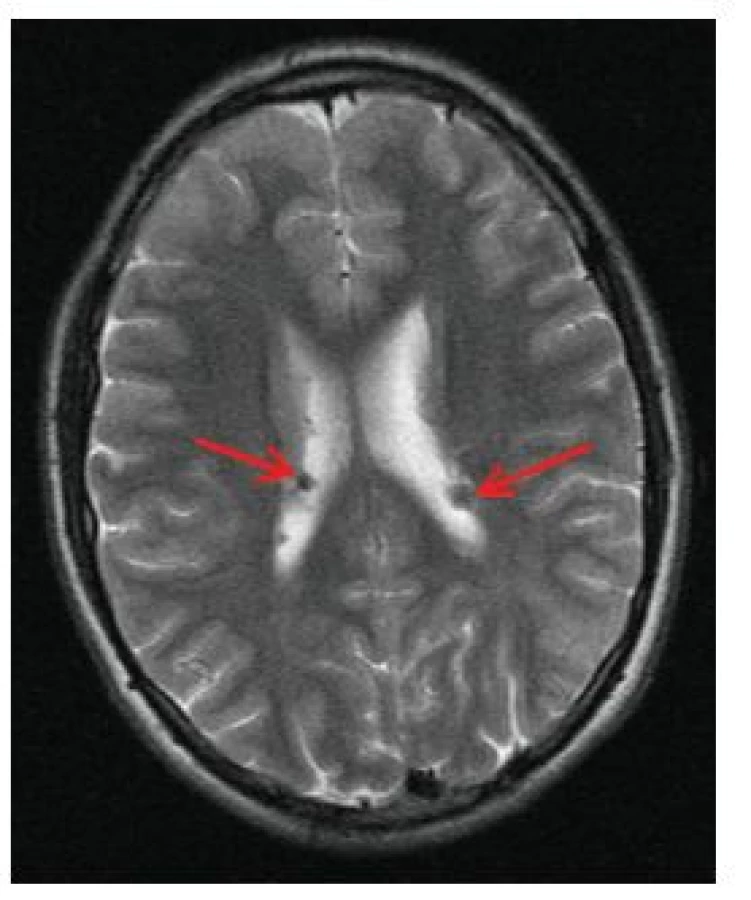
Subependymální obrovskobuněčné astrocytomy (SEGA)
Jsou benigními nádory vyrůstajícími ze subependymálních nodulů (SEN), zejména při foramen Monroi, mohou být v komorovém systému i vícečetné. Jsou popisovány až u 20 % nemocných TSC [30]. SEGA mohou být přítomny již prenatálně nebo při narození, typicky rostou v prvních 2 dekádách života. Nová manifestace SEGA po 20. roce života je výjimečná. Ačkoliv jsou SEGA obvykle pomalu rostoucími nádory, mohou způsobit závažné neurologické komplikace včetně klinického obrazu nitrolební hypertenze na podkladě obstrukčního hydrocefalu. Nálezy SEN a SEGA jsou hlavními diagnostickými kritérii TSC, histologicky jsou tyto léze podobné, relativně specifické pro TSC.
Epilepsie
Epilepsie se vyskytuje až u 90 % nemocných tuberózní sklerózou [7, 18]. U řady nemocných TSC je epileptický záchvat prvním projevem nemoci, který se přibližně v 70 % případů manifestuje již v průběhu prvního roku života [18]. U starších dětí a adolescentů frekvence záchvatů klesá, nebo zcela vymizí [7]. Vyskytují se všechny typy epileptických záchvatů, až u jedné třetiny nemocných mají epileptické záchvaty charakter infantilních spasmů (tzv. bleskové křeče). Současný výskyt infantilních spasmů, zpomalení psychomotorického vývoje a abnormit na EEG se označuje jako Westův syndrom. Dále se vyskytují parciální simplexní a parciální komplexní paroxysmy a atypické absence. U pacientů s tuberózní sklerózou byla pozorována korelace mezi časným výskytem epileptických záchvatů, stupněm abnormit na EEG a mentální retardací [7].
Mentální retardace
Vyskytuje se asi u poloviny pacientů s tuberózní sklerózou, stupeň výrazně kolísá. Zatímco jedna třetina pacientů má normální inteligenční kvocient (IQ), jedna třetina má těžkou mentální retardaci. Vývoj řeči bývá obvykle opožděn v expresivní i percepční složce.
TSC asociované neuropsychiatrické poruchy (TSC associated neuropsychiatric disorders, TAND)
Klinickou manifestací morfologických a funkčních změn mozkové tkáně u tuberózní sklerózy je kromě epilepsie a mentální retardace agresivní chování, až u 30 % nemocných jsou přítomny poruchy autistického spektra (ASD), poruchy učení, hyperaktivita s poruchou pozornosti (Attention-Deficit Hyperactivity Disorder, ADHD syndrom), poruchy spánku, depresivní a úzkostné poruchy [10].
Centrální poruchy motoriky
Při neurologickém vyšetření je u většiny nemocných TSC chudý neurologický nález. Může se vyskytovat spastická hemiparéza, kvadruparéza nebo generalizovaná hypotonie s hyporeflexí. V některých zdrojích jsou uváděny též nálezy monoparézy či paraparézy a zřídka přítomnost dyskinézy a mozečkových příznaků [7].
Syndrom nitrolební hypertenze (ICH)
Většinou je způsoben obstrukčním hydrocefalem v důsledku přítomnosti SEGA, u řady pacientů může být ICH první klinickou manifestací TSC [7]. Vzestup nitrolebního tlaku se projevuje bolestmi hlavy, zvracením, poruchami zraku, při očním vyšetření je patrno městnání na očním pozadí. Při rychlém vzestupu nitrolebního tlaku se jedná o náhlý, život ohrožující stav s nutností urgentního neurochirurgického řešení [30].
Renální nálezy
Druhou nejčastější příčinou morbidity a mortality nemocných TSC je po onemocněních CNS postižení ledvin, a to zejména v dospělém věku. Postižení ledvin je přítomno až u 85 % všech nemocných tuberózní sklerózou. Nejčastějšími nálezy jsou angiomyolipomy ledvin a epiteliální patologie – epiteliální cysty, onkocytomy a renální karcinom [30].
Angiomyolipomy ledvin (AML)
Jedná se o nejběžnější, pomalu rostoucí, expanzivně se chovající benigní mezenchymové nádory ledvin složené z abnormálních krevních cév, buněk hladkého svalu a tukové tkáně (obr. 13), častým nálezem je kalcifikace. Tyto nádory se nachází u nemocných TSC nejen v ledvinném parenchymu, ale i v dalších orgánech, např. v játrech, slezině, pankreatu, ale také na vejcovodu, v pochvě a ureteru [25].
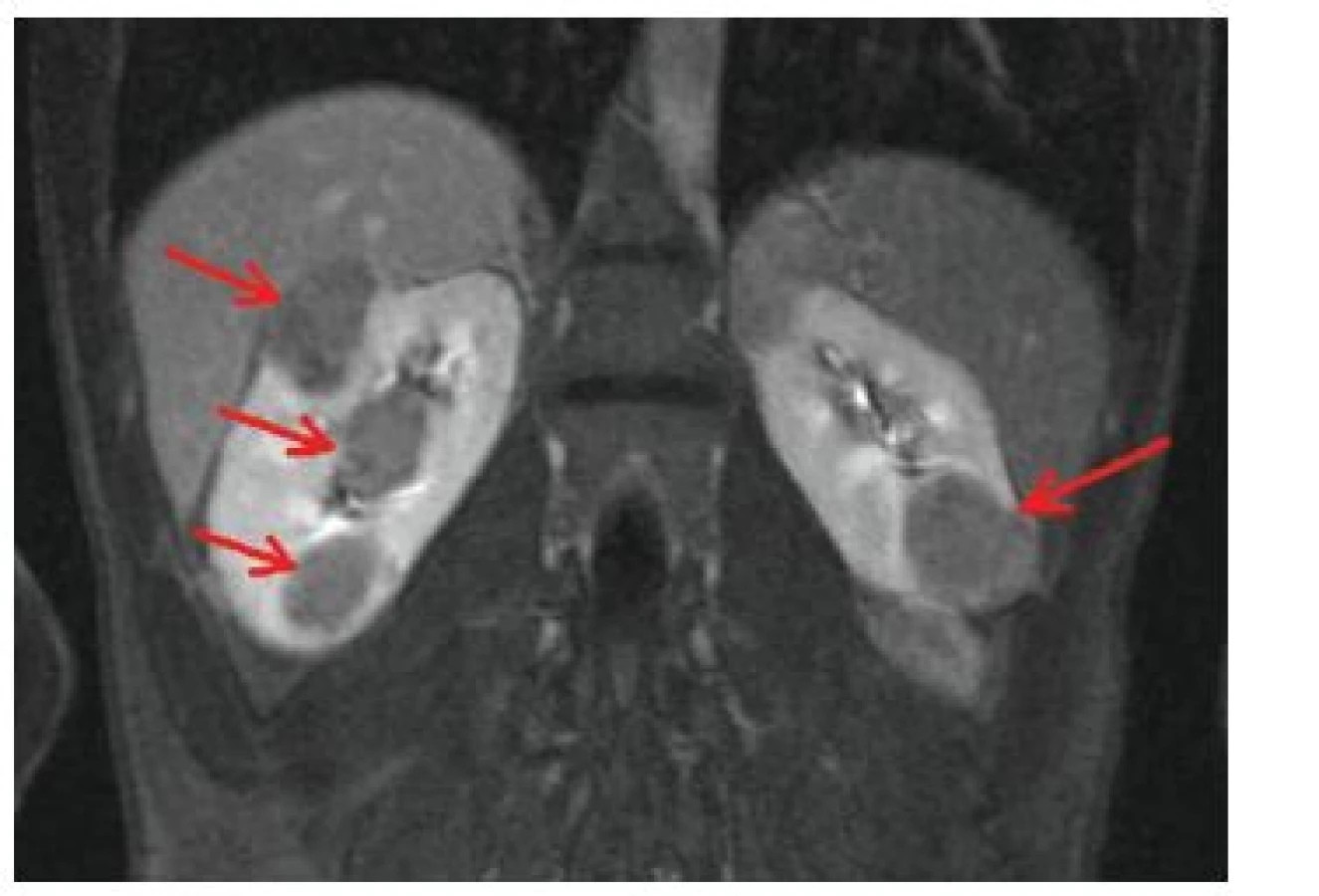
AML jsou relativně specifickým nálezem u tuberózní sklerózy. Angiomyolipomy s vysokým obsahem tuku byly pozorovány u 80 % nemocných TSC [16, 25], AML s nízkým obsahem tuku jsou také častější u pacientů s TSC, u běžné populace se vyskytují u méně než 0,1 % obyvatel. Angiomyolipomy v ledvinách u nemocných tuberózní sklerózou bývají obvykle vícečetné, bilaterální a u obou pohlaví stejně časté [18]. Objevují se od předškolního věku, byl popsán i kongenitální angiomyolipom ledvin [25]. V dětském věku jsou často asymptomatické, klinicky se manifestují většinou až v období časné dospělosti. Projevují se bolestí břicha, hematurií či hmatnou rezistencí v bederní oblasti [30]. Mohou vést kromě sekundární arteriální hypertenze také ke zhoršení funkce ledvin až k jejich selhání. Vzhledem k bohaté vaskulární složce mohou AML způsobit závažné krvácení do ledvin, močových cest a retroperitonea. Spontánní ruptura cévní složky angiomyolipomu vedoucí ke krvácení do retroperitonea postihuje až 25 % nemocných s angiomyolipomy (zejména většími než 4 cm). Rozsáhlé krvácení do retroperitonea, projevující se náhlou bolestí v břiše a rozvojem hemoragického šoku, se označuje jako Wunderlichův syndrom [11, 25].
Byly popsány případy šíření AML do venózního řečiště ledvin, renálních žil, vena cava inferior a dokonce až do pravé síně srdeční. Asi u 40 případů byla nalezena ložiska angiomyolipomů i v přilehlých uzlinách.
Poněkud vzácnější komplikací AML je útlakový syndrom, kdy tumory dosahují takové velikosti, že tlakem na splanchnickou oblast pacient není schopen přijímat potravu v běžném množství [17].
Maligní transformace do obrazu maligního epiteloidního AML je vzácná. Incidence renálního karcinomu nepřevyšuje 2 % všech případů TSC, avšak věk pacientů v okamžiku stanovení diagnózy je významně nižší než u sporadických případů (věkový průměr 28 let) [30].
Nález 2 a více angiomyolipomů ledvin je hlavním diagnostickým kritériem TSC (v minulosti byly v diagnostických kritériích uvedeny „renální angiomyolipomy“, vzhledem k možnému výskytu angiomyolipomů i v jiných orgánech bylo odstraněno slovo „renální“). Současná přítomnost renálních angiomyolipomů a LAM bez dalších příznaků TSC nesplňuje kritéria definitivní diagnózy TSC [16].
Mnohočetné cysty ledvin
V běžné populaci se vyskytují zřídka, častěji jsou nalézány u pacientů s TSC1 a TSC2 nebo jako projev delece postihující geny TSC2 a PKD1 [16].
Vyskytují se zejména u pacientů TSC2, jejich nález splňuje vedlejší kritérium tuberózní sklerózy. Mohou způsobit rychlé zhoršování funkce ledvin s nutností hemodialýzy či transplantace.
Endokrinní nálezy
V endokrinním systému se objevují různé druhy hamartomů. Až u 25 % nemocných tuberózní sklerózou se nachází angiomyolipomy nadledvin, které bývají velmi vzácně zdrojem krvácení [16]. Papilární adenom štítné žlázy u TSC obvykle nezpůsobuje poruchu funkce štítné žlázy. Byly popsány vzácné případy angiomyolipomů nebo fibroadenomů v hypofýze, pankreatu a gonádách. Tyto nálezy spadají do vedlejších diagnostických kritérií pod označením „nonrenální hamartomy“. U pacientů s tuberózní sklerózou byly popsány i gastrinom, feochromocytom a karcinoid [15]. Neuroendokrinní nádory nejsou hamartomy a nepatří mezi diagnostická kritéria TSC, jsou však u nemocných TSC častější než v běžné populaci [16].
Gastrointestinální nálezy
Angiomyolipomy jater a pankreatu se nachází u 10 až 25 % nemocných TSC. Tyto angiomyolipomy jsou zahrnuty do hlavních diagnostických kritérií. Rektální polypy byly odstraněny z diagnostických kritérií pro nedostatečnou specificitu pro TSC. Polypy sliznice tlustého střeva s rizikem maligního zvratu se vyskytují ve zvýšené míře u nemocných TSC v dospívání a v dospělém věku.
Maligní nádory u TSC
V literatuře je popsána řada zhoubných nádorů asociovaných s TSC – nádory varlat, žaludku, hepatocelulární karcinom, karcinom prsu, sarkomy, maligní melanom [8] a chordomy.
TUBERÓZNÍ SKLERÓZA V TĚHOTENSTVÍ
Dosud bylo v literatuře popsáno pouze několik případů těhotenství nemocných s tuberózní sklerózou. U poloviny pacientek byly popsány závažné komplikace u matky i plodu – akutní intraabdominální krvácení způsobené spontánní rupturou angiomyolipomu ledvin vedoucí k renálnímu selhání matky vyžadujícímu hemodialýzu. Byla pozorována těžká preeklampsie s patologicky zvětšenými ledvinami, u plodu závažné intrauterinní retardace růstu.
Postižení ledvin je nejvýznamnějším prognostickým faktorem zdravotních rizik v těhotenství pacientek s tuberózní sklerózou [1].
DIAGNOSTIKA
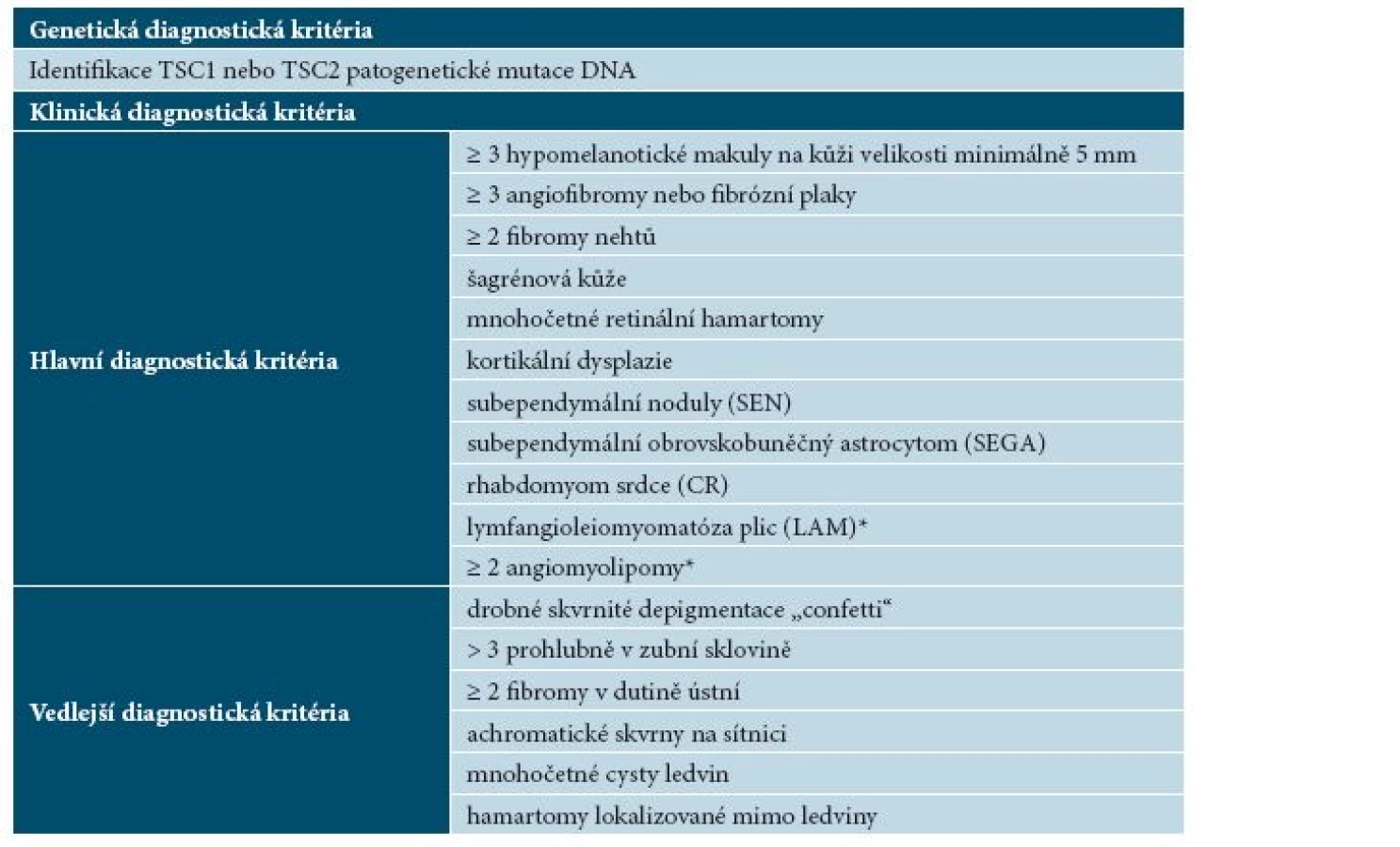
Diagnóza TSC je stanovena nejčastěji neurologem nebo dermatologem především na základě hodnocení klinického obrazu. Do 70. let dvacátého století sloužila k diagnostice TSC tzv. Vogtova triáda, tj. přítomnost křečí, psychomotorické retardace a tzv. adenoma sebaceum [30]. Diagnostická kritéria tuberózní sklerózy byla v posledních desetiletích opakovaně přepracována, poslední revize proběhla v roce 2012 na International Tuberous Sclerosis Complex Conference ve Washingtonu (tab. 3). Pro stanovení jisté diagnózy TSC je nutná přítomnost 2 nebo více hlavních nebo jednoho hlavního a 2 vedlejších kritérií, možná diagnóza TSC je při splnění jednoho hlavního nebo dvou a více vedlejších diagnostických kritérií. Kromě aktualizace hlavních a vedlejších klinických diagnostických kritérií bylo nově v roce 2012 zařazeno genetické vyšetření jako metoda vedoucí ke stanovení diagnózy TSC. Ačkoliv byly geny TSC1 a TSC2 objeveny již v roce 1998, molekulární testování tehdy nebylo široce dostupné, proto nebylo genetické vyšetření zahrnuto do starších diagnostických kritérií [16].
DIFERENCIÁLNÍ DIAGNÓZA PROJEVŮ TSC
Mnoho klinických projevů tuberózní sklerózy je nespecifických. Mohou se vyskytovat izolovaně nebo v menším počtu u zcela zdravých jedinců nebo jako projev jiného onemocnění. Řada klinických projevů je však téměř výhradním projevem TSC (tab. 4).

Kožní projevy
Hypomelanotické makuly byly pozorovány u 0,8 % novorozenců a většinou neměly klinický význam, 3 a více hypomelanotických makul bylo však pozorováno u jedinců, u kterých byla později stanovena diagnóza TSC. V diferenciální diagnostice je třeba odlišit naevus depigmentosus, naevus anaemicus, pityriasis versicolor, pozánětlivé hypopigmentace, hypomelanosis guttata idiopathica a vitiligo. Narozdíl od výše jmenovaných depigmentací je možné identifikovat HMM vyšetřením Woodovou lampou. V tabulce 5 se nachází přehled nejčastějších genodermatóz spojených s poruchami pigmentace.

Solitární obličejové angiofibromy mohou připomínat acne vulgaris, rosaceu, mnohočetné trichoepiteliomy, biopsie jednoznačně odliší tyto nálezy [16]. V diferenciální diagnostice je nutné pomýšlet na Birt-Hogg-Dubé syndrom (BHD) a mnohočetnou endokrinní neoplazii typu 1 (MEN1) [9, 16].
Diferenciálně diagnosticky je nutné odlišení šagrénové kůže od kongenitálních hamartomů hladkého svalu [7]. Malé mnohočetné kolagenomy na trupu se stejným histologickým obrazem jako u šagrénové kůže se mohou vyskytovat izolovaně nebo u dalších genetických syndromů – BHD, MEN1 a syndromu Cowdenové [16]. Tabulka 6 podává přehled výskytu névů pojivové tkáně.
![Přehled výskytu névů pojivové tkáně [21]](https://www.prelekara.sk/media/cache/resolve/media_object_image_large/media/image/5d1050c5dbd7ac1c9980171da107944c.jpg)
V diferenciální diagnostice skvrn „confetti“ je třeba odlišit vitiligo, pigmentový mozaicismus, pityriasis versicolor, pityriasis lichenoides chronica, pozánětlivé hypopigmentace, leucoderma punctatum a diseminované hypopigmentované keratózy [13]. U dospělých mohou být confetti zaměněny za idiopatickou gutátní hypomelanózu (hypopigmentace vznikající při solární expozici). K odlišení od idiopatické gutátní hypomelanózy napomůže anamnéza od dětství a asymetrie projevů na končetinách [9, 16].
Unquální fibromy je nutné odlišit od epitelových inkluzních cyst, vulgárních bradavic a infantilní digitální fibromatózy.
CNS
Kortikální tubery, subependymální noduly, subependymální obrovskobuněčné astrocytomy a radiální migrační dráhy v CNS jsou téměř výhradním projevem tuberózní sklerózy.
Ledviny
Renální cysty jsou častým nálezem v běžné populaci, vyskytují se u 1–2 %, avšak zřídka u jedinců mladších 30 let. Renální angiomyolipomy jsou vzácné nádory příležitostně objevené u jedinců bez dalších klinických potíží. Diferenciálně diagnosticky je nutné odlišení angiomyolipomu od lipomu, liposarkomu a renálního karcinomu [25].
Plíce
Některé ženy mající lymfangioleiomyomatózu a současně renální angiomyolipomy nemají žádné další klinické příznaky TSC a nesplňují tak diagnostická kritéria TSC. Sporadická lymfangioleiomyomatóza je vzácná.
Srdce
Rhabdomyom je třeba odlišit od ostatních srdečních nádorů, např. myxomu srdce, teratomu, hemangiomu a fibromu [1].
ÚLOHA DERMATOLOGA, DOPORUČENÁ VYŠETŘENÍ A DALŠÍ SLEDOVÁNÍ PACIENTŮ S TUBERÓZNÍ SKLERÓZOU
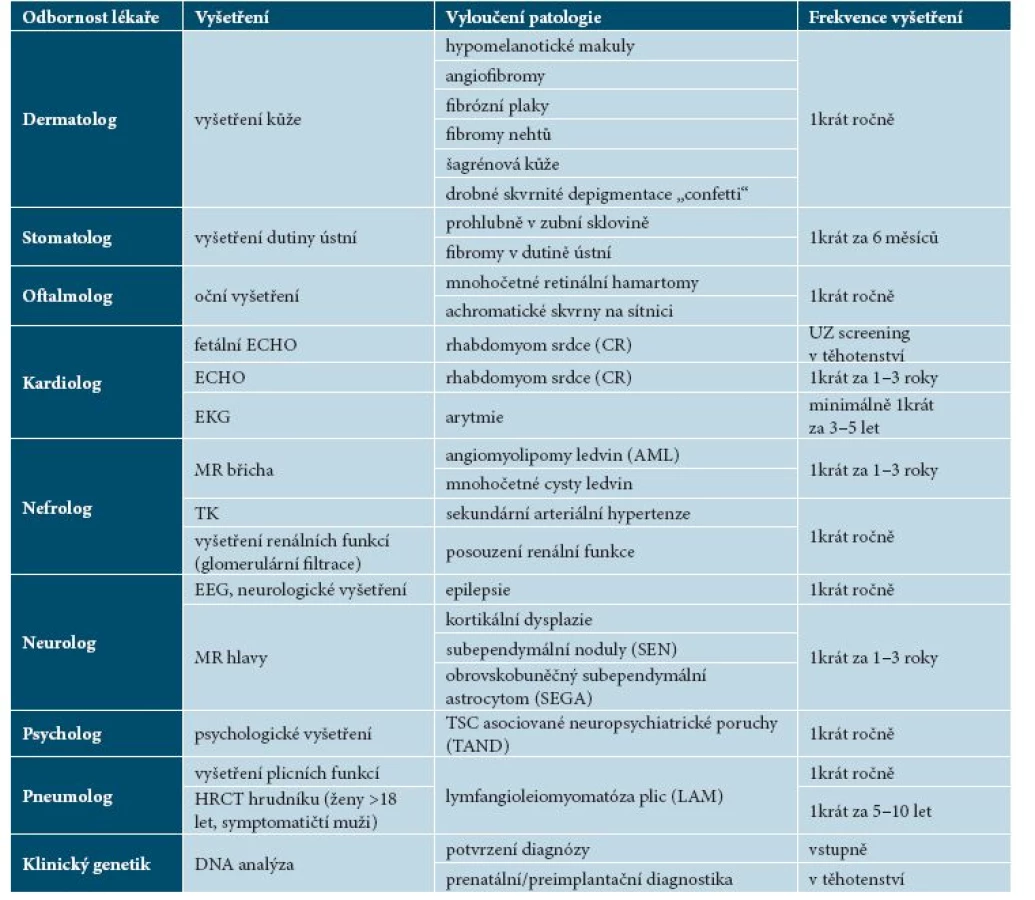
Dermatolog hraje důležitou roli v rozpoznání specifických kožních projevů tuberózní sklerózy na základě klinického vyšetření pacienta. Při podezření na onemocnění TSC odesílá pacienta v místě bydliště k vyšetření dalších projevů tuberózní sklerózy (k očnímu, neurologickému, kardiologickému, nefrologickému, plicnímu vyšetření, eventuálně ke zobrazovacím vyšetřením) a podle potřeby ke genetickému vyšetření.
Harmonogram doporučených vyšetření u nemocných tuberózní sklerózou (tab. 7) byl vypracován na základě jednání odborníků mnoha medicínských oborů na International Tuberous Sclerosis Complex Conference v roce 2012 [12].
Pacienti by měli být sledováni v místě bydliště pediatrem či obvodním lékařem a dispenzarizováni ve specializovaném centru pro vzácná onemocnění příslušnými specialisty: dermatologem, neurologem, oftalmologem, kardiologem, nefrologem, pneumologem, podle potřeby dalšími specialisty (neurochirurgem, onkologem, psychologem, psychiatrem). Magnetickou rezonanci (MR) mozku indikuje neurolog, vyšetření vnitřních orgánů další specialisté podle klinických potíží pacienta. Rozpis doporučených vyšetření uvádí tabulka 7.
Sledováni by měli být příbuzní (tab. 8) v rodinách nosičů prokázané mutace i příbuzní v rizikových rodinách, kde kauzální mutace nebyla prokázána [27].

CENTRALIZACE PÉČE O NEMOCNÉ SE VZÁCNÝMI ONEMOCNĚNÍMI
Jako vzácná onemocnění (VO) se označuje skupina chorob s výskytem méně než pět případů na 10 000 obyvatel, do této skupiny chorob patří i neurokutánní syndromy. Dosud bylo popsáno více než 6 000 vzácných onemocnění, kterými trpí až 20 milionů obyvatel Evropské unie. Centralizace péče o nemocné s vzácným onemocněním probíhá postupně v jednotlivých státech Evropy s cílem vytvořit funkční síť specializovaných pracovišť, která budou vzájemně spolupracovat. Ustanovení center pro vzácná onemocnění je v ČR řízeno Ministerstvem zdravotnictví, Meziresortní a mezioborovou pracovní skupinou pro vzácná onemocnění a odbornými společnostmi ČLS JEP. Soustředění pacientů se vzácnými onemocněními do specializovaných center je výhodné pro pacienty, jejich rodiny i z ekonomického hlediska.
Česká dermatovenerologická společnost ČLS JEP navrhla 2 dermatologická specializovaná centra pro vzácná onemocnění (VO) ve FN Motol a FN Brno. Pacienti s tuberózní sklerózou by měli být dispenzarizováni na specializovaném pracovišti pro neurokutánní onemocnění. Ministerstvem zdravotnictví však v ČR dosud nebylo uznáno žádné centrum vysoce specializované péče pro nemocné s neurokutánními syndromy.
GENETICKÉ VYŠETŘENÍ A PORADENSTVÍ
DNA diagnostika a genetické poradenství mají nezastupitelné místo v péči o nemocného a jeho příbuzné,díky metodám prenatální a preimplantační diagnostiky umožňují genetickou prevenci v rodině pacienta s TSC.
Genetické vyšetření se provádí při nejasné diagnóze TSC podle klinického obrazu, např. u dětských pacientů s dosud nerozvinutými klinickými projevy nemoci, u rodinných příslušníků nemocných s tuberózní sklerózou, kteří mají riziko nosičství mutace TSC genu a v rámci prenatální diagnostiky [29]. Cílem molekulární diagnostiky je odhalení mutace v TSC genech zodpovědné za dané onemocnění.
DNA diagnostika se provádí ze žilní krve po provedení genetického poradenství a podpisu informovaného souhlasu. DNA analýza je velmi náročná, ne vždy se podaří odhalit příčinnou mutaci [27]. U 10–25 % pacientů TSC je příčinná mutace neidentifikovatelná obvyklým genetickým testováním, normální výsledek však nevylučuje přítomnost TSC. Molekulární analýza genů TSC1 a TSC2 odhalí mutace u 75–90 % nemocných TSC splňujících klinická diagnostická kritéria [16].
Za patogenní mutaci je považována mutace, která jasně narušuje syntézu proteinu hamartinu nebo tuberinu, anebo vede k poruše funkce až k inaktivaci jejich funkce.
Analýzu genu TSC1 a TSC2 v ČR v současné době poskytuje Ústav lékařské genetiky a fetální medicíny FN Olomouc. Ústav lékařské genetiky a fetální medicíny FN Olomouc se zabývá vyhledáváním mutací u TSC pacientů více než 15 let, na základě dlouholetých zkušeností a mezinárodní spolupráce byl opakovaně stanoven optimální vyšetřovací postup při vyhledávání kauzálních mutací [29]. Od roku 2014 je využívána technika masivního paralelního sekvenování (MiSeq) obou TSC genů s cílem zrychlení zpracování vzorků [5].
Po odhalení a potvrzení mutace nebo nalezení vazby je možno definitivně potvrdit klinicky nejasnou diagnózu, nabídnout rodině vyšetření příbuzných v riziku a případně provést prenatální diagnostiku [29].
Prenatální diagnostika se provádí v rodině s odhalenou kauzální mutací nebo v rodinách s vícečetným výskytem onemocnění přímou nebo vazebnou analýzou již v 11.–13. týdnu gravidity na podkladě analýzy DNA ze vzorku choriové tkáně, výsledek vyšetření je dostupný již během několika dnů [28]. Tyto metody však nejsou použitelné v případě, kdy není známa kauzální mutace, a ani vazebnou analýzou nelze určit, který z chromozomů je zodpovědný za onemocnění [27]. V závažných případech je ve vybraných pracovištích v ČR dostupná také preimplantační diagnostika [27], která je v indikovaných případech i přes vysoké náklady uhrazena zdravotní pojišťovnou [17].
TERAPIE PROJEVŮ TUBERÓZNÍ SKLERÓZY
Léčba projevů tuberózní sklerózy je symptomatická.
Hypomelanotické makuly, fibrózní plaky, skvrny „confetti“ a šagrénová kůže nevyžadují léčbu, angiofibromy v obličeji a fibromy nehtů vyžadují léčbu nejen z kosmetických důvodů.
K odstranění angiofibromů v obličeji se v minulosti používaly metody shave excize, dermabraze, laser, kryoterapie a elektrodesikace. Tyto léčebné metody byly sice efektivní, avšak docházelo k častým recidivám s nutností opakování léčebných výkonů. Pulsed dye laser (PDL)se ukázal být účinný s obzvlášť dlouhodobým efektem, zejména u nově vznikajících angiofibromů s převažující vaskulární složkou. Prominující léze mohou být úspěšně odstraněny CO2 laserem (carbon dioxide laser) spíše než PDL, u CO2 laseru je však vyšší riziko vzniku hypertrofických jizev. K dosažení optimálního kosmetického efektu je možné kombinovat laserové a chirurgické metody. V posledních letech se zavádí lokální léčba angiofibromů inhibitory mTOR (viz dále).
Fibromy nehtů lze odstranit obdobnými léčebnými metodami jako angiofibromy – chirugickou excizí, CO2 laserem, shave excizí či fenolizací [9].
Léčba epilepsie spadá do kompetence neurologa. Vzhledem k tomu, že epileptické záchvaty se negativně podílejí na dalším psychomotorickém vývoji dítěte, je doporučováno nasazení antiepileptické terapie již po prvním epileptickém záchvatu [18].
Metodou volby subependymálních obrovskobuněčných astrocytomů je chirurgické řešení, avšak radikální resekce SEGA není pro rozsah postižení či významná rizika pooperačního neurologického deficitu ve všech případech možná. U pacientů se SEGA, kteří vyžadují léčebný zásah, ale nejsou indikováni k chirurgické resekci, je možná farmakologická léčba pomocí mTOR inhibitoru [17].
Neuropsychiatrické projevy školních dětí často vyžadují, vzhledem k omezeným schopnostem koncentrace a učení, sestavení individuálního studijního plánu a úzkou spolupráci rodiny nemocného dítěte s pedagogicko-psychologickou poradnou a s psychiatrem.
Závažnost komplikací angiomyolipomů ledvin, které bývají hodnoceny jako nejčastější příčina úmrtí pacientů s TSC, vyžaduje proaktivní přístup k ochraně reziduální funkce zdravého ledvinného parenchymu a oddálení rozvoje chronické renální insuficience a terminálního selhání ledvin. Podle klinického nálezu lze zvážit podání inhibitorů mTOR, další možností je selektivní embolizace angiomyolipomů cestou a. renalis (u tumorů nad 8 cm nebo i menších, pokud je přítomné krvácení), popř. podle klinického nálezu a přání pacienta i chirurgická resekce [17].
Léčba lymfangioleiomyomatózy plic je chirurgická, příležitostně vyžaduje transplantaci plic. Ve stadiu klinického zkoušení je terapie mTOR inhibitory a antiestrogenní terapie inhibitory aromatáz [30].
TERAPIE TUBERÓZNÍ SKLERÓZY mTOR INHIBITORY
Tuberózní skleróza je prvním onemocněním ze skupiny neurokutánních syndromů, kde byla prokázána účinnost terapie inhibitory mTOR (mammalian target of rapamycin). Od 90. let 20. století je zkoumán vliv rapamycinu na kinázu mTOR, jejíž aktivita je u mnoha lidských nádorů zvýšená a zaujímá významné místo v metabolické cestě tuberózní sklerózy.
Proteinová kináza mTOR je hlavní složkou proteinového komplexu označovaného jako mTORC1. Buněčné signální dráhy vedou prostřednictvím mTORC1 k aktivaci buněčných anabolických drah, k syntéze lipidů, proteinů a nukleových kyselin a ve svém důsledku ke kontrole buněčného růstu a proliferace. Mutace genů TSC1 nebo TSC2 je podkladem disrupce komplexu hamartin-tuberin a hyperaktivity mTORC1 [30].
Inhibitory mTOR jsou skupinou farmak souhrnně označovaných jako rapaloga, vedou k disociaci mTORC1 a tím k inhibici mTOR signální dráhy. Rapamycin neboli sirolimus je prototypem mTOR inhibitoru, dalšími rapalogy jsou everolimus a temsirolimus.
Rapaloga se používají k léčbě subependymálních obrovskobuněčných astrocytomů, angiomyolipomů ledvin a lymfangioleiomyomatózy plic, během této léčby byl pozorován jejich efekt i na angiofibromy v obličeji, hypomelanotické makuly, velikost rhabdomyomů, ale dokonce i na poruchy chování a epilepsii [10, 14, 17, 30].
RAPAMYCIN (SIROLIMUS)
Rapamycin se váže na specifický cytosolový protein FKPB-12, komplex FKPB-12-rapamycin inhibuje aktivaci mTORC1. Inhibice mTOR vede k blokádě mnoha specifických signálních transdukčních cest, výsledkem je inhibice aktivace lymfocytů, která vede k imunosupresi.
Rapamycin byl izolován z půdní bakterie Streptomyces hygroscopicus na ostrově Rapa Nui (Velikonoční ostrov ve východní části Polynésie) v roce 1975 a byl původně zkoumán jako účinné antimykotikum. Posléze byl odhalen významný antiproliferační a imunosupresivní efekt [30].
Přípravek Rapamune obsahující rapamycin je léčebně využíván u dospělých pacientů k profylaxi orgánové rejekce po transplantaci ledviny. Léčba sirolimem je zatížena relativně vysokým výskytem nežádoucích účinků (nejčastěji změny v krevním obraze, minerálový rozvrat, elevace krevních lipidů a glykémie, horečka, hypertenze, dyspepsie) [22].
Byla provedena řada klinických studií k hodnocení účinku lokální aplikace rapamycinu na angiofibromy v obličeji. Byla publikována zlepšení ve velikosti, počtu a míře erytému angiofibromů, v některých studiích však docházelo k recidivě do několika týdnů po vysazení léčby, proto byla léčba znovu nasazena až na 30 měsíců [19]. Rapamycin byl obvykle dobře snášen, v některých případech pacienti udávali pocit píchání v místě aplikace, proto byl rapamycin v případě potřeby aplikován současně s nízce potentním kortikosteroidem ke snížení iritačního účinku [19]. Pouze u 2 pacientů byly detekovatelné hladiny rapamycinu v séru při jeho lokální aplikaci, tato hladina však byla výrazně nižší, než je terapeutická hladina při systémové léčbě mTOR inhibitory.
V literatuře je možné se setkat s různými způsoby přípravy a schématy léčebného podávání rapamycinu. Koncentrace rapamycinu kolísá od 0,02 % do 1 %, lék je podáván obvykle 1–2krát denně. Lék je obvykle připravován smísením roztoku nebo rozemletých tablet rapamycinu s emolienciem. Jacks uvádí přípravu z 15 tablet o obsahu 1 mg rapamycinu smísením s emolienciem za vzniku 15 g 0,1% krému. Tato dávka, postačující na 1–2 měsíce léčby, stojí okolo 45 USD [9].
EVEROLIMUS
Everolimus se váže také na intracelulární protein FKBP - -12 a tvoří komplex, který inhibuje aktivitu mTOR komplexu - 1 (mTORC1). Inhibice signální kaskády mTORC1 interferuje s translací a syntézou proteinů redukcí aktivity ribozomální protein-S6-kinázy (S6K1) a vazebného proteinu 4E (4EBP-1) eukaryotického elongačního faktoru, které regulují proteiny zapojené do buněčného cyklu, angiogeneze a glykolýzy. Everolimus může redukovat hladinu vaskulárního endoteliálního růstového faktoru (VEGF). U pacientů s TSC zvyšuje léčba everolimem hladiny VEGF-A a snižuje hladiny VEGF-D. Everolimus je účinný inhibitor růstu a proliferace nádorových buněk, endoteliálních buněk, fibroblastů a buněk hladkého svalstva krevních cév [23].
Přípravek Votubia obsahující everolimus je indikován k léčbě dospělých pacientů s renálním angiomyolipomem u nemocných s tuberózní sklerózou, u nichž hrozí riziko komplikací na základě zhodnocení faktorů, jako je velikost tumoru, výskyt aneurysmat nebo výskyt vícečetných nebo bilaterálních tumorů nevyžadujících bezprostřední chirurgický výkon [23]. Studie s mTOR inhibitory potvrdily zpomalení růstu angiomyolipomů, dokonce regresi objemu a snížení frekvence krvácení z tumorů [17].
Přípravek Votubia je také indikován k léčbě pacientů se subependymálním obrovskobuněčným astrocytomem (SEGA) spojeným s TSC, kteří vyžadují terapeutický zásah, ale vzhledem k riziku nejsou vhodnými kandidáty chirurgického výkonu. Votubia prokázala zmenšení objemu SEGA, ale také klinicky významné zlepšení angiofibromů v obličeji.
Nejčastější nežádoucí účinky podle shromážděných bezpečnostních údajů jsou podle klesající četnosti: stomatitida, amenorea, infekce horních cest dýchacích, hypercholesterolémie, nazofaryngitida, nepravidelná menstruace, akné, sinusitida, zánět středního ucha a pneumonie [23].
V České republice je předepisován everolimus k lokální aplikaci na angiofibromy obličeje po podpisu informovaného souhlasu pacientem a schválení revizním lékařem jako off-label léčivo. Připravuje se směs everolimu ve vazelíně v 0,4% koncentraci v množství 15 gramů, podává se na angiofibromy 1krát denně večer za důsledné fotoprotekce. Tato dávka léku postačí na 3 měsíce léčby [31].
Časné podání inhibitorů mTOR zřejmě působí preventivně na vývoj angiofibromů obličeje, neboť ve studii jednovaječných dvojčat se dívce, která byla od 4 let léčena systémovým everolimem pro subependymální obrovskobuněčný astrocytom, v obličeji nikdy nevytvořily angiofibromy, její neléčené sestře se vytvořily ve věku 6 let [9].
Zavedení inhibitorů mTOR do běžné klinické praxe je limitováno značnou finanční nákladností. Narozdíl od TOsystémové terapie není lokální terapie kožních projevů TSC pomocí mTOR inhibitorů dosud v České republice ani v zahraničí komerčně dostupná, nebylo definováno optimální dávkovací schéma ani příprava léku [26]. Získání zkušeností z hlediska dlouhodobé efektivity a bezpečnosti jejich užití bude trvat ještě řadu let.
PROGNÓZA
Nejčastějí příčina úmrtí v souvislosti s TSC se liší v závislosti na věku pacienta. V novorozeneckém věku jsou rizikové rozsáhlé rhabdomyomy srdečního svalu, v kojeneckém a batolecím věku může ohrozit život pacienta farmakorezistentní epilepsie a komplikace nádorů CNS, zejména subependymálního obrovskobuněčného astrocytomu. V pubertě a dospělosti mohou být fatální selhání ledvin, náhlé vnitřní krvácení z velkých angiomyolipomů ledvin a vzácně vznik renálního karcinomu. U žen ve věku 20–40 let může být fatální postižení plic lymfangioleiomyomatózou [27, 29]. Úmrtí nemocných s tuberózní sklerózou v souvislosti s maligními nádory jsou vzácná. Z hlediska etiologie je nejčastější příčinou smrti všech nemocných tuberózní sklerózou postižení CNS (45 %). Druhá nejčastější příčina smrti (30 %) u všech pacientů TSC a zároveň nejčastější příčina smrti u dospělých pacientů s TSC je renální postižení. Lymfangioleiomyomatóza plic a bronchopneumonie vedly ve studii k úmrtí u 20 % a srdeční rabdomyomy u 5 % nemocných TSC. Celková naděje na dožití byla hodnocena jako snížená oproti běžné populaci [17].
ZÁVĚR
Tuberózní skleróza je po neurofibromatóze druhý nejčastější neurokutánní syndrom. Klinická diagnostická kritéria a genetické vyšetření umožňují jednoznačné stanovení diagnózy TSC. Časné rozpoznání nemoci, ve kterém hraje vyšetření dermatologem zásadní roli, kvalitní multioborové sledování pacientů a využití moderních léčebných metod v centrech specializované péče může vést k významnému snížení morbidity a mortality nemocných tuberózní sklerózou.
Děkuji Radiodiagnostickému oddělení Masarykovy nemocnice v Ústí nad Labem, o. z., za poskytnutí MR snímků a MUDr. Bořivoji Petrákovi, CSc., z Kliniky dětské neurologie 2. LF UK a FN Motol za poskytnutí vybrané fotodokumentace.
Do redakce došlo dne 7. 3. 2016.
Adresa pro korespondenci:
MUDr. Daniela Humhejová
Dětská klinika UJEP
Masarykova nemocnice v Ústí nad Labem, o. z.
Krajská zdravotní a. s.
Sociální péče 3316/12A
401 13 Ústí nad Labem
e-mail: daniela.humhejova@kzcr.eu
Sources
1. AGRAVAL, S. N., KULKARNI, Y. A., DESHMUKH, Y. R., JANE, S. D. Tuberous sclerosis in pregnancy. Our Dermatol. Online, 2014, 5, 2, p. 160–162.
2. BENYOUNES N., FOHLEN M., DEVYS, J-M., DELALANDE, O., MOURES, J. M., COHEN, A. Cardiac rhabdomyomas in tuberous sclerosis patients: A case report and review of the literature. Arch Cardiovascular Diseases, 2012, 105, p. 442–445.
3. CURATOLO, P. Tuberous sclerosis complex: from basic science to clinical phenotypes. Cambridge: Cam bridge University Press, 2003, p. 301, ISBN 1 898 68339 5.
4. DOLEŽEL, Z., RÁČILOVÁ, Z., PAVLOVSKÁ, D., DOSTÁLKOVÁ, D., KOCÁNOVÁ, E. Tuberózní skleróza. Pediatr. praxi, 2012, 2, s. 130.
5. FILIPOVÁ, H., VRTĚL, R. Molekulární diagnostika komplexu tuberózní sklerózy. Medical tribune, 2014, 10, 24, ISSN 1214-8911.
6. GEORGE, A., KANISH, B., BHATIA, A. Tuberous sclerosis. Indian Dermatol. Online J., 2015, 6, p. 142 – 143.
7. GURČÍK, L., TOMÁŠOVÁ, A., BENC, O., GALIK, P., GAŠPARÍKOVÁ, V., LIŠKOVÁ, S., VRTĚL, R. Tuberózna skleróza z pohľadu neurológa. Neurol. pro praxi, 2008, 3, s. 155–158.
8. GŰNALDI, M., PAYDAS, S., AFSAR, C. U., DORAN, F. Coexistence of tuberous sclerosis complex and malignant melanoma. Singapore Med. J., 2013, 11, p. 233–235.
9. JACKS, S. K., WITMAN, P. M. Tuberous Sclerosis Complex: An Update for Dermatologists. Pediatr. Dermatol., 2015, 5, p. 563–570.
10. JÜLICH, K., SAHIN, M. Mechanism-based Treatment in Tuberous Sclerosis Complex. Pediatr. Neurol., 2014, 4, p. 290–296.
11. KLÉZL, P., RICHTEROVÁ, R., ŠTANC, O., KLEČKA, J., ZÁŤURA, F. Objemný angiomyolipom pravé ledviny u mladé ženy. Urol. List, 2013, 4, s. 45–48.
12. KRUEGER, D. A., NORTHRUP, H. Tuberous Sclerosis Complex Surveillance and Management: Recommendations of the 2012 International Tuberous Sclerosis Complex Consensus Conference. Pediatr. Neurol., 2013, 4, p. 255–265.
13. LOQUAI, C., METZE, D., NASHAN, D., LUGER, T. A., BŐHM, M. Confetti-like lesions with hyperkeratosis: a novel ultraviolet-induced hypomelanotic disorder? Br. J. Dermatol., 2005, 1, p. 190–193.
14. MOAVERO, R., CONIGLIO, A., GARACI, F., CURATOLO, P. Is mTOR inhibition a systemic treatment for tuberous sclerosis ? Ital. J. Pediatr., 2013, 39, p. 57.
15. NORTHRUP, H., KOENIG, M. K., PEARSON, D. A. et al. Tuberous Sclerosis Complex. 1999 Jul 13 [Updated 2015 Sep 3]. In Pagon, R. A., Adam, M. P., Ardinger, H. H. et al., editors. GeneReviews® [Internet].
Seattle (WA): University of Washington, Seattle; 1993–2016. Dostupné na: http://www.ncbi.nlm.nih.gov/books/NBK1220/.
16. NORTHRUP, H., KRUEGER, D. A. International Tuberous Sclerosis Complex Consensus Group. Tuberous Sclerosis Complex Diagnostic Criteria Update: Recommendations of the 2012 International Tuberous Sclerosis Complex Consensus Conference. Pediatr. Neurol., 2013, 49, p. 243–254.
17. NOVOTNÝ, T. Pokroky v diagnostice a léčbě tuberózní sklerózy. Medical tribune, 2015, 11, 4, ISSN 1214-8911.
18. PETRÁK, B., GABERA, A., FILIPOVÁ, H., TOMEK, V., PUCHMAJEROVÁ, A., MRÁZKOVÁ, L., JAHODOVÁ, A., MALÍKOVÁ, M., ČERNÝ, M., VRTĚL, R. Tuberózní skleróza u dětí sledovaných od novorozeneckého věku pro prenatální nález rhabdomyomů srdce – dvě kazuistiky. Cesk Slov Neurol, 2013, 6, s. 763–768.
19. PYNN, E. V., COLLINS, J., HUNASEHALLY, P. R. Y., HUGHES, J. Successful Topical Rapamycin Treatment for Facial Angiofibromata in Two Children. Pediatr. Dermatol., 2015, 3, p. 120–123.
20. RAYER, P. F. O. Traité des maladies de la peau/atlas. Paris: JB Baillière, 1835, p. 88.
21. SLOAN, S. B. Connective Tissue Nevus. Dostupné na: http://emedicine.medscape.com/article/1056490-overview, 2014.
22. Státní ústav pro kontrolu léčiv (SÚKL). Rapamune 1 mg/ml. Souhrn údajů o přípravku. Dostupné na: http:// www.sukl.cz/modules/medication/detail.php? - code=0027235&tab=texts.
23. Státní ústav pro kontrolu léčiv (SÚKL). Votubia 5 mg. Souhrn údajů o přípravku. Dostupné na: http:// www.sukl.cz/modules/medication/detail.php? - code=0168458&tab=texts.
24. TEY, H. L. A Practical Classification of Childhood Hypopigmentation Disorders. Acta Derm. Venereol., 2010, 90, p. 6–11.
25. ŰRGE, T., HORA, M., HES, O., CHUDÁČEK, Z. Renální angiomyolipom, histologie, diagnostika a terapie. Urol. Praxi, 2005, 6, s. 270–271.
26. VASANI, R. J. Facial Angiofibromas of Tuberous Sclerosis Treated with Topical Sirolimus in an Indian Patient. Indian J. Dermatol., 2015, 2, p. 165–169.
27. VRTĚL, R., FILIPOVÁ, H., VODIČKA, R., ŠANTAVÁ, A., CURTISOVÁ, V., FORETOVÁ, L. Tuberózní skleróza. Klin. Onkol., 2009, 22 (Suppl), s. 50–53.
28. VRTĚL, R., VODIČKA, R., ŠANTAVÁ, A., ŠANTAVÝ, J., KREJČIŘÍKOVÁ, E. Prenatální diagnostika tuberózní sklerózy založená na znalosti kauzální mutace. Čas. Lék. čes, 2006, 145, s. 130–132.
29. VRTĚL, R., VOUTSINAS, G., VODIČKA, R., FILIPOVÁ, H., KOTLÁROVÁ, P., SMUTNÁ, V., ŠIMKOVÁ, D., KONVALINKA, D., ŠANTAVÁ, A., ŠANTAVÝ, J. Tuberózní skleróza: optimalizace postupu její DNA diagnostiky. Cesk Slov Neurol, 2008, 4, s. 478–482.
30. ZITTERBART, K. mTOR inhibice: nové možnosti farmakologické léčby nádorů u pacientů s tuberózní sklerózou. Postgr. Med., 2014, 16, s. 880–886.
31. ZITTERBART, K. Nové možnosti farmakologické léčby nádorů spojených s tuberózní sklerózou. (přednáška) Brno: 19. konference dětské dermatologie, 23. 10. 2015.
Labels
Dermatology & STDs Paediatric dermatology & STDsArticle was published in
Czech-Slovak Dermatology

2016 Issue 2
Most read in this issue
- Tuberózní skleróza
- Pityriasis lichenoides et varioliformis acuta popis dvou – případů a přehled literatury
- Spinocelulární karcinom dolního rtu: naše zkušenosti a přehled literatury