
Wiskottův-Aldrichův syndrom – onemocnění vyžadující včasnou transplantaci kmenových buněk krvetvorby
Wiskott-Aldrich Syndrome – Disease Requiring Early Transplantation of Hemopoietic Stem Cells
Wiskott-Aldrich syndrome (WAS) is a rare disease characterized by thrombocytopenia with small platelets, combined immunodeficiency and eczema. The disease is caused by the WASP gene, which is localized on chromosome X and is coding multifunctional intracellular protein WASp. The disease becomes clinically manifest by skin hemorrhage manifestations, epistaxis events, as well as bleeding from gingival and intestines exanthema and recurrent respiration infections. Increased occurrence of autoimmune diseases and malignancies was observed in patients with WAS.
WAS is a very rare disease and because of that the diagnostics is not easy even at the present time. The diagnosis of WAS is to be considered in all cases of thrombocytopenia in combination with eczema and relapsing respiratory infections, small thrombocytes are typical for diagnosis. In the case of suspected WAS the child patient should be admitted to hematological center as early as possible and the center should confirm the diagnosis in collaboration with an immunological workplace together with molecular genetic examination and initiates the search for HLA-identical donor. The problem of necessary correct and early establishment of diagnosis is documented on case histories of 6 patients handed over to our workplace and indicated to allogenic transplantation of hemopoietic stem cells (SCT) in the years 1996-2007. SCT is presently the only possibility to cure up WAS and the successfulness is markedly dependent on early establishment of diagnosis and efficient prophylaxis and directed therapy of infectious complications.
Key words:
Wiskott-Aldrich syndrome, WASP gene, thrombocytopenia, eczema, primary immunodeficiency, transplantation of hemopoietic stem cells
Authors:
R. Formánková 1; P. Sedláček 1; T. Freiberger 3,4; J. Bartůňková 2; A. Šedivá 2; E. Mejstříková 1; P. Keslová 1; B. Ravčuková 3; V. Vávra 1; J. Litzman 4; E. Pařízková 5; Y. Jabali 6; H. Schneiderová 7; J. Starý 1
Authors‘ workplace:
Klinika dětské hematologie a onkologie UK 2. LF a FN Motol, Praha
přednosta prof. MUDr. J. Starý, DrSc.
1; Ústav imunologie UK 2. LF a FN Motol, Praha
přednostka prof. MUDr. J. Bartůňková, DrSc.
2; Genetická laboratoř Centra kardiovaskulární a transplantační chirurgie, Brno
vedoucí MUDr. T. Freiberger, PhD.
3; Ústav klinické imunologie a alergologie LF MU a FN u Sv. Anny, Brno
přednosta prof. MUDr. J. Litzman, CSc.
4; Dětská klinika LF UK a FN, Hradec Králové
přednosta prof. MUDr. M. Bayer, CSc.
5; Dětské oddělení Nemocnice České Budějovice
primář MUDr. V. Smrčka
6; II. dětská klinika FN Brno
přednosta prof. MUDr. Z. Doležel, CSc.
7
Published in:
Čes-slov Pediat 2009; 64 (3): 106-114.
Category:
Original Papers
Overview
Wiskottův-Aldrichův syndrom (WAS) je vzácné X-vázané recesivně dědičné onemocnění charakterizované trombocytopenií s malými destičkami, kombinovanou imunodeficiencí a ekzémem. Příčinou onemocnění jsou mutace genu WASP, lokalizovaného na chromozomu X a kódujícího multifunkční intracelulární protein WASp. Klinicky se onemocnění manifestuje kožními krvácivými projevy, epistaxemi, krvácením z dásní a střev, objevuje se exantém a rekurentní respirační infekce. U pacientů s WAS byl popsán zvýšený výskyt autoimunních onemocnění a malignit.
WAS je onemocněné velmi vzácné a z toho důvodu jeho diagnostika ani v současné době není snadná. Na diagnózu WAS je třeba myslet vždy v případě trombocytopenie v kombinaci s ekzémem a recidivujícími respiračními infekty, patognomická je přítomnost malých trombocytů. V případě podezření na WAS musí být dětský pacient co nejdříve předán do hematologického centra, které ve spolupráci s imunologickým pracovištěm potvrdí diagnózu, zajistí molekulárně genetické vyšetření a zahájí vyhledávání HLA identického dárce.
Problematiku nutnosti správného a včasného stanovení diagnózy autoři prezentují na kazuistikách 6 pacientů předaných na jejich pracoviště a indikovaných k alogenní transplantaci kmenových buněk krvetvorby (SCT) v letech 1996–2007. SCT je v současné době jedinou možností vyléčení WAS a její úspěšnost je velmi závislá na včasném stanovení diagnózy a na efektivní profylaxi a cílené léčbě infekčních komplikací.
Klíčová slova:
Wiskottův-Aldrichův syndrom, gen WASP, trombocytopenie, ekzém, primární imunodeficience, transplantace kmenových buněk krvetvorby
Úvod
Wiskottův-Aldrichův syndrom (WAS) je vzácné X-vázané recesivně dědičné onemocnění charakterizované trombocytopenií s malými destičkami, rekurentními infekcemi na podkladě kombinované imunodeficience a ekzémem. U pacientů s WAS byl popsán zvýšený výskyt autoimunních onemocnění a malignit, především lymfomů a leukemií [1, 2, 3]. Incidence WAS se odhaduje na 1–10 na 1 milion narozených dětí [4]. Klinicky se onemocnění manifestuje zpravidla záhy po narození, jedinou kauzální léčbou je v současné době transplantace kmenových buněk krvetvorby (SCT) [5].
Jako klinický syndrom popsal onemocnění poprvé dr. Wiskott v roce 1937 u tří bratrů s trombocytopenií, krvavými průjmy, ekzémem a rekurentními otitidami [1]. V roce 1954 demonstroval dr. Aldrich X-vázanou dědičnost syndromu na rodině s 16 postiženými chlapci, nikoliv dívkami v 6 generacích [2]. V 60. letech byl WAS zařazen mezi primární imunodeficity postihující buněčnou i protilátkovou imunitu [6]. V roce 1994 byl identifikován gen zodpovědný za WAS (WASP) a mutace WAS proteinu (WASp) byla prokázána nejen u pacientů s klasickým WAS [7], ale také u dětí s klinicky mírnější formou onemocnění s příznivější prognózou – X-vázanou trombocytopenií (XLT) [8].
Klinické a laboratorní nálezy
Klinické příznaky
Klasická forma onemocnění se manifestuje zpravidla v novorozeneckém nebo časném kojeneckém období. U většiny chlapců nacházíme pozitivní rodinnou anamnézu (podobné příznaky se vyskytují u mužských příbuzných z matčiny strany), pouze u jedné třetiny pacientů se jedná o mutaci de novo.
Nízký počet typicky malých destiček vede ke kožním krvácivým projevům, epistaxím, krvácením z dásní a střev u 84 % pacientů s WAS, život ohrožující krvácení se vyskytuje u 30 % pacientů (jedná se zpravidla o krvácení do gastrointestinálního traktu, pouze u 2 % pacientů je to krvácení do CNS) [4].
Typickým projevem onemocnění je ekzém, od mírných po velmi těžké formy, tranzientní či perzistující bývá popisován u 81 % pacientů s WAS. Těžké formy špatně odpovídající na léčbu mohou být komplikovány kožními bakteriálními infekcemi nebo virovými infekcemi herpes simplex a molluscum contagiosum [3, 4].
Během prvního půl roku života se objevují zpravidla i rekurentní respirační bakteriální, virové a mykotické infekce, nejčastěji purulentní otitidy nebo pneumonie, méně často se mohou vyskytnout sepse či meningitidy [3, 4, 9].
K manifestaci autoimunního onemocnění dochází asi u 40 % pacientů s WAS. Nejčastěji je popisována autoimunní hemolytická anémie (AIHA), dále vaskulitidy, nefropatie, Henoch-Schönlein-like purpura, zánětlivá střevní onemocnění, méně časté jsou neutropenie, dermatomyozitidy, angioedémy, uveitidy a cerebrální vaskulitidy [3, 4, 9].
Maligní onemocnění se může manifestovat již v dětství, mnohem častěji se ale vyskytuje u adolescentů a mladých dospělých s klasickým WAS genotypem. Nejčastější malignitou provázející WAS je B-lymfom, často EBV pozitivní, v souladu s poruchou imunitního systému. Malignity asociované s WAS patří k prognosticky velmi nepříznivým [3, 4].
Pro klasifikaci onemocnění podle stupně závažnosti byl stanoven jednoduchý skórovací systém [10, 11]:
- 0,5: intermitentní trombocytopenie;
- 1: trombocytopenie s malými trombocyty;
- 2: trombocytopenie a mírný ekzém nebo občasné infekce horních cest dýchacích;
- 2,5: trombocytopenie a na léčbu odpovídající těžký ekzém nebo respirační infekce vyžadující antibiotickou léčbu;
- 3: ekzém a zároveň respirační infekce vyžadující antibiotickou léčbu;
- 4: ekzém vyžadující trvalou léčbu a/nebo těžké až život ohrožující infekce;
- 5: autoimunní onemocnění nebo malignita u pacienta s XLT/ WAS.
Laboratorní nálezy
Typickým nálezem v krevním obraze je trombocytopenie s malými destičkami, relativní lymfopenie a často normochromní anémie. Počet trombocytů kolísá mezi jednotlivými rodinami, chlapci v jedné rodině i u samotných pacientů v rozmezí od 5 x 109/l do 50 x 109/l, střední objem trombocytu (MPV) je zpravidla poloviční než u zdravých jedinců (3,8–5,0 fl) [4, 12]. Trombocytopenie je částečně způsobená zvýšenou fagocytózou a destrukcí trombocytů ve slezině a ostatních retikuloendotelových orgánech (v důsledku zvýšené exprese fosfatidylserinu na povrchu cirkulujících trombocytů), částečně sníženou produkcí trombocytů. V kostní dřeni je normální počet megakaryocytů s četnými jadernými abnormalitami [13].
Závažnost imunodeficitu závisí na typu mutace a expresi proteinu, funkce imunitního systému je narušena jak ve složce humorální (B lymfocyty), tak ve složce buněčné (T lymfocyty). V kojeneckém věku může být počet cirkulujících lymfocytů normální nebo jen lehce snížený. Lymfopenie v důsledku akcelerace apoptózy T lymfocytů se zpravidla manifestuje do 6 let věku u dětí s klasickou formou WAS (nikoliv u pacientů s XLT), počet B lymfocytů může být nadále normální nebo lehce snížený. Sérová hladina IgG je zpravidla v mezích normy, hladina IgM bývá snížená, ale může být i normální nebo zvýšená, hladiny IgA a IgE jsou často zvýšené. Signifikantním nálezem jsou nízké titry izohemaglutininů, výrazně snížená protilátková odpověď na polysacharidové antigeny a řadu proteinových antigenů (difteriový a tetanový toxoid, Haemophilus influenzae B, pneumokoky), naproti tomu protilátková odpověď na živé protivirové vakcíny bývá většinou normální [4, 9, 14]. Žádný z imunologických nálezů není specifický pouze pro toto onemocnění. Diagnostika spočívá v současné době v průkazu absence proteinu WASp [15] nebo v molekulárně genetickém vyšetření mutací genu WASP [4, 7].
Molekulárně genetická analýza
Gen WASP je lokalizován v regionu Xp11.22-Xp11.3 [16]. WASP kóduje intracelulární protein WASp složený z 502 aminokyselin a exprimovaný fyziologicky na všech hematopoetických buňkách včetně kmenových CD34+. Protein působí jako signalizační molekula a účastní se procesu organizace cytoskeletu v lymfoidní i myeloidní řadě a při biogenezi trombocytů. Jeho defekt vede k poruše interakce megakaryocytů s kolagenem a k absenci podosomů trombocytů bohatých na aktin. Protrombocyty jsou formovány předčasně a z megakaryocytů potom uvolňovány atypicky [4, 7, 17, 18].
V souboru 270 evropských a severoamerických nepříbuzných rodin s postižením WAS nebo XLT bylo identifikováno 158 různých mutací v genu WASP [11, 18]. Nejčastěji byly detekovány missense mutace v exonech 1–3, splice-site (sestřihové) mutace, krátké delece a nonsense mutace, méně často inzerce a komplexní mutace. U 25 % vyšetřovaných rodin byla nalezena mutace v jedné ze šesti oblastí častého vzniku mutací. Tři z těchto mutačních „hot spots“ leží v kódující oblasti a další 3 postihují místa sestřihu RNA (splice-sites). Mutace c.168C>T (p.T45M), c.290C>N/c.291G>N (p.R86S/G/C/H/L) a IVS6+5g>a byly shodně nalezeny u WASp pozitivních pacientů s mírnějšími fenotypovými projevy onemocnění (XLT), zatímco 3 zbylé mutace c.665C>T (p.R211X), IVS8+1g>n a IVS8+1_+4del gtga byly dokumentovány u pacientů WASp negativních s těžkým fenotypem WAS [11, 18].
Korelace genotyp – fenotyp onemocnění
Mutace genu WASP vede minimálně ke čtyřem fenotypově odlišným formám onemocnění [4] (tab. 1):
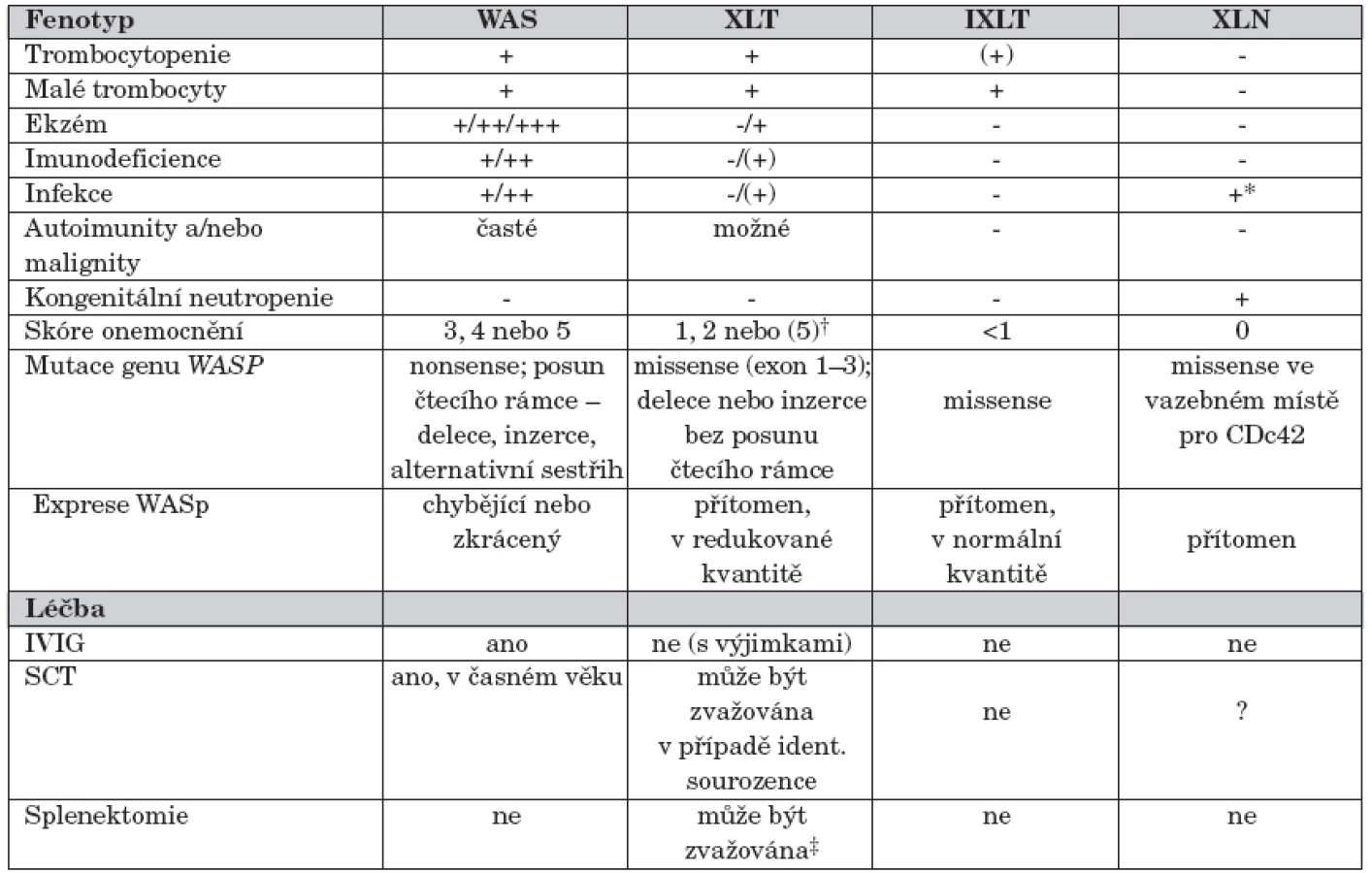
- klasická forma WAS s trombocytopenií s malými destičkami, rekurentními infekcemi a ekzémem, klinicky nejzávažnější, často komplikovaná autoimunními onemocněními a malignitami;
- X-vázaná trombocytopenie [8];
- intermitentní trombocytopenie [17];
- kongenitální X-vázaná neutropenie bez ostatních klinických nálezů charakteristických pro WAS/XLT [19].
U pacientů s klasickou formou WAS (skóre 3–4) lymfocyty neexprimují WASp nebo pouze zkrácený WASp (WASp negativní), zatímco u pacientů s XLT fenotypem (skóre 1–2) je mutovaný WASp zpravidla exprimován v normální velikosti, často ve snížené kvantitě (WASp pozitivní). Progrese do skóre 5 (onemocnění komplikované autoimunitou nebo malignitou) byla popsána u obou fenotypů, častěji ale u pacientů s klasickým WAS (skóre 3–4) [11, 20]. U pacientů s X-vázanou neutropenií je onemocnění zpravidla podmíněno missense mutací ve vazebném místě pro Cdc42 [19] (tab. 1).
Léčebné možnosti
Transplantace kmenových buněk krvetvorby je v současné době u pacientů s WAS jedinou kurativní metodou. Filipovich et al. [5] prezentovala v souboru 170 pacientů transplantovaných pro WAS v letech 1968–1996 celkové přežití 70 %. Výsledky byly signifikantně lepší u pacientů transplantovaných od HLA identického sourozence (87 % 5leté přežití) než u nepříbuzenských transplantací (52 % 5leté přežití). Celkové přežití pacientů transplantovaných od nepříbuzného dárce bylo pouze 56 %, ale až 84 % v podskupině dětí mladších 5 let. V průběhu posledních let se výsledky transplantací u pacientů s WAS zlepšují zejména v důsledku včasného stanovení diagnózy, velký důraz je kladen na výběr vhodného dárce a efektivní léčbu potransplantačních komplikací. Relativně častou příčinou úmrtí byla u pacientů transplantovaných pro WAS (zejména v případě T-depletovaných štěpů nebo režimů s redukovanou intenzitou) EBV vázaná lymfoproliferace [5]. V léčbě této komplikace je v posledních letech úspěšně používán rituximab (anti-CD20 monoklonální protilátka) [21].
Úspěšnost SCT je stejně jako u jiných kombinovaných imunodeficitů do značné míry závislá na včasném stanovení diagnózy a indikaci pacienta k transplantaci a na efektivní profylaxi a cílené léčbě infekčních komplikací [4]. U pacientů s lymfopenií je nutná profylaxe pneumocystové pneumonie a pravidelná substituce IVIG. Pacienti nesmí být očkováni živými vakcínami, kontraindikované je podávání derivátů kyseliny acetylsalicylové a léků ze skupiny nesteroidních antirevmatik. Léčba ekzému vyžaduje často podávání lokálních kortikoidů a v některých případech i krátkodobou systémovou léčbu kortikosteroidy, měla by být vedena zkušeným dermatologem. Transfuze trombocytů je vyhrazena k léčbě závažných krvácení (gastrointestinální nebo intrakraniální hemoragie). Krevní deriváty je nutno podávat ozářené. Splenektomie snižuje riziko krvácení v důsledku zvýšení počtu cirkulujících destiček, na druhé straně však významně zvyšuje riziko infekčních komplikací (život ohrožujících sepsí) a pacienti po splenektomii vyžadují doživotní ATB profylaxi (penicilin).
Alternativním postupem v léčbě WAS by se měla stát již brzy genová terapie, v posledních několika letech úspěšně ověřovaná na zvířecích modelech [22, 23]. Na základě těchto preklinických studií byla v nedávné době provedena první transplantace retrovirem transdukovaných autologních kmenových buněk krvetvorby u 2 pacientů s WAS [18]. První klinická studie u pediatrických pacientů by měla být zahájena v první polovině roku 2009 v Miláně (prof. A. Aiuti – IEWP Meeting 2008, ústní prezentace).
Kazuistiky
Pacient č. 1
Chlapec, diagnóza WAS stanovena ve věku 16 měsíců, prokázána mutace v genu WASP.
RA: pozitivní (bratr matky zemřel v 5 měsících na bronchopneumonii, dokumentována trombocytopenie, malabsorpce). OA: Od 2 měsíců ekzém, trombocytopenie (64–38 x 109/l, enteroragie, recidivující infekce horních cest dýchacích, otitidy, artritida. Ve 3 letech krvácení do CNS (trombocyty 8 x 109/l), provedena splenektomie, ve 4 letech recidiva krvácení do CNS (trombocyty 21 x 109/l). Laboratorní nálezy: Lymfocyty 3216/μl, CD3 16 %, CD4 12 %, CD8 2 %, NK 43 %, CD19 13 %, IgG 33,0 g/l, IgA 2,96 g/l, IgM 1,78 g/l, malé trombocyty (5,9 fl), kolísající počet. Léčen kortikoidy, pravidelně IVIG (prevence infektů).
SCT v 52 měsících, dárce: HLA haploidentická matka, periferní kmenové buňky (PBSC), T deplece štěpu (TCD). Přípravný režim: Busulfan 16 mg/kg, Cyklofosfamid 200 mg/kg, Thiotepa 300 mg/m2, antithymocytární globulin (ATG). Prevence reakce štěpu proti hostiteli (GVHD): Cyklosporin A (CsA). Přihojení štěpu: granulocyty den (D) +15, trombocyty D +17, D +21 dosaženo kompletního dárcovského chimerismu. Komplikace: cytomegalovirová (CMV) infekce D +21 – gastroenteritida, intersticiální pneumonie, D +25 krvácení do plic, exitus letalis.
Pacient č. 2
Chlapec, diagnóza WAS stanovena ve věku 8 měsíců, prokázána mutace v genu WASP.
RA: negativní. OA: Od 3 měsíců ekzém, od 5. měsíce recidivující respirační infekty (bronchitidy, bronchopneumonie), v 8 měsících trombocytopenie, kožní hemoragická diatéza, enteroragie, neprospívání, růst pod 3. percentilem. Laboratorní nálezy: Lymfocyty 3440/μl, CD3 70 %, CD4 18 %, CD8 40 %, NK 5,5 %, CD19 19 %, IgG 15,7 g/l, IgA 2,9 g/l, IgM 2,8 g/l, trombocyty 80–12 x 109/l, malé trombocyty (4 fl). Od 8 měsíců profylakticky trimetoprim, pravidelné podávání IVIG, opakovaně transfuze ozářených trombocytů a erymasy.
Od 10 měsíců věku (5/1997) hledán dárce pro transplantaci kmenových buněk krvetvorby, neúspěšně. Ve 38 měsících manifestace AIHA, léčen kortikoidy. Ve věku 42 měsíců provedena splenektomie, přechodně vzestup trombocytů na 300 x 109/l. Ve 46 měsících enteroragie, exitus letalis.
Pacient č. 3
Chlapec, diagnóza WAS stanovena ve věku 3 měsíce, prokázána mutace v genu WASP.
RA: pozitivní (otec matky zemřel ve 35 letech na krvácení do CNS, bratr otce matky zemřel v 5 letech na krvácení do CNS, matka přenašečka WAS). OA: Od novorozeneckého věku enteroragie, ekzém, ve 12 měsících intersticiální bronchopneumonie. Laboratorní nálezy: Lymfocyty 4128/μl, CD3 70 %, CD4 63 %, CD8 5 %, NK 23 %, CD19 5 %, IgG 6,6 g/l, IgA 0,41 g/l, IgM 0,30 g/l, isohemaglutininy negativní, malé trombocyty (6,8 fl), kolísající počet 40–100 x 109/l.
SCT ve 13 měsících, dárce: HLA identický nepříbuzný muž, kostní dřeň (KD). Přípravný režim: Busulfan 16 mg/kg, Cyklofosfamid 200 mg/kg, ATG. Prevence GVHD: CsA, metotrexát (MTX). Přihojení štěpu: granulocyty D +17, trombocyty D +17, od D +21 kompletní dárcovský chimerismus. Komplikace: hemoragická cystitida, tachypnoe, sepse Pseudomonas aeruginosa D +60, recidivující AIHA od 9 měsíců po SCT (terapie kortikoidy, IVIG, rituximab). Smíšený chimerismus znovu detekován od 6 do 48 měsíců po transplantaci (7 až 13 % autologní krvetvorby). Poslední sledování: 84 měsíců po transplantaci, kompletní dárcovská krvetvorba, Coombs negativní, sledován na pneumologii pro chronický kašel převážně na podkladě významného GER, podle spirometrie normální funkce plic, nadále závislý na substituci IVIG, hypotyreóza korigovaná substituční léčbou, růstová dynamika v normě. Lymfocyty: 1485/μl, CD3 68 %, CD4 35 %, CD8 30 %, NK 6 %, CD19 25 %. BTL (blastická transformace lymfocytů) a specifická protilátková odpověď v normě. IgG 4,29 g/l, IgA <0,07 g/l, IgM 0,40 g/l. Pozitivní protilátky proti štítné žláze (A-TPO 74,8 kU/l; A-TG negativní), ostatní autoprotilátky negativní.
Pacient č. 4
Chlapec, diagnóza WAS stanovena ve věku 50 měsíců, prokázána mutace v genu WASP.
RA: negativní. OA: Od novorozeneckého věku kožní krvácivé projevy, trombocytopenie, protilátky proti trombocytům negativní. Opakovaně léčen kortikoidy a IVIG s přechodným efektem. Opakovaně podkožní abscesy a paronychia (Staphylococcus aureus), enteritidy s enteroragiemi, ekzém, zachyceny malé destičky. Zvažována diagnóza WAS, imunologické vyšetření neprokázalo kombinovaný imunodeficit, sledován v hematologické ambulanci, susp. X-vázaná trombocytopenie. Ve 4 letech recidivující bronchopneumonie, enteritida (Adenovirus) s následným výrazným poklesem počtu trombocytů (12 x 109/l), kožní a slizniční hemoragickou diatézou. Laboratorní nálezy: Lymfocyty 2436/μl, CD3 40 %, CD4 33 %, CD8 6 %, NK 5 %, CD19 54 %, IgG 11,6 g/l, IgA 1,50 g/l, IgM 0,47 g/l, trombocyty 12–6 x 109/l, malé trombocyty (4,9 fl), HGB 92–86 g/dl, zjištěna IgG senzibilizace erytrocytů (pozitivní PAT), pozitivní nespecifické tepelné autoprotilátky. Léčen bolusem kortikoidů, IVIG. Podle vývoje imunologických nálezů i klinického stavu diagnóza uzavřena jako WAS (následně potvrzena i molekulárně genetickým vyšetřením), indikován k alogenní SCT.
SCT ve věku 54 měsíců, dárce: HLA identický nepříbuzný muž, KD. Přípravný režim: Busulfan 16 mg/kg, Cyklofosfamid 200 mg/kg, ATG. Prevence GVHD: CsA, MTX. Přihojení štěpu: granulocyty D +21, trombocyty D +15, od D +21 smíšený chimerismus s převahou dárcovské krvetvorby (autologní do 5 %). Komplikace: akutní GVHD II. st., aktivace EBV. Poslední sledování: 60 měsíců po SCT, trvá smíšený chimerismus (4 % autologní krvetvorby), hladiny imunoglobulinů v normě bez substituce IVIG, buněčná imunita, BTL i specifická protilátková odpověď v normě. Nemocnost nezvýšená, normální růstová dynamika.
Pacient č. 5
Chlapec, diagnóza WAS stanovena ve věku 36 měsíců, prokázána mutace v genu WASP.
RA: negativní. OA: Od 1 měsíce věku ekzém, od 3 měsíců kožní krvácivé projevy, častější infekce horních cest dýchacích, opakovaně léčen ATB. V 7 měsících zachycena trombocytopenie (20 x 109/l), léčen Prednisonem s přechodným nevýrazným efektem, dále podáván 3 měsíce CsA, bez efektu. V 36 měsících provedeno podrobnější imunologické vyšetření a molekulárně genetické vyšetření a stanovena diagnóza WAS, indikován k alogenní SCT. Laboratorní nálezy: Lymfocyty 2480/μl, CD3 44 %, CD4 30 %, CD8 16 %, NK 36 %, CD19 18 %, IgG 10,7 g/l, IgA 8,81 g/l, IgM 0,35 g/l, trombocyty 38–20 x 109/l, MPV 7,6 fl.
SCT ve 48 měsících, dárce: HLA identická nepříbuzná žena, KD. Přípravný režim: Busulfan 20 mg/kg, Cyklofosfamid 200 mg/kg, ATG 40 mg/kg. Prevence GVHD: CsA, MTX. Přihojení štěpu: granulocyty D +18, trombocyty D +21, od D +21 smíšený chimerismus s postupným poklesem podílu autologní krvetvorby (15–5 %), kompletního dárcovského chimerismu dosaženo poprvé D +67. Komplikace: akutní GVHD II. st. Poslední sledování: 60 měsíců po SCT, hladiny imunoglobulinů v normě bez substituce IVIG, buněčná imunita, BTL i specifická protilátková odpověď v normě. Nemocnost nezvýšená, normální růstová dynamika.
Pacient č. 6
Chlapec, diagnóza WAS zvažována od 10 měsíců, potvrzena molekulárně genetickým vyšetřením (mutace v genu WASP) ve 13 letech.
RA: pozitivní (strýc matky trombocytopenie, stav po splenektomii, exitus na horečnaté onemocnění v 18 letech; bratranec matky trombocytopenie, ekzém, v dospělosti stanovena dg. XLT s potvrzenou mutací WASP genu). OA: Od 5 měsíců ekzém, občasné kožní krvácivé projevy, epistaxe, zjištěna trombocytopenie (50 x 109/l), MPV 5,8 fl. Léčen Prednisonem bez většího efektu. Recidivující bronchitidy, bronchopneumonie, otitidy (Pseudomonas aeruginosa), angíny, sinusitidy. Ve 4 letech manifestace AIHA s tepelnými protilátkami, zvládnuta kortikoidy a IVIG. V 9 letech vaskulitida (tranzientní hematurie, otoky a bolestivost kolen, nártů, lýtka, pozitivní ANCA, ANF). Ve 12 letech CMP v povodí a. cerebri media l. dx. (levostranná hemiparéza a následně sekundární fokální epi-paroxyzmy) v důsledku vaskulitidy a hypertenzního inzultu, perikardiální a pleurální výpotky, pankarditida, hemoragická retinopatie. Recidivující herpes simplex labialis i herpes zoster. Ve 12,5 letech bronchopneumonie s incipientním septickým šokem. Laboratorní nálezy: Lymfocyty 2511/μl, CD3 72 %, CD4 35 %, CD8 30 %, NK 16 %, CD19 4 %, IgG 9,82 g/l (substit.), IgA 3,53 g/l, IgM 0,35 g/l, trombocyty 40 až 50 x 109/l, malé trombocyty (5,8 fl).
SCT ve 13 letech, dárce: HLA identický bratr, vzhledem k velkému váhovému nepoměru dárce-příjemce kombinace KD + PBSC. Přípravný režim: Busulfan 16 mg/kg, Cyklofosfamid 120 mg/kg, Fludarabin 160 mg/m2. Prevence GVHD: CsA, MTX. Přihojení štěpu: granulocyty D +19, trombocyty D +20, od D +21 kompletní dárcovská krvetvorba. Komplikace: engraftment syndrom (exantém), v průběhu prvního roku po transplantaci 3krát bronchopneumonie l. sin. (Staph. koaguláza neg.). Poslední sledování: 36 měsíců po SCT, hladiny imunoglobulinů v normě bez substituce IVIG, buněčná imunita, BTL i specifická protilátková odpověď v normě, výrazně pozitivní protilátky proti štítné žláze (A-TG 279,4; A-TPO >1300 kU/l), ostatní autoprotilátky negativní. Nemocnost nezvýšená, normální růstová dynamika, hypotyreóza korigovaná substituční léčbou.
Diskuse ke kazuistikám
V letech 1996–2007 bylo na naše pracoviště referováno a indikováno k alogenní transplantaci kmenových buněk krvetvorby 6 dětí s WAS (tab. 2).
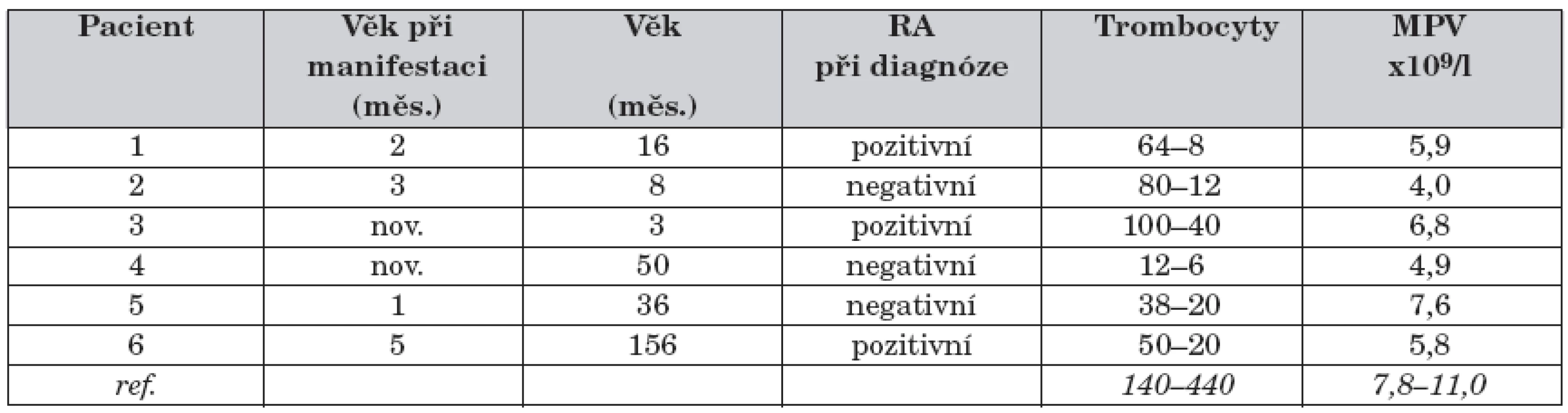
Pozitivní rodinná anamnéza (úmrtí nebo onemocnění chlapců z matčiny strany) byla zaznamenána u 3 ze 6 pacientů. První příznaky se objevily u všech 6 pacientů do 5 měsíců věku, u 2 dětí se onemocnění manifestovalo již v novorozeneckém věku. U 5 pacientů byl prvním klinickým projevem onemocnění ekzém, u 1 chlapce kožní hemoragická diatéza. U všech 6 pacientů byly dokumentovány recidivující respirační infekty. Enteroragie se vyskytla u 4 pacientů, kožní hemoragická diatéza rovněž u 4 pacientů, krvácení do CNS pouze u 1 chlapce ve věku 3 let. K manifestaci autoimunního onemocnění došlo u 3 pacientů ve věku 3–4 let, u všech se jednalo o autoimunní hemolytickou anémii, u 1 z nich došlo v dalších letech k rozvoji těžké autoimunní vaskulitidy. U žádného ze 6 pacientů nedošlo k manifestaci maligního onemocnění. Diagnóza WAS byla stanovena ve věku 3 až 50 měsíců, medián 12 měsíců.
Mutace genu WASP byla prokázána u všech 6 pacientů (tab. 3), zpočátku byl materiál na molekulárně genetické vyšetření odesílán do specializovaných zahraničních center, od roku 2004 probíhá vyšetřování v genetické laboratoři CKTCH v Brně.
Ze 6 pacientů s diagnózou WAS zemřel jeden netransplantován ve věku 46 měsíců. Pět pacientů podstoupilo alogenní transplantaci kmenových buněk krvetvorby (tab. 4). Čtyři chlapci byli transplantováni do 5 let věku (13–54 měsíců), 1 pacient byl transplantován až ve 13 letech. Dárcem kmenových buněk byl HLA identický sourozenec (1), nepříbuzný dárce (3) nebo haploidentický rodinný dárce (1). Zdrojem kmenových buněk byla kostní dřeň (3x), T-depletované periferní kmenové buňky (1x; haploidentický rodinný dárce) a kombinace KD a PBSC (1x; velký váhový nepoměr dárce-příjemce).
Všech 5 transplantací bylo provedeno po myeloablativním přípravném režimu. U žádného pacienta nedošlo k rejekci či selhání štěpu. Tři pacienti dosáhli kompletní dárcovské krvetvorby již D +21 po transplantaci, 1 pacient D +67 a u 1 chlapce trvá 5 let po transplantaci stabilní smíšený chimerismus (do 5 % autologní krvetvorby). Akutní GVHD (2. stupně) se vyskytla u 2 z 5 pacientů, chronická GVHD nebyla zaznamenána u žádného pacienta. Dva pacienti byli po transplantaci léčeni rituximabem (1x recidivující AIHA, 1x EBV lymfoproliferace). Dva pacienti mají hypotyreózu vyžadující substituční léčbu. Jeden pacient zemřel D +25 po transplantaci na cytomegalovirovou infekci. Čtyři chlapci žijí v dobrém klinickém stavu (37, 60, 64 a 84 měsíců po transplantaci), jeden z nich dosud závislý na substituci IVIG (84 měsíců po transplantaci; pacient po opakovaných aplikacích rituximabu pro recidivující AIHA po transplantaci). Tři pacienti mají normální parametry buněčné i humorální imunity, nemocnost je u nich nezvýšená a růstová dynamika v normě.
Závěr
WAS je onemocnění velmi vzácné a právě z toho důvodu jeho diagnostika i v současné době jistě není snadná.
Na diagnózu WAS je třeba myslet vždy v případě trombocytopenie v kombinaci s ekzémem a recidivujícími respiračními infekty. V iniciálních fázích onemocnění však imunodeficience a ekzém mohou chybět, zejména tito pacienti mohou být chybně diagnostikováni a léčeni pod diagnózou idiopatické trombocytopenické purpury. Patognomická pro WAS je přítomnost malých trombocytů. V případě podezření na WAS musí být dětský pacient co nejdříve předán do hematologického centra, které ve spolupráci s imunologickým pracovištěm potvrdí diagnózu, zajistí molekulárně genetické vyšetření mutací genu WASP a zahájí vyhledávání HLA identického dárce.
Transplantace kmenových buněk krvetvorby je v současné době jedinou možností vyléčení WAS. Úspěšnost SCT je stejně jako u jiných kombinovaných imunodeficitů velmi závislá na včasném stanovení diagnózy a indikaci pacienta k transplantaci a na efektivní profylaxi a cílené léčbě infekčních komplikací
Tato práce byla podpořena VZ FNM MZ 000 64203.
Došlo: 26. 11. 2008
Přijato: 2. 2. 2009
MUDr. Renata Formánková, PhD.
Klinika dětské hematologie a onkologie
UK 2. LF a FN Motol
V Úvalu 84
150 06 Praha 5
e-mail: renata.formankova@lfmotol.cuni.cz
Sources
1. Wiskott A. Familial congenital Morbus Werlhoffi? Monatsschr. Kinderheilkd. 1937;68 : 212–216.
2. Aldrich RA, Steinberg AG, Campbell DC, et al. Pedigree demonstrating a sex-linked recessive condition characterized by draining ears, eczematoid dermatitis and bloody diarrhea. Pediatrics 1954;13 : 133–139.
3. Sullivan KE, Mullen CA, Blaese RM, et al. A multiinstitutional survey of the Wiskott-Aldrich syndrome. J. Pediatr. 1994;125 : 876–885.
4. Ochs HD, Trasher J. The Wiskott-Aldrich syndrome. J. Allergy Clin. Immunol. 2006;117 : 725–738.
5. Filipovich AH, Stone JV, Tomany SC, et al. Impact of donor type on outcome of bone marrow transplantation for Wiskott-Aldrich syndrome: collaborative study of the International Bone Marrow Transplant Registry at the National Marrow Donor Program. Blood 2001;97 : 1598–1603.
6. Cooper MD, Chae HP, Lowman JT, et al. Wiskott-Aldrich syndrome: an immunological deficiency disease involving the afferent limb of immunity. Am. J. Med. 1968;44 : 499–513.
7. Derry JM, Ochs HD, Francke U. Isolation of a novel gene mutated in Wiskott-Aldrich syndrome. Cell 1994;78 : 635–644.
8. Villa A, Notarangelo L, Macchi P, et al. X-linked trombocytopenia and Wiskott-Aldrich syndrome are allelic diseases with mutations in the WASP gene. Nat. Genet. 1995;9 : 414–417.
9. Ochs HD, Notarangelo LD. Structure and function of the Wiskott-Aldrich syndrome protein. Curr. Opin. Hematol. 2005;12 : 284–291.
10. Ochs HD. The Wiskott-Aldrich syndrome. Isr. Med. Assoc. J. 2002;4 : 379–384.
11. Jin Y, Mazza C, Christie JR, et al. Mutation of the Wiskott-Aldrich syndrome protein (WASP): hotspots, effect on transcription and translation and phenotype/genotype correlation. Blood 2004;104 : 4010–4019.
12. Ozsahin H, Cavazzana-Calvo M, Notarangelo LD, et al. Long-term following hematopoietic stem cell transplantation in Wiskott-Aldrich syndrome: collaborative study of the European Society for Immunodeficiencies and European Group for Blood and Marrow Transplantation. Blood 2008;111 : 439–445.
13. Burns S, Cory GO, Vainchenker W, et al. Mechanisms of WASp-mediated hematologic and immunologic disease. Blood 2004;104 : 3456–3462.
14. Park JY, Kob M, Prodeus AP, et al. Early deficit of lymphocytes in Wiskott-Aldrich syndrome: possible role of WASP in human lymphocyte maturation. Clin. Exp. Immunol. 2004;136 : 104–110.
15. Yamada M, Ohtsu M, Kobayashi I, et al. Flow cytometric analysis of Wiskott-Aldrich syndrome (WAS) protein in lymphocytes from WAS patients and their familial carriers. Blood 1999;93 : 756–757.
16. Kwan SP, Lehner T, Hagemann T, et al. Localization of the gene for the Wiskott-Aldrich syndrome between two flanking markers, TIMP and DXS255, on Xp11.22-Xp11.3. Genomics 1991;10 : 29–33.
17. Notarangelo LD, Mazza C, Viliami S, et al. Missense mutations of WASP gene cause intermitent X-linked trombocytopenia. Blood 2002;99 : 2268–2269.
18. Notarangelo LD, Miao CH, Ochs HD. Wiskott-Aldrich syndrome. Current Opinion in Hematology 2008;15 : 30–36.
19. Ancliff PJ, Blundell MP, Cory GO, et al. Two novel activating mutations in the Wiskott-Aldrich syndrome protein result in congenital neutropenia. Blood 2006;108 : 2182–2189.
20. Imai I, Morio T, Zhu Y, et al. Clinical course of patients with WASP gene mutations. Blood 2004;103 : 456–464.
21. Cohen JM, Sebire NJ, Harvey J, et al. Successful treatment of lymphoproliferative disease complicating primary immunodeficiency/immunodysregulatory disorders with reduced-intensity allogeneic stem-cell transplantation. Blood 2007;110 : 2209–2214.
22. Charrier S, Dupre L, Scaramuzza S, et al. Lentiviral vectors targeting WASp expression to hematopoietic cells, efficiently transduce and correct cells from WAS patients. Gene Ther. 2007;14 : 415–428.
23. Dupre L, Marangoni F, Scaramuza S, et al. Efficacy of gene therapy for Wiskott-Aldrich syndrome using a WAS promoter/cDNA-containing lentiviral vector and nonlethal irradiation. Hum. Gene Ther. 2006;17 : 303–313.
Labels
Neonatology Paediatrics General practitioner for children and adolescentsArticle was published in
Czech-Slovak Pediatrics

2009 Issue 3
- What Effect Can Be Expected from Limosilactobacillus reuteri in Mucositis and Peri-Implantitis?
- The Importance of Limosilactobacillus reuteri in Administration to Diabetics with Gingivitis
Most read in this issue
- Inzulinóm – príčina recidivujúcich hypoglykémií u 16-ročného pacienta
- Wiskottův-Aldrichův syndrom – onemocnění vyžadující včasnou transplantaci kmenových buněk krvetvorby
- Porucha růstu a vývoje u chlapce s X-vázanou ichtyózou, protrahovaným porodem a nízkou hladinou estriolu u matky v průběhu těhotenství
- Anafylaxe u dětí vyvolaná potravinami