
Deficit pyruvátkinázy v dětském věku
Pyruvate kinase deficiency in children
Purpose of the study:
Pyruvate kinase deficiency (PK) is the second most common enzymatic defect leading to hemolytic anemia. The aim is to demonstrate the clinical and laboratory findings in PK deficiency in the first pediatric patients in the Czech Republic who were defined at the molecular level.
Patients and methods:
Four children (10 months – 7 years) were examined for severe nonspherocytic hemolytic anemia with hemoglobin levels 64–97 g/l. All the children required transfusions and phototherapy after delivery, two of them even an exchange transfusion. The clinical finding was dominantly characterized by pallor, subicterus of sclerae and skin. Erythrocyte membrane disorder, hemoglobinopathies and congenital dyserythropoietic anemia were excluded using standard methods. PK activity was determined and sequencing of the gene encoding PK was done. Because of increased ferritin levels, hepcidin level was measured.
Results:
All children have macrocytic anemia with bilirubin values from 38 to 89 micromol/l. PK activity is decreased in all children (23–32%). Two patients are mixed heterozygotes for mutations c.1529C>A (p.Arg510Gln) and c.1594C>T (p.Arg532Trp) and one for the c.1493G>A (p.Arg498His) and c.1529C>A (p.Arg510Gln) mutations. In one case, a homozygous deletion in exon 11, which has not yet been described, was found. This patient with the most severe course of the disease has a high ferritin level, but a reduced level of hepcidin, which may contribute to iron overload. Coincidence with type I hemochromatosis was excluded.
Conclusion:
PK deficiency in the newborn may cause severe anemia and hyperbilirubinemia sometimes requiring an exchange transfusion and repeated administration of transfusions in infancy; their frequency is usually gradually reduced. PK deficiency may also be associated with iron overload and the need for chelation therapy. In the Czech population this disease is still underdiagnosed.
Key words:
glycolysis, pyruvate kinase, nonspherocytic hemolytic anemia, iron overload, hepcidin
Authors:
B. Ludíková 1; R. Mojzíková 2; P. Pospíšilová 2; J. Houda 1; L. Sulovská 1; M. Divoká 3; J. Hak 4; D. Procházková 5; V. Divoký 2; D. Pospíšilová 1
Authors‘ workplace:
Dětská klinika Lékařské fakulty Univerzity Palackého a Fakultní nemocnice, Olomouc
přednosta prof. MUDr. V. Mihál, CSc.
1; Ústav biologie Lékařské fakulty Univerzity Palackého, Olomouc
ředitel doc. RNDr. V. Divoký, Ph. D.
2; Hemato-onkologická klinika Lékařské fakulty Univerzity Palackého a Fakultní nemocnice, Olomouc
přednosta prof. MUDr. K. Indrák, DrSc.
3; Dětská klinika Lékařské fakulty Univerzity Karlovy a Fakultní nemocnice, Hradec Králové
přednosta prof. MUDr. M. Bayer, CSc.
4; Dětské oddělení, Krajská zdravotní, a. s. – Masarykova nemocnice, Ústí nad Labem
primář MUDr. J. Škvor, CSc.
5
Published in:
Čes-slov Pediat 2012; 67 (3): 152-159.
Category:
Original Papers
Overview
Účel studie:
Deficit pyruvátkinázy (PK) je druhý nejčastější enzymatický defekt vedoucí k hemolytické anemii. Cílem práce je demonstrovat klinické a laboratorní nálezy u deficitu PK u prvních dětských pacientů v České republice definovaných na molekulární úrovni.
Pacienti a metody:
Čtyři děti (10 měsíců – 7 let) byly vyšetřeny pro závažnou nesférocytární hemolytickou anemii s hladinou hemoglobinu 64–97 g/l. Všechny děti vyžadovaly po porodu fototerapii a transfuzi, dvě dokonce výměnnou transfuzi. V klinickém nálezu dominovala bledost, subikterus sklér a kůže. Standardními metodami byla vyloučena porucha membrány erytrocytu, hemoglobinopatie a kongenitální dyserytropoetická anemie. Byla stanovena aktivita PK a provedena sekvenace genu kódujícího PK. Pro zvýšení hladin feritinu byla vyšetřena hladina hepcidinu.
Výsledky:
Všechny děti mají makrocytární anemii. Hodnoty bilirubinu se pohybují v rozmezí 38–89 μmol/l. Aktivita PK je u všech dětí snížená (23–32 %). Dva pacienti jsou smíšení heterozygoti pro mutace c.1529C>A (p.Arg510Gln) a c.1594C>T (p.Arg532Trp) a jeden pro mutace c.1493G>A (p.Arg498His) a c.1529C>A (p.Arg510Gln). V jednom případě se jedná o homozygotní deleci v exonu 11, která nebyla dosud popsána. Tento pacient s nejtěžším průběhem nemoci má vysokou hladinu feritinu, avšak sníženou hladinu hepcidinu, což se může podílet na přetížení železem. Byla vyloučena koincidence s hemochromatózou typu I.
Závěr:
Deficit PK může být u novorozence příčinou závažné anemie a hyperbilirubinemie vyžadující někdy výměnnou transfuzi a opakované podání transfuzí v kojeneckém věku, jejichž frekvence se většinou postupně snižuje. Deficit PK může být provázen i přetížením železem s nutností chelatační léčby. V české populaci je onemocnění zatím poddiagnostikováno.
Klíčová slova:
glykolýza, pyruvátkináza, nesférocytární hemolytická anemie, přetížení železem, hepcidin
ÚVOD
Deficit pyruvátkinázy (PK) je nejčastější enzymatickou abnormalitou glykolytické dráhy a spolu s deficitem glukózo-6-fosfátdehydrogenázy (G6PD) nejčastější příčinou nesférocytární hemolytické anemie [1, 2]. Poprvé byl popsán počátkem šedesátých let minulého století Valentinem a spolupracovníky [3]. Jedná se o vrozené autozomálně recesivně dědičné metabolické onemocnění s prevalencí 1 : 20 000 [2].
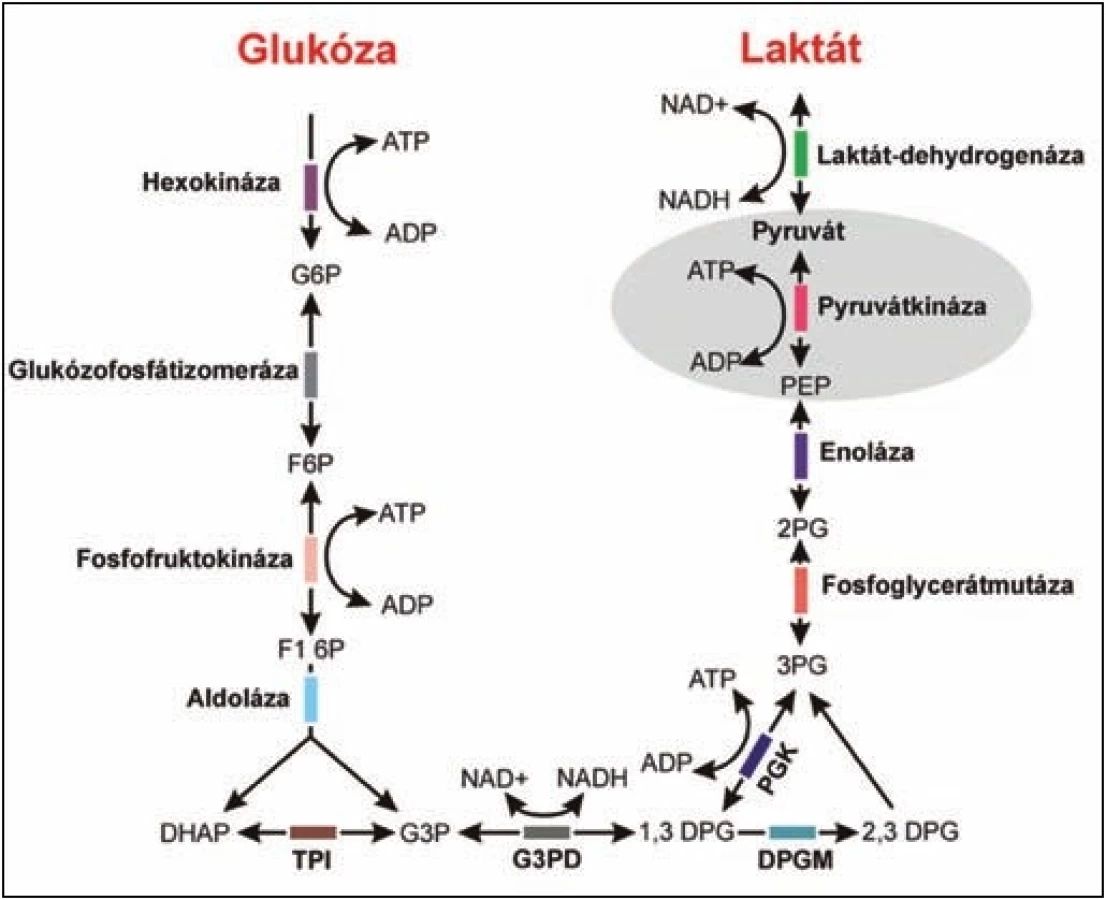
Pyruvátkináza hraje klíčovou roli v glykolytické dráze. Katalyzuje jeden ze dvou kroků při tvorbě adenozintrifosfátu (ATP).
Vzhledem k tomu, že zralé červené krvinky neobsahují mitochondrie, je glykolýza jedinou dráhou generující ATP, který je nezbytný pro další metabolické pochody, pro zpětnou regulaci glykolýzy, membránový transport a pro zachování flexibility a integrity membrány erytrocytu. Mimo jiné zde dochází ke generaci redukované formy nikotinamidadenindinukleotidu (NADH) nezbytného pro redukci methemoglobinu na hemoglobin a 2,3 difosfoglycerátu (2,3-DPG), regulátoru afinity hemoglobinu ke kyslíku. Deficit PK se tak projeví neefektivní glykolýzou vedoucí ke snížené životaschopnosti erytrocytů a extravaskulární hemolýze.
PK je aktivní ve formě tetrameru. U savců byly popsány čtyři různé izoenzymy PK (M1, M2, L, R), které jsou specifické pro jednotlivé tkáně [4], z nichž pouze R forma se vyskytuje výhradně v červených krvinkách [5]. Jaterní (PK-L) a erytrocytární (PK-R) izoenzymy jsou kódovány genem PKLR, který se nachází na chromozomu 1 (1q21) [4]. Kódující oblast je rozdělena do dvanácti exonů [6]. Exon 1 je specifický pro transkripci erytrocytární formy, exon 2 pro jaterní formu, zbývající exony pak jsou společné. Dva další izoenzymy PK-M1 a PK-M2 jsou kódovány genem PKM nacházejícím se na 15. chromozomu. PK-M1 se vyskytuje převážně v srdci, mozku a kosterním svalstvu, PK-M2 ve fetálních tkáních, leukocytech a trombocytech. Dosud bylo popsáno přes 180 mutací PKLR genu, které jsou asociovány s defektem PK. Většinou se jedná o mutace měnící smysl kodonu a způsobující jednoduchou aminokyselinovou záměnu, která má za následek snížení enzymové aktivity na různých úrovních [7]. Mezi nejčastější mutace patří: 1) c.1529G>A typická pro USA a severní a střední Evropu, 2) c.1456C>T častá v jižní Evropě a 3) c.1468C>T vyskytující se v Asii [2, 3, 7].
Klinické symptomy deficitu pyruvátkinázy se objevují u jedinců s homozygotní mutací nebo u složených heterozygotů. Heterozygoti jsou přenašeči bez projevů onemocnění, výjimečně se u nich mohou projevit příznaky mírné hemolýzy. Průběh onemocnění je velmi variabilní, od kompenzované anemie bez klinických projevů přes lehkou anemii s exacerbacemi hemolýzy při infekcích až po těžkou anemii s nutností opakovaného podávání transfuzí. Vždy je přítomna retikulocytóza. U novorozenců s deficitem PK se poměrně často objevuje významná anemie a hyperbilirubinemie s rizikem rozvoje jádrového ikteru, která si v některých případech může vyžádat výměnnou transfuzi. Byly popsány i případy hydropsu plodu [8] a život ohrožující anemie u novorozence. U nemocných s chronickou anemií se mohou vyvinout žlučové kameny, a to již v první dekádě života. U pacientů s deficitem PK byly popsány i tranzientní aplastické krize způsobené parvovirem B19. K vzácným komplikacím patří chronické vředy na dolních končetinách, akutní pankreatitidy, abscesy slinivky, útlak míchy extramedulární hematopoetickou tkání a tromboembolická onemocnění [9]. Hemolýza nebývá provokována léky, někteří autoři však popisují zvýšení hemolýzy po podání vyšších dávek salicylátů.
Podezření na enzymový deficit obecně podporuje obraz normocytární, méně často makrocytární anemie. Biochemické parametry, jako je zvýšení nekonjugovaného bilirubinu a laktátdehydrogenázy (LD) a snížení hladiny haptoglobinu, odpovídají známkám extravaskulární hemolýzy. Při morfologickém hodnocení erytrocytů v nátěru periferní krve bývá přítomná polychromazie, anizocytóza, v některých případech ojedinělé normoblasty (jaderné erytroidní buňky), vzácně echinocyty a akantocyty.
Vzhledem k absenci jednoznačného klinického i laboratorního diagnostického znaku, který by odpovídal deficitu PK, je vždy nutné nejprve vyloučit běžně se vyskytující vrozené hemolytické anemie, zejména hereditární sférocytózu, případně nestabilní hemoglobinopatie. K odlišení sférocytózy je možné použít test kryohemolýzy a test s eosin-5 - -maleimidem, jejichž výsledky jsou u enzymového defektu normální. K vyloučení hemoglobinopatií je vhodné provést elektroforézu hemoglobinu, isopropanolový test či test tepelné stability erytrocytů, které jsou u enzymopatií negativní. Výskyt Heinzových tělísek může odpovídat nestabilní hemoglobinopatii nebo souviset s enzymopatiemi v metabolismu glutathionu (G6PD aj.). Pomocným vyšetřením může být test autohemolýzy, který je u většiny pacientů patologický a v případě deficitu PK se upravuje pouze po přidání ATP, nikoli však glukózy. K přesné diagnóze deficitu PK však stejně tak jako ostatních erytrocytárních enzymopatií slouží průkaz snížené kinetické aktivity enzymu. Diagnózu definitivně potvrzuje molekulárně genetické vyšetření na DNA/RNA úrovni prokazující kauzální mutaci genu PKLR.
Terapie onemocnění je symptomatická, kauzální léčba zatím neexistuje. Někteří nemocní jsou závislí na transfuzní léčbě. Splenektomie je vyhrazena pro nejtěžší případy onemocnění s těžkou anemií, u kterých může vést k omezení nutnosti transfuzní léčby a ke stabilizaci hodnot hemoglobinu. U části pacientů s deficitem PK se rozvíjí přetížení organismu železem, které si může vyžádat léčbu chelátory. Byla popsána i úspěšná transplantace kostní dřeně u malého dítěte [10]. Je zkoumána možnost genové léčby.
V české populaci bylo popsáno jen několik případů dospělých pacientů s deficitem PK s prokázanou mutací genu pro PK [11]. V tomto článku prezentujeme klinický a laboratorní profil prvních dětských pacientů s průkazem kauzální mutace pro PK.
PACIENTI
Soubor pacientů tvoří čtyři děti ve věku 10 měsíců – 7 let s diagnózou chronické hemolytické anemie nejasné etiologie. U všech pacientů byla při základním vyšetření krevního obrazu a markerů hemolýzy zjištěna různě závažná anemie s projevy extravaskulární hemolýzy.
Pacienti č. 1 a 2
První dva pacienti jsou sourozenci, děvče a chlapec ve věku 4 roky a 10 měsíců. Obě děti se narodily v termínu, dívka byla hypotrofická s porodní hmotností 2100 g a porodní délkou 44 cm, chlapec se narodil s hmotností 3130 g a délkou 51 cm.
Obě děti měly protrahovaný novorozenecký ikterus s nutností fototerapie, u chlapce bylo dokonce nutné provést výměnnou transfuzi. Makrocytární anemie s výraznou retikulocytózou je u obou dětí provázena mírnou hyperbilirubinemií a v prvním roce života si u obou vyžádala opakované podání transfuze erytrocytární masy. V klinickém nálezu u dětí dominuje bledost a subikterus sklér a kůže, mírná hepatosplenomegalie a systolický šelest v prekordiu bez průkazu vrozené srdeční vady při echokardiografickém vyšetření.
Po prvním roce věku byly u dívky podávány transfuze erytrocytární masy již méně často, většinou při poklesu hemoglobinu v souvislosti s infekcí. U dívky a obou rodičů nebyly překvapivě při screeningovém vyšetření aktivity G6PD a PK na jiném pracovišti prokázány patologické hodnoty. Rodiče dětí jsou zdraví, netrpí žádným hematologickým ani metabolickým onemocněním.
Pacient č. 3
Třetím nemocným je 11měsíční chlapec, který byl po propuštění z novorozeneckého oddělení odeslán na hematologickou ambulanci pro suspektní vrozenou hemolytickou anemii. Je z prvního těhotenství šestnáctileté matky. Má závažnou perinatální anamnézu. Byl narozen v termínu s porodní hmotností 3020 g, po porodu byl těžce hypoxický a byla nutná intubace a napojení na umělou plicní ventilaci po dobu 6 dnů. Ihned po porodu byla zjištěna vysoká hladina bilirubinu (90 µmol/l), těžká anemie a trombocytopenie. Byla provedena výměnná transfuze. V dalším průběhu byla nutná po dobu 5 dní fototerapie a opakovaně byla podána transfuze erytrocytární masy.
Při klinickém vyšetření v hematologické ambulanci byl nápadný mírný ikterus s šedým nádechem kůže dítěte, lehce ikterické skléry a výrazná hepatosplenomegalie. Dítě bylo přijato k hospitalizaci k vyšetření pro anemii a pro podezření na vrozenou metabolickou vadu. Otec nežije s rodinou a o jeho zdravotním stavu nelze získat dostačující informace.
Pacient č. 4
Poslední pacient je 7letý chlapec odeslaný k dovyšetření anemie nejasného původu. Chlapec byl anemický již v novorozeneckém věku, po porodu měl časnou nekonjugovanou hyperbilirubinemii, bez sérologického konfliktu, která si vyžádala osmidenní fototerapii. Vzhledem k postupné anemizaci v měsíci věku byla nutná další transfuzní léčba. Koncentrace hemoglobinu (Hb) se pohybovala kolem 60 g/l, hyperbilirubinemie (224 µmol/l) přetrvávala. K vyloučení poruchy membrány erytrocytu byl proveden test kryohemolýzy s negativním nálezem. Byl vyšetřen test stability Hb, fetální hemoglobin (HbF) alkalickou denaturací a autohemolýza, vše s negativním výsledkem.
Pacient v roce věku podstoupil punkci kostní dřeně, při které byla zjištěna aktivovaná erytropoeza bez dalších specifických patologických nálezů. V dalším sledování měl středně těžkou makrocytární anemii s hladinou Hb kolem 100 g/l, retikulocytózu a lehkou nekonjugovanou hyperbilirubinemii (30 µmol/l). Při screeningu enzymopatií na jiném pracovišti rovněž nebylo potvrzeno snížení aktivity PK nebo G6PD. Matka 7letého chlapce dříve užívala preparáty železa pro sideropenickou anemii.
METODY
U všech dětí bylo provedeno opakované standardní vyšetření krevního obrazu včetně nátěru periferní krve a biochemické vyšetření markerů hemolýzy a metabolismu železa. Test kryohemolýzy, elektroforéza hemoglobinů, průkaz Heinzových tělísek a test tepelné stability byly provedeny podle standardních postupů.
Stanovení aktivity pyruvátkinázy a G6PD
Aktivita G6PD byla stanovena spektrofotometricky pomocí komerčního kitu (The Microplate Neonatal G6PD Screening Assay, Bio-Rad Laboratories, UK).
Aktivita PK byla stanovena následovně: Pro přípravu enzymových lyzátů byla použita periferní krev (heparin) s následným odstraněním leukocytů a destiček filtrací přes směs mikrokrystalické celulózy s α-celulózou. Po odstranění plazmy centrifugací byla připravena buněčná suspenze ve fyziologickém roztoku (1 : 1). Výsledný enzymový lyzát byl připraven přidáním buněčné suspenze do stabilizačního roztoku obsahujícího EDTA a merkaptoethanol (1 : 9).
Aktivita pyruvátkinázy (E.C.2.7.1.40) byla stanovena spektrofotometricky v temperované směsi (37 °C) obsahující laktátdehydrogenázu, ADP, fosfoenolpyruvát a NADH sledováním poklesu absorbance při 340 nm (Infinite 200 NanoQuant, Tecan, Švýcarsko). Specifická aktivita (U/g), která se uvádí v literatuře, byla vztažena k množství hemoglobinu v lyzátu stanoveném rovněž spektrofotometricky při 414 nm [12].
Sekvenace genů PKLR a HFE1
Genomická DNA byla izolována z periferní krve pomocí QIAamp DNA kitu (Qiagen, USA). Všechny exony byly amplifikovány pomocí PCR (PCR primery a podmínky jsou k dispozici na vyžádání). Pro sekvenační reakci odpovídajících PCR fragmentů byl použit BigDye terminator kit v 1.1 (Applied Biosystem, USA) a sekvenační analýzy byly provedeny na sekvenátoru ABI Prism 3100 (Applied Biosystems, USA).
Stanovení hladiny hepcidinu
Hladiny hepcidinu (bioaktivní forma hepcidinu--25) byly stanoveny v plazmě. Část plazmy určená k měření hladiny hepcidinu byla rozdělena do alikvotů, aby se předešlo opakovanému rozmrazování, a zamražena při -80 °C. K měření hladin hepcidinu byl použit komerční ELISA kit (DRG Instruments, Německo).
Parametry metody podle výrobce byly následující: dynamický rozsah 0,9–140 ng/ml, senzitivita této metody byla stanovena na 0,9 ng/ml, reprodukovatelnost – intra assay CV 4,86 %, inter assay test CV 11,42 %.
VÝSLEDKY
U 3 dětí (pacienti 1, 2 a 4) se jednalo o makrocytární, v jednom případě o přechodně normocytární anemii (výsledek zkreslen předchozí aplikací erytrocytární masy – pacient č. 3). Hladina Hb se pohybovala v rozmezí 62 až 100 g/l a retikulocyty byly v rozmezí 10–19 %. Při biochemickém vyšetření byla u všech pacientů výrazně zvýšená hladina LD a jen lehce vyšší hladina nekonjugovaného bilirubinu (tab. 1). U pacientů č. 2 a 3 byla prokázána výrazně zvýšená hladina feritinu: 716 µg/l a 4853 µg/l.
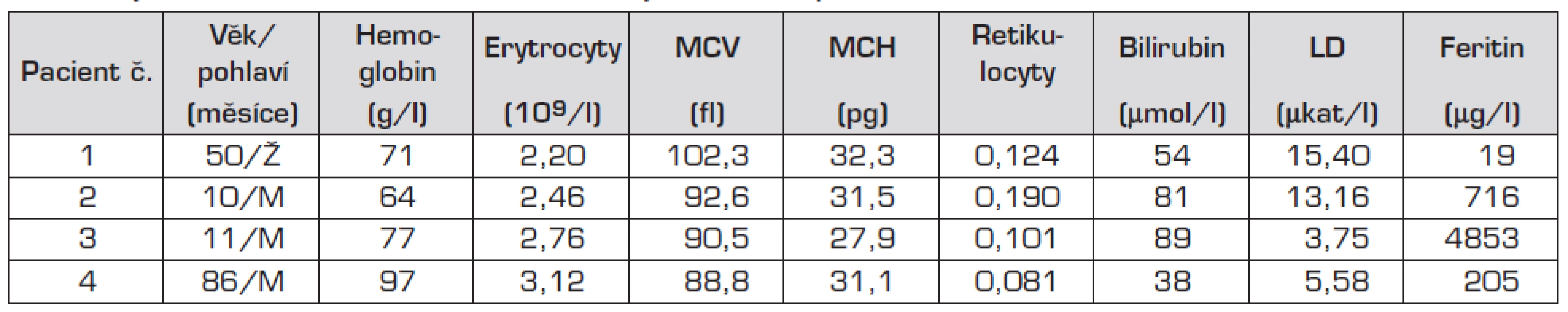
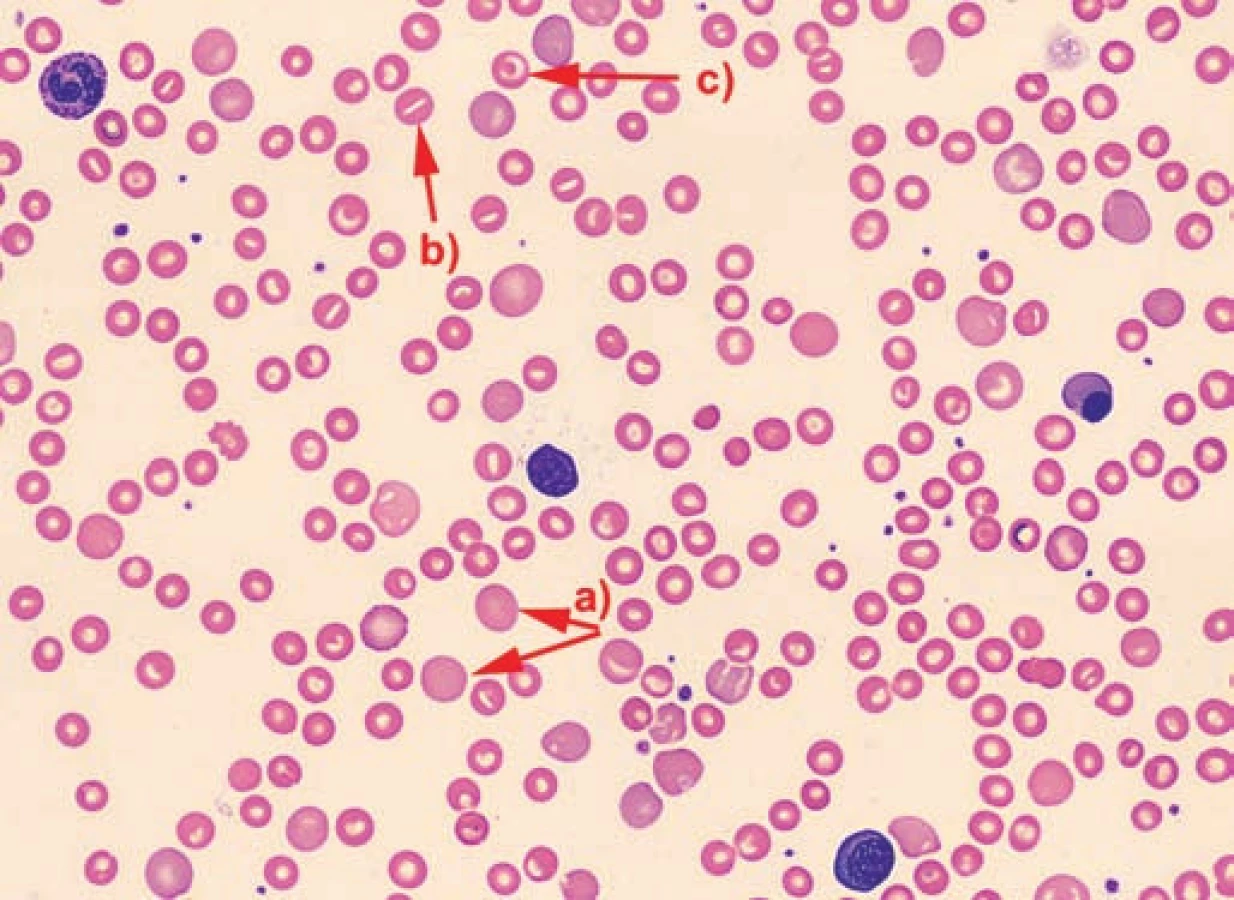
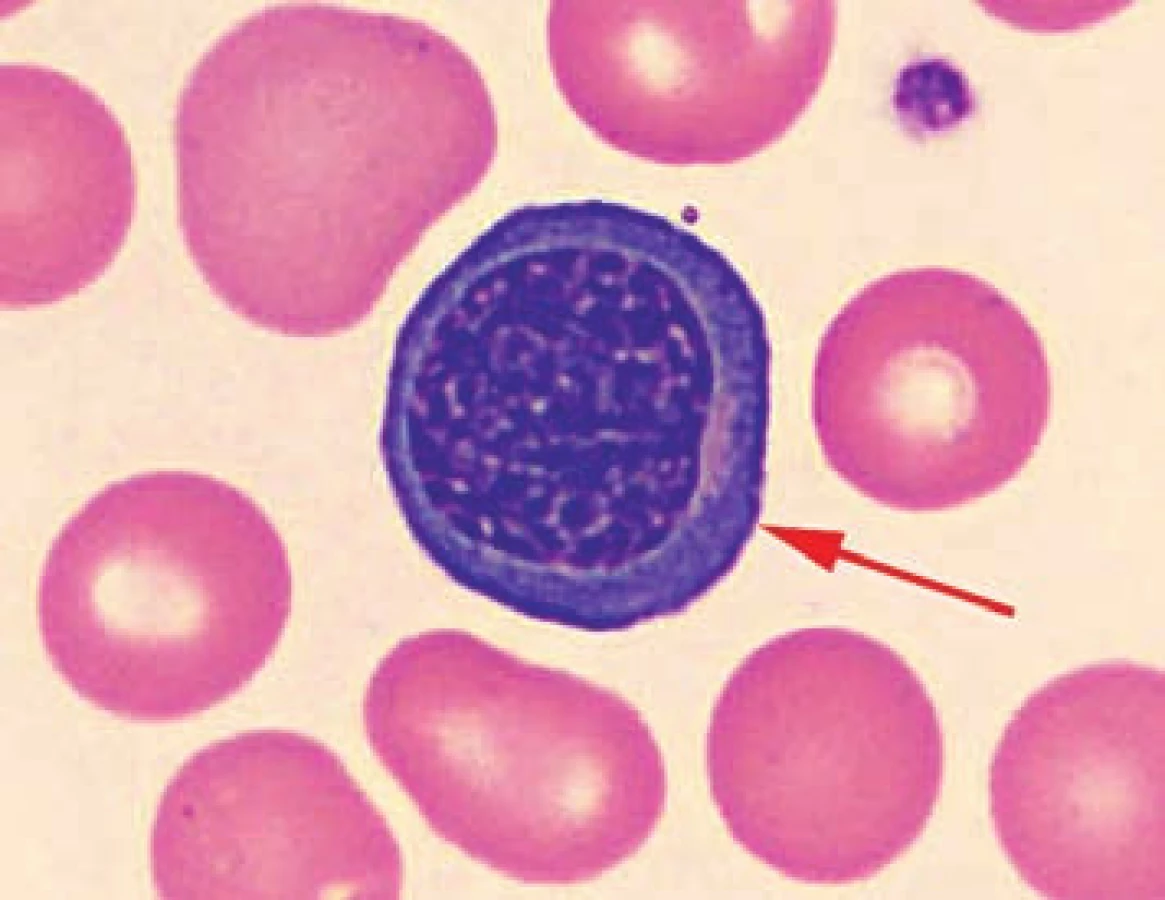
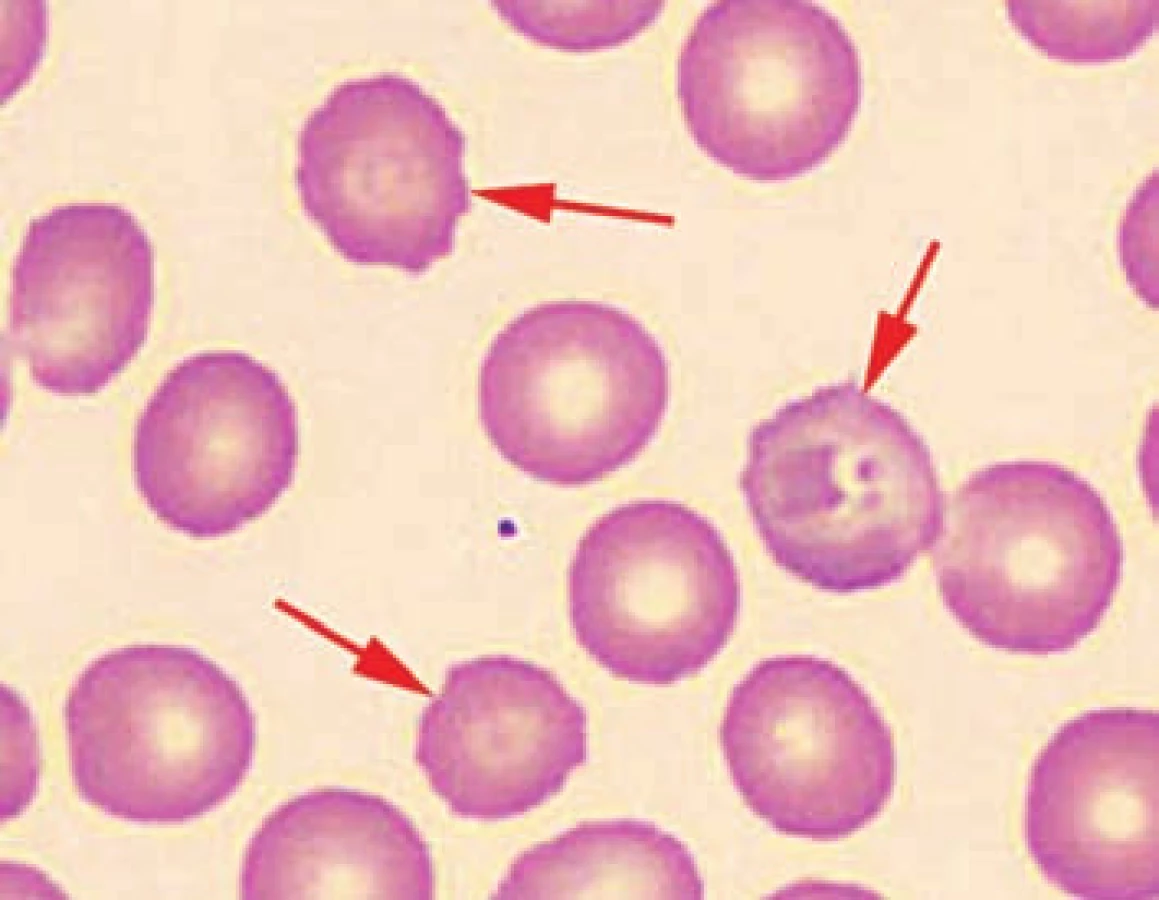
V nátěru periferní krve byly u všech pacientů patrné nespecifické změny morfologie erytrocytů: polychromazie, anizocytóza a u pacientů č. 2 a 3 rovněž terčovité erytrocyty a stomatocyty. U pacienta č. 3 byly navíc přítomny echinocyty a ojedinělé normoblasty.
Porucha membrány erytrocytů a nestabilní hemoglobinopatie byly u všech dětí vyloučeny testem kryohemolýzy, elektroforézou hemoglobinů, průkazem Heinzových tělísek a testem tepelné stability, které byly ve všech případech negativní.
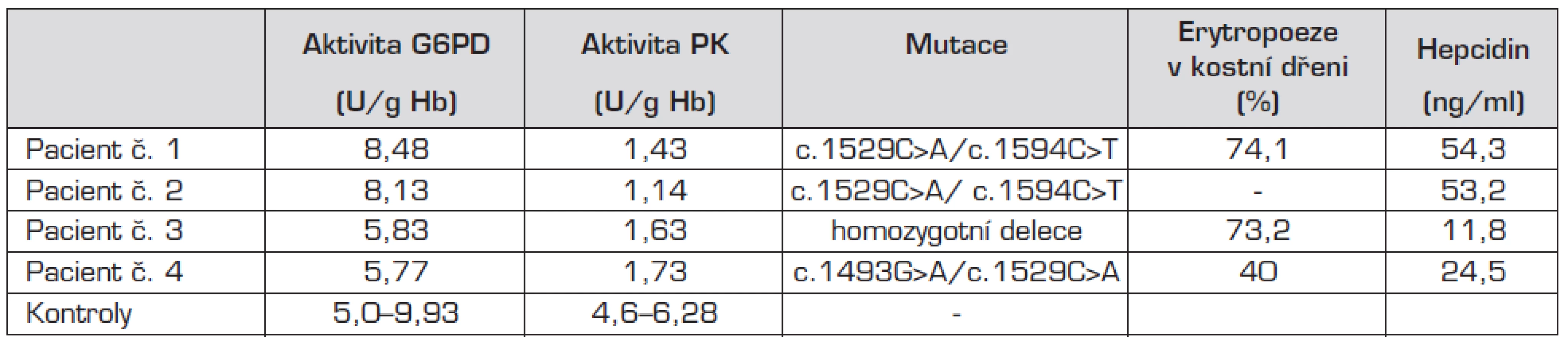
Vyšetření kostní dřeně u pacientů č. 1, 3 a 4 prokázalo výraznou akcentaci prekurzorových buněk erytropoezy (40 až 74,1 %) se známkami mírné dysplazie, avšak bez nálezu zvýšeného procenta vícejaderných erytroblastů a prstenčitých sideroblastů. Byla tedy vyloučena kongenitální dyserytropoetická anemie i sideroblastická anemie.
U všech nemocných byla stanovena aktivita pyruvátkinázy, která byla ve všech případech snížená a odpovídala přibližně 30 % hodnoty kontrolních aktivit. Sekvenováním genu pro PK byla u všech 4 pacientů prokázána kauzální mutace. Oba sourozenci (pacienti č. 1 a 2) jsou smíšenými heterozygoty pro známé mutace c.1529C>A (p.Arg510Gln) a c.1594C>T (p.Arg532Trp) nacházející se v exonu 11. U pacienta č. 3 se jedná o dosud nepopsanou homozygotní aminokyselinovou deleci v exonu 11, která vede k posunu čtecího rámce a tvorbě proteinu, kterému chybí část aktivační domény. Tato mutace nebyla doposud popsána a její funkční studie dále probíhají. Pacient č. 4 je také smíšený heterozygot pro dvě již popsané mutace, a to mutaci c. 1493G>A (p.Arg498His) a opět c.1529C>A (p.Arg510Gln).
Vzhledem ke zvýšení hladiny feritinu byla u všech pacientů vyšetřena hladina hepcidinu, která byla u pacienta č. 3 snížená (11,3 ng/ml), u pacienta č. 2 na dolní hranici normy. U pacientů č. 1 a 4 byla hodnota hepcidinu normální.
Vzhledem k neobvykle vysoké hladině feritinu bylo u pacienta č. 3 provedeno i molekulárně genetické vyšetření k vyloučení současně přítomné mutace pro hemochromatózu typu 1 (HFE), aby se vyloučila nebo potvrdila možná koincidence deficitu PK s některou z dalších poruch metabolismu železa – vrozenou hemochromatózou. Nejčastější mutace spojené s HFE1 – c.845C>A (p.Cys 282Tyr), c.187C>G (p.His63Asp), c.193A>T (p.Ser65Cys) – však nebyly sekvenační analýzou prokázány.
DISKUSE
Na popsaném souboru pacientů poukazujeme na různorodost klinických projevů při deficitu PK. Zajímavé je, že mutace c.1529C>A, kterou jsme detekovali u 3 pacientů, je svým nálezem sice typická pro střední a severní Evropu, ale ve slovanské populaci, pro kterou je charakteristický nález mutace c.1594C>T, se objevuje jen zřídka [11].
I přesto, že není známa přímá korelace mezi genotypem a fenotypem u různých mutací, dá se předpokládat, že mutace měnící čtecí rámec, promotorovou oblast nebo větší delece, které vedou k výrazné redukci aktivity PK, mohou vést k závažnému fenotypu, jako je intrauterinní smrt plodu nebo život ohrožující anemie u novorozence. Na základě studie italských autorů [7], která se zabývá vztahem mezi závažností anemie a konkrétními mutacemi, se dá předpokládat, že některé homozygotní mutace, které mohou být i důsledkem konsanguinity, odpovídají lehkému, střednímu nebo těžkému stupni anemie. U pacientů s mutací c.1456C>T a c.1594C>T jde tak často o mírnou anemii (více než 100 g/l Hb). Mutace c.1529C>A by měla podle uvedených autorů vést ke středně těžké anemii (Hb 80–100 g/l, 10 a méně transfuzí) [3, 7]. U smíšených heterozygotů se dá fenotyp odhadnout jen obtížně, což potvrzují i klinické projevy u obou sourozenců (pacienti č. 1 a 2). I přesto, že oba jsou smíšenými heterozygoty pro mutace c.1529C>A a c.1594C>T, které jsou zmiňované spíše s mírnějšími projevy anemie, oba sourozenci mají těžkou formu anemie s nutností opakovaných transfuzí.
Na patogenezi rozvoje přetížení železem u pacientů s deficitem PK se mohou podílet opakované transfuze, hemolýza, ale i neefektivní erytropoeza. K přetížení železem může přispívat i koincidence deficitu PK s vrozenou poruchou metabolismu železa. Až 20 % evropské populace má mírnou alteraci v metabolismu železa spojovanou s heterozygotním přenašečstvím mutací c.187C>G (p.His63Asp) a c.845C>A (p.Cys282Tyr) v HFE genu způsobujících vrozenou hemochromatózu. Poslední studie prokázaly abnormality v HFE genu u 35 % pacientů s PK deficitem, kteří nejsou závislí na transfuzích [13]. Tyto abnormality jsou spojeny se zvýšením hladiny železa (Fe) v séru a feritinu [14]. Přetížení železem i bez mutace genu pro hemochromatózu bylo popsáno u polytransfundovaných pacientů a pacientů s PK deficitem po splenektomii [14].
Diskutuje se i o možné roli snížení hladiny hepcidinu, klíčové molekuly regulující přísun železa do organismu [15]. Hladina hepcidinu je kontrolována zejména erytropoetickou aktivitou prostřednictvím signálních molekul GDF15 a TWSG1, které reagují na anemii. Stav erytropoezy je pravděpodobně nadřazeným faktorem všem ostatním mechanismům ovlivňujícím metabolismus Fe a hladinu železa v organismu. Hladina hepcidinu se snižuje u anemií, především u stavů se zvýšenou potřebou Fe, jako je sideropenická anemie, což vede ke zvýšenému vstřebávání železa ze střeva. Zvyšuje se naopak u stavů s vysokou hladinou železa v organismu, např. u vrozených hemochromatóz nebo chorob spojených se sekundárním přetížením železem a nezávisle u akutního zánětu. Bylo prokázáno, že u deficitu PK se kombinují oba faktory regulující protichůdně produkci hepcidinu: na jedné straně anemie s vyššími nároky na přísun železa při větším obratu erytropoezy a na straně druhé zvýšená nabídka železa s vysokou hladinou feritinu. Těžká anemie je v tomto případě nadřazena signálům o přetížení organismu železem a produkce hepcidinu je negativně regulována cestou aktivace GDF 15. Výsledkem je potom snížení jeho hladiny s následným zvýšením resorpce Fe i přes jeho zvýšené zásoby, což může přispívat ke zvyšování zásobního železa a k rozvoji přetížení železem. U pacienta č. 3 jsme také prokázali snížení hladiny hepcidinu, která se mohla spolupodílet na neobvykle vysoké hladině feritinu. U pacienta č. 2 byla hladina hepcidinu na dolní hranici normy i při vysoké hladině feritinu. Při přetrvávající výrazné hyperferitinemii bude potřeba u pacienta č. 3 zvážit sekvenování HFE2A genu kódujícího hemojuvelin a HFE3 genu kódujícího transferinový receptor 2 [16].
I když je deficit pyruvátkinázy nejčastější enzymatickou abnormalitou glykolytické dráhy, která způsobuje dědičnou nesférocytární hemolytickou anemii, bylo v České republice u dětí zatím popsáno pouze několik případů. A to i přesto, že podle celosvětové prevalence 1 : 20 000 [2, 7] by v České republice mělo být teoreticky diagnostikováno 500 osob trpících touto nemocí. Jednou z příčin může být určení nesprávné diagnózy, a to z několika možných důvodů. U polytransfundovaných pacientů může být stanovená aktivita PK zkreslená, zejména pokud se stanovuje v odběru bezprostředně po transfuzi nebo v krátkém intervalu od transfuze, protože se tak měří vlastně i aktivita PK dárcovských erytrocytů. Je proto důležité, aby se v těchto případech aktivita PK stanovovala až ve vzorku odebraném těsně před další transfuzí, kdy už prakticky nehrozí „kontaminace“ dárcovskými erytrocyty. Dalším faktorem zkreslujícím vyšetření může být přítomnost leukocytů a trombocytů ve vyšetřovaném materiálu, protože jak leukocyty, tak trombocyty obsahují jinou izoformu enzymu (PK-M2), která má až 300krát vyšší aktivitu než erytrocytární forma PK-R [7]. Vzhledem k tomu, že aktivita řady enzymů souvisí se stářím populace červených krvinek, může mít i zvýšený počet retikulocytů za následek zvýšení aktivity některých enzymů, zejména hexokinázy a pyruvátkinázy.
ZÁVĚR
Cílem tohoto sdělení, které popisuje soubor prvních dětských pacientů s průkazem kauzální mutace genu PKLR, je upozornit na variabilitu klinických i laboratorních příznaků u pacientů s deficitem PK, která může být vzácně provázena závažným přetížením železem, a na úskalí diagnostiky této enzymopatie.
U pacientů s deficitem PK a s rozvojem závažného přetížení železem je vždy nutno zvážit možnost výskytu přidružené vrozené hemochromatózy. Je třeba pečlivě monitorovat stav zásob železa a v indikovaných případech včas zahájit chelatační léčbu jako prevenci rozvoje cirhózy jater při neléčené hemosideróze jater [14].
Práce byla podporována grantem NS9951 [IGA MZ ČR], NT11208 [IGA MZ ČR] a Studentskými projekty LF-2011-006 a LF-2011-011 Univerzity Palackého v Olomouci.
Došlo: 30. 11. 2011
Přijato: 30. 3. 2012
MUDr. Barbora Ludíková
Na Zákopě 15
772 00 Olomouc
e-mail: ludikova.b83@gmail.com
Sources
1. Beutler E, Gelbart T. Estimation the prevalence of pyruvate kinase deficiency from the gene frequency in the general white population. Blood 2000; 95 : 3585–3588.
2. Zanella A, Fermo E, Bianchi P, et al. Red cell pyruvate kinase deficiency: molecular and clinical aspects. Br J Hematol 2005; 130 : 11–25.
3. Valentine WN,Tanaka KR, Miwa S. A specific erythrocyte gylcolytic enzyme defekt [pyruvate kinase] in free subjects with congenital non-spherocytic hemolytic anemia. Transactions of the Association of American Physicians 1961; 74 : 100–110.
4. Satoh H, Tani K, Yoshida MC, et al. The human liver-type pyruvate kinase [PKL] gene is on chromosome 1 at band q21. Cytogenet Cell Genet 1988; 47 : 132–133.
5. Takegawa S, Fuji H, Miwa S. Change of pyruvate kinase isozymes from M2 - to L-type during development of the red cell. Br J Hematol 1983; 54 : 467–474.
6. Tani K, Yoshida MC, Satoh H, et al. Human M2-type pyruvate kinase: cDNA cloning, chromosomal assignment and expression in hepatoma. Gene 1988; 20 : 509–516.
7. Zanella A, Fermo E, Bianchi P, et al. Pyruvate kinase deficiency: The genotype-phenotype association. Blood Rev 2007; 21 : 217–231.
8. Ferreira P, Morais L, Costa R, et al. Hydrops fetalis associated with erythrocyte pyruvate kinase deficiency. Eur J Pediatr 2000; 159 : 481–482.
9. Tanaka KR, Zerez CR. Red cell enzymopathies of the glycolytic pathway. Semin Haematol 1990; 27 : 165–185.
10. Tanphaichitr VS, Suvatte V, Issaragrisil S, et al. Successful bone marrow transplantation in a child with red blood cell pyruvate kinase deficiency. Bone Marrow Transplant 2000; 26 : 689–690.
11. Lenzner C, Nurnberg P, Thiele BJ, et al. Mutations in the pyruvate kinase L gene in patients with hereditary hemolytic anemia. Blood 1994; 83 : 2817–2822.
12. Beutler E, Blume KG, Kaplan JC, et al. International committee for standardization in haematology: recommended methods for red-cell enzyme analysis. Br J Haematol 1977; 35 : 331–340.
13. Zanella A, Bianachi P, Iurlo A, et al. Iron status and HFE genotype in erytrocyte pyruvate kinase deficiency: Study of Italian cases. Blood Cells Mol Dis 2001; 27 : 653–661.
14. Hilgard P, Gerken G. Liver cirrhosis as consequence of iron overload caused by hereditary nonspherocytic hemolytic anemia. World J Gastroenterol 2005; 11 (8): 1241–1244.
15. Viatte L, Vaulont S. Hepcidin, the iron watcher. Biochimie 2009; 91 : 1223–1228.
16. Finkenstedt A, Bianchi P, Theurl I, et al. Regulation of iron metabolism through GDF15 and hepcidin in pyruvate kinase deficiency. Br J Haematol 2009; 144 : 789–793.
Labels
Neonatology Paediatrics General practitioner for children and adolescentsArticle was published in
Czech-Slovak Pediatrics

2012 Issue 3
- What Effect Can Be Expected from Limosilactobacillus reuteri in Mucositis and Peri-Implantitis?
- The Importance of Limosilactobacillus reuteri in Administration to Diabetics with Gingivitis
Most read in this issue
- Neobvyklé manifestace infekce parvovirem B19 u dětí
- Deficit pyruvátkinázy v dětském věku
- Variabilita klinické manifestace norovirové infekce u novorozence – od perakutní nekrotizující enterokolitidy po asymptomatický průběh
- Benigní infantilní křeče asociované s norovirovou gastroenteritidou