
Molekulárna genetika, fenotypová variabilita a súčasné trendy v personalizovanej medicíne fenylketonúrie
Molecular genetics, phenotypic diversity and recent trends of personalized medicine in phenylketonuria
Phenylketonuria is a rare inborn error of amino acid metabolism presenting with autosomal recessive pattern. In the majority of cases, PKU is caused by mutations in the phenylalanine hydroxylase gene (PAH), which catalyses the essential hydroxylation of phenylalanine (Phe) to tyrosine (Tyr) in the presence of its natural cofactor - tetrahydrobiopterin (BH4). In this paper we review the recent knowledge of molecular genetics of PKU, trends in research and we also discuss the molecular diagnostic approaches in this disease. Intensive research has been conducted in the last decade in order to determine the relationship between patients' genotype and clinical picture of the patient, as well as to most accurately assign the relationship of genotype and patients' responsiveness to tetrahydrobiopterin treatment.
In this article we review the recent knowledge of genotype-phenotype correlations and we discuss possible causes of inconsistencies observed in many studies. In addition, we cite the latest findings regarding personalized therapy in PKU regarding cofactor treatment and we also discuss the limitations of genotype-based predictions of BH4 responsiveness. Besides, we bring up the latest data from molecular-genetic studies in Slovakia as well as the spectrum of most frequent mutations and genotypes identified in the Slovak population. This data could be useful for all clinical workplaces in Slovakia.
Key words:
PKU, PAH, HPA, BH4 – responsiveness, mutations, genotype-phenotype correlations
Authors:
E. Polák 1; O. Ürge 2; Ľ. Kádaši 1,3
Authors‘ workplace:
Katedra molekulárnej biológie, Prírodovedecká fakulta Univerzity Komenského, Bratislava
vedúci prof. RNDr. J. Turňa, CSc.
1; Klinika pre deti a dorast A. Gentlíka, Slovenská zdravotnícka univerzita
NsP sv. Cyrila a Metoda, Bratislava
prednostka doc. MUDr. K. Furková, CSc., mim. prof.
2; Molekulárno-medicínske centrum, Slovenská akadémia vied, Bratislava
riaditeľ MUDr. M. Vlček, PhD.
3
Published in:
Čes-slov Pediat 2015; 70 (6): 333-341.
Category:
Review
Overview
Fenylketonúria (PKU) je vzácna dedičná porucha metabolizmu aminokyselín s autozomálne recesívnym typom dedičnosti. PKU spôsobujú vo väčšine prípadov mutácie v géne pre fenylalanín hydroxylázu(PAH), ktorá v pečeni katalyzuje esen-ciálnu hydroxyláciu fenylalanínu (Phe) na tyrozín (Tyr) v prítomnosti svôjho prirodzeného kofaktora – tetrahydrobiopterínu (BH4). V tomto prehľadnom článku sa zaoberáme aktuálnymi poznatkami z molekulárnej genetiky PKU, trendmi vo výskume ochorenia ako aj metodickými prístupmi v jeho molekulárnej diagnostike. Intenzívny výskum PKU sa v poslednej dekáde upriamuje najmä na determináciu vzťahu genotypu a klinického obrazu pacienta, ako aj na čo najpresnejšiu determináciu vzťahu genotypu a responzivity pacienta na liečbu tetrahydrobiopterínom.
V tejto publikácii prinášame aktuálne poznatky o genotypovo-fenotypových koreláciách a diskutujeme možné príčiny inkonzistencií evidovaných v mnohých štúdiách. Taktiež uvádzame najnovšie poznatky z oblasti personalizovanej terapie v súvislosti s liečbou kofaktorom a diskutujeme limitácie genotypovej predikcie responzivity pacienta na túto liečbu. Okrem spomínaného uvádzame aktualizované dáta z molekulárno-genetických štúdií na Slovensku, ako aj spektrum najčastejších mutácií a genotypov identifikovaných v slovenskej populácii. Tieto dáta môžu byť užitočné pre klinické a diagnostické pracoviská na Slovensku.
Kľúčové slová:
PKU, PAH, HPA, BH4 – responzivita, mutácie, genotyp-fenotyp korelácie
Úvod
Fenylketonúria (PKU) a s ňou asociovaná hyperfenyl-alaninémia (HPA) sú heterogénna skupina ochorení s autozomálne recesívnym typom dedičnosti. Sú primárne spôsobené deficienciou hepatálneho enzýmu fenylalanín hydroxylázy (PAH), ktorý je dôležitým článkom v metabolickej dráhe aminokyseliny fenylalanínu (Phe). PAH (EC 1.14.16.1) v normálnej ľudskej pečeni katalyzuje hydroxyláciu L-fenylalanínu (Phe) na L-tyrozín (Tyr) za prítomnosti kyslíka, železa a svojho kofaktora tetrahydro-biopterínu (BH4) [1]. Sekundárnou príčinou vzniku ochorenia je defekt v tvorbe alebo recyklácii kofaktora BH4 [2]. Strata aktivity tohto enzýmu je spôsobená mutáciami v géne PAH a je zodpovedná za akumuláciu Phe v krvi, kyseliny pyruvátovej a jej metabolitov v moči, ktoré vznikajú ako dôsledok úsilia organizmu kompenzovať metabolickú blokádu a odbúrať hromadiaci sa fenylalanín týmto spôsobom. V dôsledku patologických koncentrácií týchto látok dochádza k neurotoxickému poškodeniu centrálnej nervovej sústavy (CNS), čo v najvážnejších prípadoch vedie k ťažkej mentálnej retardácii jedinca. Postihnutiu možno predchádzať len včasnou restrikciou dietického fenylalanínu v neonatálnom veku.
Klinické príznaky fenylketonúrie sa v prípade neliečených pacientov začnú prejavovať asi v 2. mesiaci života. Medzi najčastejšie symptómy patrí psychomotorická retardácia, bledá pokožka, častý výskyt ekzémov, charakteristický myší zápach potu a moču, nápadné svetlé vlasy, prípadne svetlejší odtieň v porovnaní s rodičmi alebo nepostihnutými súrodencami [3]. V kaukazoidnej populácii je priemerná frekvencia približne 1 : 10 000, pričom varíruje medzi jednotlivými subpopuláciami [4]. V Českej republike je to 1 : 7234 [5] a na Slovensku bola stanovená na 1 : 5753 vo výročnej správe Skríningového centra novorodencov Slovenskej republiky z roku 2014 (http://dfnbb.sk/). Frekvencia prenášačov (heterozygótov) v kaukazoidnej populácii je veľmi vysoká, až 1 : 50 [6].
Diagnostika a fenotypová klasifikácia ochorenia
Fenylketonúria je diagnostikovaná v novorodeneckých skríningových programoch mnohých krajín sveta. Referenčnú metódu v niektorých krajinách predstavuje Guthrieho bakteriologický inhibičný test. Umožňuje semikvantitatívne stanovenie hladiny Phe z krvnej škvrny odoberanej ihneď po narodení, väčšinou z päty novorodenca [7]. Zavedenie tandemovej hmotnostnej spektrometrie umožňuje spoľahlivú kvantifikáciu koncentrácií aminokyselín z malého množstva krvi alebo plazmy [8]. Táto metóda produkuje podstatne menej falošne pozitívnych alebo negatívnych výsledkov, keďže stanovuje hladinu oboch aminokyselín (Phe aj Tyr) a taktiež poskytuje pomer Phe/Tyr, čo je podstatne kvalitatívnejšia informácia. Navyše umožňuje detegovať aj iné dedičné poruchy metabolizmu paralelne v jednej analýze [9]. V súčasnosti sa v skríningových centrách po celom svete využívajú rôzne iné metódy na diagnostiku PKU. Najpresnejším spôsobom pri oddiferencovaní PKU pacientov od zdravých jedincov je DNA diagnostika, ktorá je už dobre etablovaná v laboratóriách mnohých európskych krajín [10].
Všetci novorodenci by mali byť skrínovaní pre PKU v prvých dňoch života, kvôli včasnému nasadeniu eliminačnej diéty a predídeniu neurologického poškodenia. Boli vyslovené obavy, že príliž skorá diagnostika môže byť falošne negatívna, nakoľko zvýšenie hladiny Phe v krvi môže nastať až po niekoľkých dňoch po prísune exogénneho Phe pri kojení. Avšak podľa súčasných doporučení prebieha skríning v rozsahu 48–72 hodín po narodení [10]. Pri pozitívnom výsledku je vhodné stanoviť hladinu Phe prípadne pomer Phe/Tyr opakovane, nakoľko maximálna hladina nemusela byť dosiahnutá. Taktiež je to dôležité pre stanovenie kategórie ochorenia. Niektorí novorodenci, obzvlášť tí predčasne narodení, môžu byť pozitívny v skríningu, nakoľko systémy metabolizmu aminokyselín nemusia byť ešte dobre vyvinuté. Výsledky je preto dôležité interpretovať opatrne, čo platí taktiež u chorých (napr. infekcia) novorodencov. Druhý test je preto nevyhnutný. Hraničné hodnoty Phe v krvi sú rôzne v závislosti od kritérií jednotlivých skríningových programov. Bežne používaná „cut-off“ hladina je 120–130 µmol/L (pomer Phe/Tyr >2) [9].
Skríning fenylketonúrie na Slovensku je zavedený od roku 1976 a uskutočňuje sa v troch recall centrách Skríningového centra novorodencov Slovenskej republiky: v Bratislave pre západné Slovensko, v Banskej Bystrici pre stredné Slovensko a v Košiciach pre východné Slovensko. Hladiny fenylalanínu sa stanovujú fluorimetrickou metódou a hmotnostnou tandemovou spektrometriou a cut-off limit Phe je stanovený na 110 µmol/L a zároveň pomer Phe/Tyr 1,5 (http://dfnbb.sk/).
Fenylketonúria je heterogénne ochorenie so širokou škálou metabolického fenotypu v závislosti od závažnosti hyperfenylalaninémie. V súčasnosti neexistuje univerzálny konsenzus pre klasifikáciu tohto ochorenia a je možné brať do úvahy niekoľko parametrov [9]. Iniciálnym a dodnes používaným parametrom pri kategorizácii PKU je skríningová hladina Phe. Na základe tohto údaju sa PKU najčastejšie rozdeľuje do troch kategórií: cPKU – classic phenylketonuria (skríningové hladiny Phe vyššie ako 1200 µmol/l), mierna mPKU – moderate phenylketonuria (Phe od 600 do 1200 µmol/l) a MHP – mild hyperphenylalaninemia (Phe do 600 µmol/l). V rámci tejto klasifikácie je možné pre presnejšie rozdelenie rozlišovať dva subtypy mPKU – miernu (Phe od 900–1200 µmol/l) a ľahkú (Phe 600–900 µmol/l) [11]. Hoci je tento parameter nenahraditelný pri fenotypizácii PKU, je závislý na niekoľkých premenných, ako napríklad načasovanie stanovenia Phe v krvi, metabolizmus novorodenca a príjem exogénneho Phe v strave. Je však dodnes používaný pre fenotypizáciu PKU približne v 80 % skríningových centier [9].
Denná Phe tolerancia sa preukázala ako stabilný parameter pri klasifikácii hyperfenylalaninémií. Stanovuje sa vo veku 5 rokov a indikuje denné množstvo dietického Phe, ktoré nezvýši hladinu Phe v krvi nad kritickú hodnotu 360 µmol/l. Najčastejšie sa podľa tohto ukazovateľa rozlišujú tri fenotypy: cPKU: tolerancia menej ako 20 mg na kg telesnej hmotnosti za deň, mPKU: 20–50 mg/kg//deň a MHP, u ktorej je tolerancia vyššia ako 50 mg/kg//deň. Nedávno bolo dokázané, že predikcia tolerancie je možná aj vo veku 2–3 rokov, čo následne korelovalo s Phe toleranciou vo veku 5 a 10 rokov [12]. Phe tolerancia predstavuje dobrý parameter pre klasifikáciu PKU, avšak musí podliehať prísne stanovenému protokolu [9]. Nedávne štúdie spájajú tieto dve kritériá v nasledovných fenotypových kategóriách [13–15]: Klasická fenylketonúria (cPKU) Phe >1200 µmol/l, Phe tolerancia je <20 mg/kg/deň; mierna fenylketonúria (mPKU) Phe 600–1200 µmol/l, Phe tolerancia je 20–25 mg/kg/deň; benígna hyperfenylalaninémia (MHP) Phe <600 µmol/l, Phe tolerancia je >50 mg//kg/deň. MHP nevyžaduje eliminačnú diétu, v niektorých prípadoch len miernu restrikciu Phe [16].
Okrem spomínaného, dôležitú úlohu pri klasifikácii fenotypu PKU má samozrejme aj klinický obraz pacienta. To znamená, akú má pacient fluktuáciu hladiny Phe a pomeru Phe/Tyr v krvi počas bežnej infekcie, alebo nedodržiavaní nízkoproteínovej diéty, aké zmeny správania a nálady sú pozorované a iné. Inými slovami, pravidelná kontrola príslušným lekárom je u fenylketonurických pacientov nevyhnutná [9].
Približne 2 % pacientov s vysokými hladinami Phe nemajú defekt v PAH, ale v tvorbe alebo recyklácii kofaktora BH4. Z tohto dôvodu je nevyhnutná diferenciálna diagnostika [17]. Pri BH4 deficiencii nie je eliminačná diéta efektívna a liečba sa v porovnaní s PKU podstatne líši. Hladina pterínov u takýchto pacientov je vyššia ako u zdravých ľudí a taktiež ako u PAH deficientných pacientov. Stanovenie pterínov v krvnej škvrne alebo moči, ako aj meranie aktivity dihidropteridín reduktázy (DHPR) je preto esenciálne pre potvrdenie tejto diagnózy a malo by sa vykonať čo najskôr [18]. V tomto smere je dôležitá aj DNA diagnostika, ktorá umožňuje definitívne potvrdenie PKU. Na základe informácií zo Skríningového centra novorodencov SR (RNDr. Mária Knapková – osobná komunikácia), kvantifikácia pterínov pre potvrdenie BH4 deficiencií nie je zaradená v slovenskom skríningovom programe (http://dfnbb.sk/).
Molekulárna genetika fenylketonúrie
PAH gén
Gén pre ľudskú fenylalanín hydroxylázu (PAH; E.C.1.14.16.1) bol lokalizovaný na distálnej časti dlhého ramienka chromozómu č. 12 v oblasti q22 až q24.1. Pokrýva približne 90 kb (kilobáz) genómu a jeho kódujúca oblasť pozostáva z trinástich pomerne krátkych exónov a dvanástich dlhých intrónov [19, 20]. Maturovaná mRNA je dlhá približne 2,6 kb a kóduje polypeptid o veľkosti 452 aminokyselín [6]. Promótorová oblasť má dĺžku približne 9 kb, no len prvých 154 báz 5‘ od štartovacieho kodónu postačuje na minimálnu hladinu transkripcie. Zaujímavosťou je, že promótor tohto génu neobsahuje TATA box [21, 22]. PAH je konštitutívne exprimovaný najmä v pečeni, avšak expresia bola potvrdená aj v obličkách [23]. Regulácia expresie génu je pomerne zložitý proces, keďže spočíva v synchronizovanej akcii niekoľkých transkripčných faktorov [24]. Taktiež bolo v géne PAH identifikovaných množstvo SNP (Single Nucleotide Polymorphism), RFLP (Restriction Fragment Lenght Polymorphism) a multi-alelických polymorfizmov v intrónoch a neprekladaných oblastiach s doposiaľ neznámou funkciou.
Mutácie v PAH géne
Na PAH lokuse sa vyskytujú takmer všetky typy mutácií. Mutácie meniace zmysel (missense) sú najčastejšie zastúpené s početnosťou až 63 %. Jednobázové delécie, delécie kodónu, prípadne časti exónov tvoria približne 13 %. Podiel mutácií, ktoré postihujú zostrihové miesta, je okolo 11 %. Zdánlivo tiché mutácie tvoria približne 7 % [6]. Tu je však nutné poznamenať, že boli identifikované mutácie, ktoré nespôsobujú aminokyselinovú zámenu, avšak spôsobujú aberantný zostrih mRNA. Takáto mutácia je napríklad p.G10G [25]. Nezmyselné (nonsense) mutácie vedú k predčasnému ukončeniu translácie a tvoria približne 5 %. Mutácie vedúce k posunu čítacieho rámca (frameshift) sú dôsledkom zostrihových mutácií alebo delécií časti génu, prípadne veľmi vzácne sa vyskytujúcich inzercií a predstavujú približne 1 %. Taktiež boli identifikované mutácie, ktoré ovplyvňujú stabilitu a processing mRNA a veľké delécie exónov v dôsledku tzv. „preskočenia exónu“ (exon-skipping efect) [26]. Pôvodne sa myslelo, že veľké delécie, inzercie a celogénové prestavby sa vyskytujú len vzácne, avšak ich priemerná frekvencia je približne 3 % [27].
Bolo popísaných niekoľko molekulárnych mechanizmov, akými mutácia poškodí PAH [28]. Avšak spoločný menovateľ väčšiny mutácií je destabilizácia enzýmu s následnou degradáciou bunkou a nesprávne poskladanie do aktívnych štruktúr (diméry a tetraméry enzýmu) [29, 30]. V závislosti od typu a lokalizácie mutácie v géne, môže byť enzým úplne inaktívny, alebo si zachová čiastkovú tzv. reziduálnu aktivitu (RA), ktorá sa stanovuje ako percentuálny podiel aktivity PAH divého typu v in vitro podmienkach. Treba poznamenať, že boli identifikované aj mutácie s vyššou aktivitou ako 100 %. Vďaka intenzívnemu výskumu PKU bola charakterizovaná aktivita všetkých najčastejších mutácií PAH a tieto dáta sú v súčasnosti dostupné v dvoch internetových databázach – PAHdb (http://www.pahdb.mcgill.ca) a PAHvdb (www.biopku.org/pah). Okrem toho, tieto databázy taktiež disponujú množstvom užitočných informácií o PAH géne, haplotypoch a polymorfizmoch tohto lokusu. PAHvdb (PAH Variation Database) databáza je pomerne nová a taktiež pravidelne aktualizovaná. Ku dnešnému dátumu (Júl 2015) bolo v tejto databáze evidovaných viac ako 880 rôznych variant PAH génu.
Metódy molekulárnej diagnostiky PAH génu
Najperspektívnejší nástroj v molekulárnej diagnostike PKU predstavuje priame automatické sekvenovanie DNA. Poskytuje kvalitatívne podstatne obsiahlejšiu informáciu ako skríningové metódy, ktoré zväčša len určia prítomnosť resp. neprítomnosť variácie v analyzovanom úseku (exóne) génu. Pribúdajúce populačné štúdie jednotlivých európskych krajín však naznačujú, že skríningové metódy dokážu stále šetriť náklady a čas a ich aplikácia v týchto programoch je užitočná [13, 14, 31]. V súčasnosti sú najviac používanými skríningovými metódami pre detekciu mutácií v PAH géne predovšetkým DHPLC (Denaturating High Performance Liquid Chromatography), DGGE (Denaturating Gel Gradient Electrophoresis), HRM (High Resolution Melting) a MLPA (Multiplex Ligation-dependent Probe Amplification) využívaná najmä na detekciu veľkých génových delécií a duplikácií.
Treba však poznamenať, že prichádzajúce technológie sekvenovania druhej a tretej generácie sa vďaka neustálemu vývoju a inováciám stávajú viac výkonnými, predovšetkým však viac dostupnými pre rutinné diagnostické použitie. Niet pochýb, že v blízkej budúcnosti nahradia súčasné skríningové metódy ako aj Sangerovo sekvenovanie a stanú sa referenčnou metódou číslo jedna.
Liečba
Najrozšírenejší a aj najlepšie etablovaný spôsob terapie PKU celosvetovo je eliminačná diéta, ktorej podstatou je zníženie prísunu exogénneho Phe do takej miery, ktorá je nevyhnutná pre anabolizmus proteínov. Nasadenie diéty je nevyhnutné v prvom mesiaci po narodení a je dôležitá počas celého života, keďže niekoľko štúdií poukazuje na možnú regresiu neurokognitívnych funkcií asociovaných s dysfunkciou katecholamínov u pacientov, ktorí diétu v dospelosti vysadili [32, 33]. Názory na krvné hladiny Phe u detí aj dospelých sa celosvetovo líšia a taktiež závisia od kritérií jednotlivých metabolických centier. Na Slovensku je doporučené udržiavať hladinu Phe do 200 µmol/l pre deti do 1 roku života, do 12 rokov <360 µmol/l, od 12–18 rokov <500 µmol/l a nad 18 rokov do 900 µmol/l. Eliminačná diéta síce predstavuje významný míľnik v liečbe genetických ochorení, avšak jej sociálne a ekonomické aspekty nie sú vôbec zanedbateľné. Z tohto dôvodu boli vyvinuté rôzne ďaľšie alternatívne a menej zaťažujúce prístupy liečby, ktoré sú rozsiahlo diskutované v publikácii Procházkovej z roku 2010 [34]. Za zmienku však stojí významný míľnik v liečbe PKU – terapia kofaktorom PAH, tetrahydrobiopterínom (BH4). Objav tejto liečby totiž vniesol nové svetlo do regulácie PAH enzýmu a mnohých súvisiacich molekulárnych mechanizmov.
BH4 terapia
V roku 1999 Kure et al. zistili, že u PAH deficientných pacientov došlo po orálnej administrácii BH4 k zníženiu hladiny Phe v krvi. Od tohto roku sa stala BH4 terapia stredobodom pozornosti vedeckých výskumov, ale aj relevantnou adíciou či dokonca náhradou zaťažujúcej eliminačnej diéty. Táto skutočnosť taktiež viedla k definícii nového subtypu PKU – BH4 responzívnej HPA [35]. Avšak na tento terapeutický prístup odpovedá len približne 30 % pacientov. Pribúdajúce množstvo klinických skúšok odhalilo silnú koreláciu medzi pozitívnou reakciou a miernymi mutáciami, teda s takými, ktoré majú vysokú reziduálnu aktivitu [9, 36]. Veľká časť z nich aj bola charakterizovaná v in vitro expresných štúdiách. Dnes vieme, že približne 80 % pacientov s MHP je responzívnych, 50 % s mPKU a len 10 % s cPKU odpovedá na liečbu s BH4 [9].
Presný molekulárny mechanizmus pôsobenia BH4 na PAH nie je doposiaľ plne pochopený a má multifaktoriálnu podstatu. Hlavný mechanizmus jeho pôsobenia však spočíva v pôsobení BH4 ako molekulárneho šaperónu – chemickej látky, ktorá stabilizuje proteíny a napomáha ich skladaniu do trojrozmerných štruktúr [37]. Okrem toho, BH4 obnovuje rovnováhu dimérov a tetramérov PAH v prípade mutácií, ktoré túto rovnováhu svojim patologickým účinkom narúšajú [38].
Syntetická forma BH4 sa nazýva sapropterín dichlorid a je registrovaná pod názvom Kuvan® (Merck Serono). V súčasnosti neexistuje jeden štandardizovaný protokol univerzálny pre testovanie pacienta na BH4 responzívnosť. Odporúča sa však sledovať parameter 30%ného poklesu Phe v krvi. Vtedy je pacient považovaný za pozitívneho respondéra. Pre identifikáciu pacientov responzívnych k liečbe kofaktorom sa používajú zpravidla 8-, 16-, 24 - a 48-hodinové alebo aj týždňové záťažové testy. Odporúčaná iniciačná dávka liečiva je 10–20 mg na 1 kg telesnej hmotnosti. Pred a po administrácii sa potom v danom časovom intervale pacientom meria hladina Phe v krvi. Medzi pacientami sa rozlišujú rýchli respondéri (pokles do 8 hodín), strední (do 24) a pomalí (48 a viac). Je treba poznamenať, že často je vhodný aj týždňový test, ktorý môže odhaliť veľmi pomalých respondérov a naopak, niektorí pacienti sú falošne pozitívny v záťažovom teste a dlhodobá korekcia Phe u nich nie je efektívna. Z tohto dôvodu majú význam testy dlhšie ako 24 hodín. U responzívnych pacientov je dávkovanie sapropterínu stanovené na základe Phe tolerancie. Dávky liečiva bývajú následne prispôsobené od 5–20 mg/kg [9]. Nedávna štúdia jednoznačne poukazuje, že liečba sa-propterínom predstavuje bezpečný prístup, ako dlhodobo korigovať hyperfenylalaninémiu a nemá takmer žiadne nežiadúce účinky [39]. Nezanedbateľným nedostatkom tejto liečby však ostáva jej cena, nakoľko sa pohybu-je rádovo v desaťtisícoch EUR za rok na jedného pacienta.
Skutočnosť, že PKU sa dá liečiť farmakologickými šaperónmi, vniesla nový pohľad na terapiu ochorenia. Pozornosť sa uberá práve týmto smerom a v blízkej dobe môžme očakávať prvé výsledky klinických skúšok nových farmakologických šaperónov pre liečbu PKU, ale aj iných dedičných metabolických porúch [40].
Genotypovo-fenotypové korelácie
Optimálny prístup, ako korelovať genotyp s metabolickým fenotypom HPA, je túto koreláciu uskutočniť u takých pacientov, kde jedna mutácia determinuje celkovú reziduálnu aktivitu oboch alel. Teda u homozygotov alebo u pacientov, ktorí majú na jednom chromozóme mutáciu s nulovou aktivitou – u tzv. funkčných hemizygotov. Tento prístup bol použitý vo veľkej multicentrickej štúdii v druhej polovici 90. rokov minulého storočia. Na základe fenotypovej charakteristiky bolo 297 funkčne hemizygótnych pacientov rozdelených do štyroch kategórií: cPKU, mierna PKU, ľahká PKU a MHP. U každého z nich boli identifikované obe kauzatívne mutácie. Na základe tejto kategorizácie bola stanovená závažnosť 105 kauzatívnych mutácií. Ku každej z nich bola priradená AV hodnota (arbitrary assigned value) nasledovným spôsobom: 1 pre mutácie v kategórii cPKU, 2 pre mutácie spôsobujúce miernu PKU, 4 pre ľahkú PKU a 8 pre MHP mutácie. Výsledný fenotyp bol potom kalkulovaný ako súčet AV hodnôt dvoch mutácií a jednotlivé fenotypové kategórie PKU mali nasledovné hodnoty: 2 – klasická PKU, 3 a 4 mierna PKU, 5–8 ľahká PKU a 9–16 MHP. Tento jednoduchý model bol následne otestovaný u 184 pacientov, ktorí neboli funkčne hemizygotní a očakávaný fenotyp zodpovedal skutočnému fenotypu v 79 % prípadov vo všetkých kategóriách. Pre mutácie spôsobujúce MHP bola korelácia 100 %. Výsledok štúdie definitívne potvrdil existenciu silnej korelácie medzi PAH genotypom a fenotypom, a teda, že genotyp je hlavným určujúcim faktorom metabolického fenotypu u väčšiny pacientov s fenylketonúriou [11].
Nenahraditeľné uplatnenie pri predikcii fenotypu našli aj in vitro expresné štúdie mutovaných PAH enzýmov a stanovenie ich reziduálnej aktivity. Pre najčastejšie mutácie bola stanovená predpokladaná reziduálna aktivita (PRA), vypočítaná ako priemer reziduálnej aktivity získaný z rôznych in vitro experimentov v eukaryotických bunkách. Tieto dáta sú evidované v PAHvdb databáze a umožnili štatisticky korelovať priemernú PRA jednotlivých genotypov s fenotypovými kategóriami HPA. Z týchto korelácií vyplynulo, že kombinácie mutácií, ktoré umožňujú reziduálnu aktivitu v genotype menšiu ako 15 %, spôsobujú cPKU. Kombinácie mutácií, ktoré umožňujú reziduálnu aktivitu vyššiu ako 20%, zodpovedajú miernejším fenotypom [9]. Novší prístup okrem PRA zahŕňal aj in silico charakterizáciu mutácií, najmä algoritmy stability enzýmu. Predikcia fenotypu na základe týchto parametrov dosahovala až 100 % pri genotypoch s aktivitou menšou ako 3 %, takmer 95 % pri homozygotných a hemizygotných genotypoch, ale len 77,9 % pri zložených heterozygotoch [41].
Jeden z fenoménov, ktorý komplikuje predikciu fenotypu u heterozygotných pacientov, je interalelová komplementácia (IC). Ide o jav, kedy mutovaný PAH z jednej alely interaguje s monomérom exprimovaným z alely druhej. Keďže väčšina pacientov s HPA sú heterozygoti a teda sú nositeľmi dvoch rôznych mutácií, výsledkom expresie génu z oboch mutovaných alel je potom heterotetramérny PAH. To môže viesť ku konformačným zmenám v enzýme, k zmene jeho stability alebo kinetiky a prejaviť sa v zmene aktivity. Vďaka tomuto javu sa môže aktivita aj závažnej mutácie zvýšiť (pozitívna IC), alebo naopak aktivita miernej mutácie znížiť (negatívna IC) [42, 43].
Inkonzistencie pozorované u homozygotov alebo hemizygotov v mnohých celopopulačných štúdiách [13–15, 31] môžu mať niekoľko iných vysvetlení. Hladina Phe v krvi u pacientov mohla byť v čase diagnózy blízko hraničnej hodnoty príslušnej fenotypovej kategórie, alebo v čase merania nedosiahla maximálnu hodnotu Phe v krvi a tak mohlo dôjsť k nesprávnej fenotypovej klasifikácii. Za zmienku stojí aj evidencia niekoľkých pacientov, u ktorých boli identifikované viac ako 2 mutácie v géne PAH [44]. V takýchto prípadoch sú dve mutácie v cis konformácii (lokalizované na jednom chromozóme) a fenotyp determinuje tretia mutácia. Táto skutočnosť zdôrazňuje dôležitosť molekulárnej analýzy celého génu. Okrem spomínaného, existencia modifikujúcich génov je taktiež možná [45]. V roku 2010 Stojiljkovic et al. identifikovali regulačnú oblasť v intróne 8, v oblasti veľmi dobre známeho RFLP polymorfizmu. Autori demonštrovali až trojnásobné zosilnenie transkripcie reportérového génu touto oblasťou v experimente na hepatálnej bunkovej línii. To vnieslo celkom nový pohľad na možnú reguláciu PAH modulátormi – polymorfizmami lokalizovanými v nekódujúcich oblastiach génu [46]. Pochopenie mechanizmu génovej regulácie PAH môže byť kľúčom k pochopeniu fenotypovej komplexnosti ochorenia. Identifikácia a evaluácia podobných variant roztrúsených po celom géne môže umožniť vznik molekulárno-diagnostického algoritmu, na základe ktorého bude možná presná predikcia fenotypu. Súčasné technológie to totiž umožňujú ako po technickej, tak aj po ekonomickej stránke.
Genotyp ako nástroj v personalizovanej liečbe fenylketonúrie
Jedným z hlavných cieľov biomedicínskych vied, obzvlášť molekulárnej genetiky, je implementácia vedeckých poznatkov do liečby ochorenia. S nastupujúcou érou DNA sekvenovania novej generácie sa súčasným trendom stáva prediktívna a personalizovaná biomedicína. Nie je tomu inak ani v prípade PKU. Objav liečby pomocou kofaktora, no näjma intenzívny výskum, ktorý v posledných dvoch dekádach po tomto objave nasledoval, odhalil silnú koreláciu niektorých genotypov s responzivitou pacientov na liečbu. Preto sa pozornosť upriamila na možnosť predikcie responzivity pacienta na základe genotypu.
Podobne ako pri genotypovo-fenotypových koreláciách aj v tomto prípade našla uplatnenie funkčná in vitro charakterizácia mutácií resp. ich predpokladaná reziduálna aktivita (PRA). Autori prvej veľkej klinickej štúdie brali do úvahy len jednu mutáciu v genotype, ktorá ak mala aktivitu väčšiu ako 10 % enzýmu divého typu, genotyp bol považovaný za responzívny [47]. Klinické skúšky síce ukázali signifikantnú koreláciu, avšak nie dostatočnú na spoľahlivú predikciu v rutinnej klinickej praxi. Následujúce štúdie už zohľadňovali PRA oboch mutácií, keďže sa ukázalo, že kompletný genotyp má oveľa lepši prediktívny charakter [31, 48, 49]. V najlepších prípadoch bola predikcia responzivity správna až v 85 % [50], no nedávna slovínska štúdia zaznamenala aj negatívne výsledky v genotypoch s vysokou PRA [49]. Najväčšia limitácia tohto princípu predikcie spočíva v tom, že nezohľadňuje spomínanú interalelovú komplementáciu, ktorá má nepochybne vplyv ako na aktivitu oboch mutácií v genotype, tak aj na väzbu kofaktora v aktívnom mieste enzýmu. Okrem toho, na stanovenie aktivít jednotlivých mutácií nebol použitý jeden univerzálny in vitro expresný systém, ale PRA sa vypočítala ako priemer z rôznych experimentov v rôznych expresných systémoch. Navyše, všetky experimenty prebiehajú pri presne stanovených koncentráciach substrátu a kofaktora, čo ani zďaleka neodzrkadľuje in vivo situáciu. Okrem spomínaného, do úvahy je nutné brať aj potencionálnu existenciu genetických modulátorov ako aj individuálny metabolizmus pacienta.
Fundamentálny pokrok v tomto smere uskutočnil nemecký výskumný tím pod vedením profesorky Annie C. Muntau. Najprv vyvinuli jednoduchú ale presnú fluo-rescenčnú metódu, ktorá umožnila v reálnom čase sledovať in vitro katalýzu Phe na Tyr PAH enzýmom pri variabilných koncentráciách substrátu aj kofaktora [51]. Tým v podstate demonštrovali veľmi dobrý in vitro model na simuláciu in vivo situácie v pečeni pacienta (fluktuácia Phe, rôzne dávkovanie a intestinálne vstrebávanie BH4). Potom túto metódu použili pri vybraných mutáciách ako aj enzýme divého typu a výsledky meranií pomocou softvérov transformovali do „polí aktivity“ – schématického zobrazenia aktivity enzýmu pri rôznych koncentráciách substrátu a kofaktora (obr. 1). Takto zistili, že PAH enzýmy (mutované aj divý typ) majú svoje optimálne koncentrácie obidvoch komponentov, pri ktorých sú najaktívnejšie. Autori taktiež zaznamenali, že každá mutácia má určitú minimálnu hladinu Phe, ktorá je nevyhnutná pre aktiváciu PAH a teda štart jeho katalytickej aktivity. Toto zistenie poukazuje na dôležitosť hladiny Phe u pacienta pred záťažovým testom BH4, nakoľko nedostatočná hladina môže viesť k slabej alebo žiadnej aktivácii PAH v pečeni.
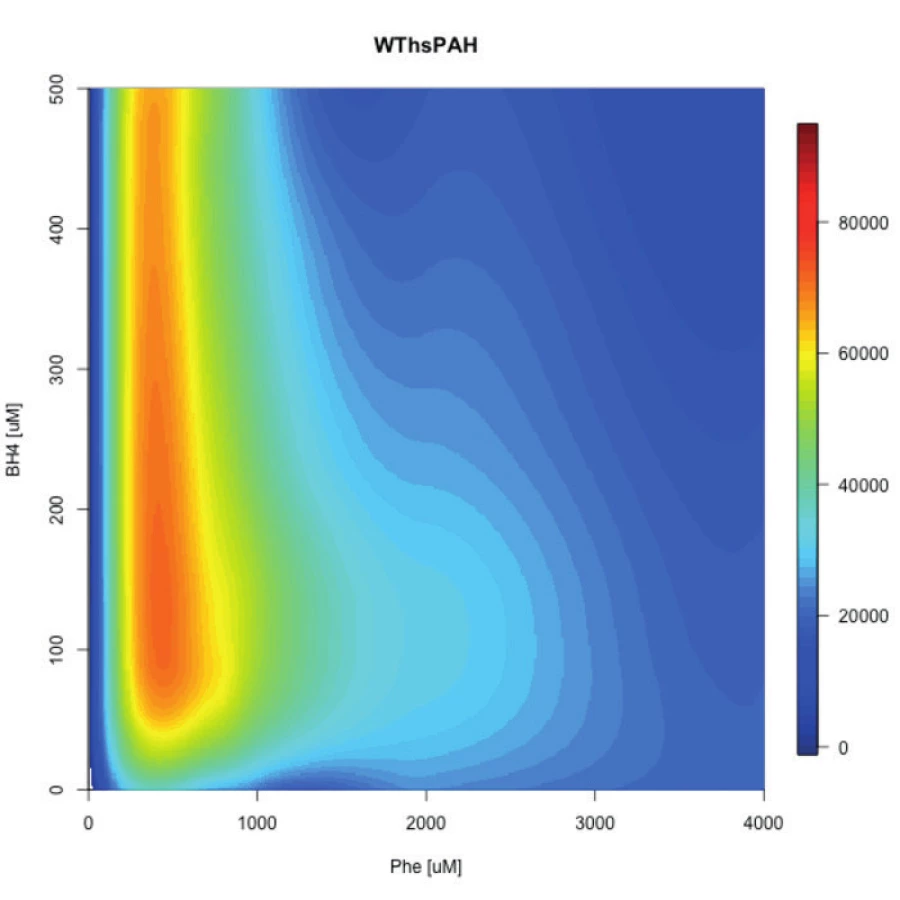
Extrapoláciou týchto experimentálnych výsledkov do in vivo situácie by sme mohli konštatovať, že BH4 responzívnosť pacienta je teda súhrou genotypu (reziduálna aktivita mutácií), metabolického fenotypu pacienta (hladina Phe) a farmakologickej dávky liečiva (koncentrácie kofaktora) [52]. V ich najnovšej štúdii použili bialelický eukaryotický expresný systém, ktorý umožňuje paralelne merať aktivitu oboch mutácií a teda simulovať genotyp pacienta. Následne podobným spôsobom ako v predošlej štúdií determinovali maximálnu aktivitu 30 najčastejších európskych genotypov a pre každý z nich stanovili „polia aktivity“, teda optimálne metabolické (koncentrácia Phe) a terapeutické (koncentrácia BH4) podmienky. Zistili teda, že niektoré genotypy vyžadujú pre aktivitu mutovaných enzýmov vyššiu koncentráciu fenylalanínu, iné zase zvýšenú potrebu kofaktora. Keď tieto dáta korelovali s klinickými dátami z publikácií, vyplynulo nasledovné: Genotypy s maximálnou reziduálnou aktivitou menšou ako 3,5 % boli asociované s klasickou fenylketonúriou a nie sú responzívne ku farmakologickej liečbe sapropterínom; zvýšená potreba kofaktora bola asociovaná s genotypmi s variabilnými fenotypmi a slabou odpoveďou na kofaktor v klinických skúškach; reziduálna aktivita nad 5 % bola asociovaná s farmakologicky liečiteľnými, miernymi fenotypmi bez potreby zvýšenia koncentrácie kofaktora.
Tento model predstavuje doposiaľ najlepšiu in vitro simuláciu genotypu, zohľadňuje interalelovú komplementáciu a poskytuje podstatne kvalitatívnejšiu informáciu ako dáta z in vitro experimentov jednotlivých mutácií [53]. Okrem toho, vytvorili internetovú aplikáciu, ktorá poskytuje všetky tieto dáta, garantuje pravidelnú aktualizáciu a poskytuje stanovenie parametrov pre konkrétne genotypy pacientov na požiadanie (http://pah-activitylandscapes.org/). V klinickej praxi to môže znamenať, že genotyp spolu s klinickým fenotypom poskytne informáciu pre stanovenie optimálnej farmakologickej dávky prípravku Kuvan pre čo najefektívnejšiu liečbu, teda personalizovanú terapiu. Tieto údaje taktiež umožnia dlhodobú predikciu fenotypu pacienta. Ovšem otázkou ostáva, ako experimentálne dáta implementovať do praxe? Vzhľadom na rapídny pokrok vo výskume PKU sa však domnievame, že implementácie sa dočkáme v blízkej budúcnosti. Používanie uvedenej internetovej aplikácie môže však poskytnúť klinickým pracovníkom kvalitatívnu informáciu o genotype pacienta a teda aspoň približnú informáciu, či je potrebné zvýšiť dávku liečiva alebo prirodzených proteínov v strave pre efektívnu liečbu kofaktorom.
Fenylketonúria na Slovensku
Pilotnú štúdiu, v ktorej boli charakterizované najčastejšie mutácie v slovenskej populácii, uskutočnili Kádaši et al. v roku 1995 metódami priamej DNA diagnostiky [54]. Feráková et al. (1992) charakterizovali prevalentnú mutáciu p.R252W u slovenských Rómov, kde predstavuje prakticky 100 % všetkých mutácií. Na základe väzby s unikátnym RFLP haplotypom bola charakterizovaná ako mutácia s efektom zakladateľa [55]. O niekoľko rokov neskôr Polák et al. (2008) nadviazali na rozpracovanú problematiku PKU a rozšírili mutačné spektrum v slovenskej populácii o 16 mutácií pomocou DHPLC (Denaturating High Performance Liquid Chromatography) a selektívneho sekvenovania. V tejto štúdii autori zaznamenali pomerne nízku detekčnú schopnosť a bolo identifikovaných len 84,3 % všetkých alel v danom súbore pacientov u slovenských nerómskych PKU pacientov. Metóda DHPLC sa v tejto štúdii nepreukázala byť dostatočne efektívnou po finančnej ani detekčnej stránke. Avšak umožnila implementovať výsledky mutačnej analýzy do diagnostickej praxe [56]. V roku 2013 Polák et al. uskutočnili celopopulačnú štúdiu, kde charakterizovali PAH mutačné spektrum slovenskej populácie, uskutočnili genotypovo-fenotypové korelácie a na základe identifikovaných genotypov predikovali responzívnosť pacientov na administráciu liečiva Kuvan. Okrem známych mutácií boli identifikované aj tri nové v katalytickej doméne PAH. Z uvedenej štúdie vyplynulo, že viac ako 70 % alel zodpovedných za manifestáciu PKU je možné detegovať priamym sekvenovaním exonov 7 a 12. Ako nosnú skríningovú metódu pre detekciu bodových mutácií, krátkych inzerčno-delečných mutácií a SNP polymorfizmov použili HRM. Pre identifikáciu veľkých génových prestavieb bola použitá metóda MLPA [13]. Dáta z tejto štúdie taktiež poskytli informácie pre diagnostické pracoviská na Slovensku a umožnili vytvoriť algoritmus na efektívnu molekulárnu diagnostiku PAH génu.
Do súčasnosti bolo v slovenskej populácii identifikovaných 54 rôznych mutácií a 93 rôznych genotypov v súbore pacientov z 229 nepríbuzných rodín, čo predstavuje charakterizáciu mutačného spektra u viac ako 80 % všetkých registrovaných PKU rodín na Slovensku. Prevalentná mutácia pre slovenskú populáciu je p.R408W s relatívnou frekvenciou 46,5 %. Ďaľšie časté mutácie sú p.R158Q v exóne 5, p.R252W v exóne 7, IVS12+1G>A v intróne 12, p.A403V v exóne 12 a p.R261Q v exóne 7. Okrem bodových mutácií a krátkych inzerčno-delečných variantov boli identifikované aj dve delécie exónov 3 a 5, ktorých frekvencia je v slovenskej populácii nižšia ako priemerná, s prevalenciou 2,2 %. Spektrum najčastejších mutácií identifikovaných v slovenskej populácii je uvedené v tabuľke 1. Prehľad najčastejších genotypov s príslušnými fenotypovými kategóriami a predikciou responzívnosti ku administrácii kofaktora uvádza tabuľka 2.
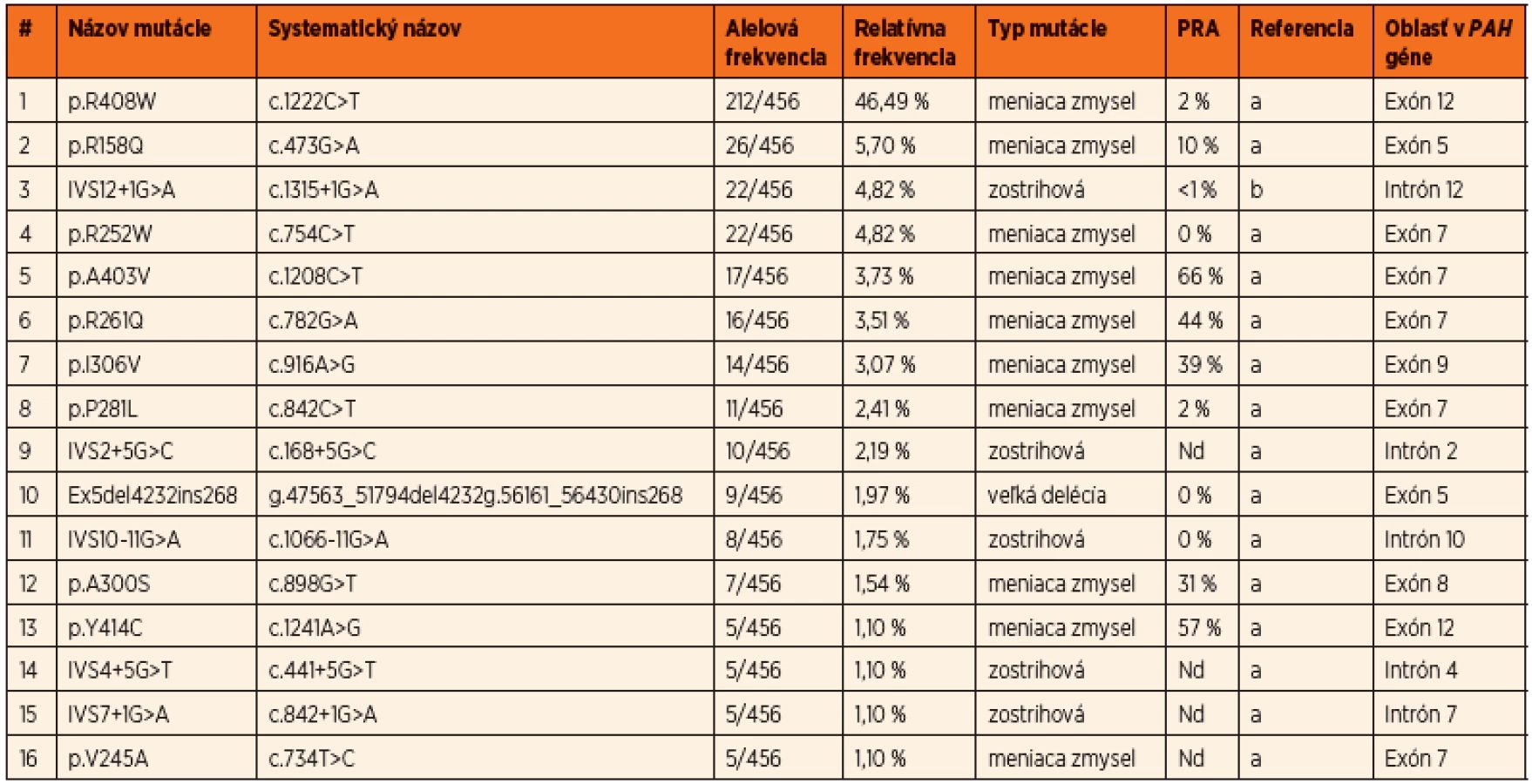
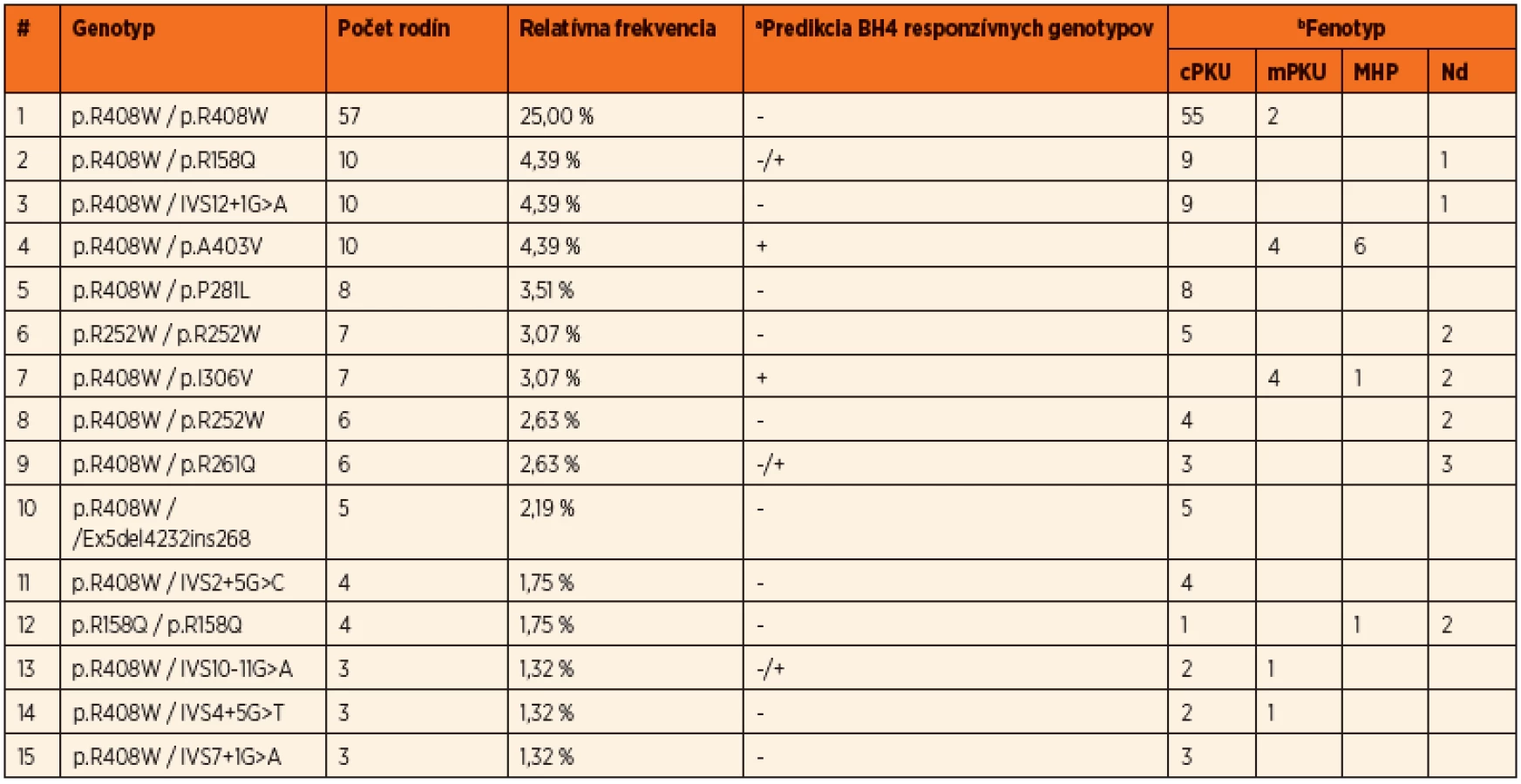
Na základe všetkých troch štúdií možno konštatovať, že takmer 67 % slovenských HPA pacientov má klasickú PKU, 13 % miernu PKU, 14 % pacientov je postihnutých ľahkou hyperfenylalaninémiou a u približne 6 % pacientov nie sú k dispozícii dáta k fenotypovej klasifikácii. Viac ako tretina pacientov sú homozygoti a dve tretiny pacientov sú zložení heterozygoti. Vysoká prevalencia mutácií s nulovou aktivitou ako aj vysoká frekvencia genotypov homozygotných pre mutáciu p.R408W v slovenskej populácii odzrkadľujú, že viac ako polovica PKU rodín veľmi pravdepodobne nebude responzívna na BH4 terapiu. Napriek tomu, vysoká PAH alelová heterogenita v slovenskej populácii umožňuje odhadnúť, že takmer 25 % PKU rodín môže potencionálne reagovať na administráciu Kuvanu.
Záver
Fenylketonúria je stredobodom pozornosti rozsiahlych biochemických a molekulárno-genetických výskumov niekoľko desaťročí. Predstavuje istú „paradigmu“ genetického ochorenia, ktoré je možné efektívne liečiť. Od identifikácie prvej PKU alely sa za posledných 20 rokov rapídne zlepšila diagnostika, vyvinuli sa alternatívne a menej zaťažujúce spôsoby liečby a posledné výskumy smerujú k personalizovanej terapii. Fenylketonúria však nie je jednoduché mendelistické ochorenie a mnoho genetických a negenetických faktorov sa podieľa na jej komplexnej podstate. Preto niekoľko otázok ostáva stále nezodpovedaných. Vzhľadom však k exponenciálnej rýchlosti vývoja technológií, ako aj k intenzite výskumu PKU môžeme v blízkej budúcnosti pravdepodobne očakávať identifikáciu molekulárno-genetického algoritmu, ktorý umožní spoľahlivú predikciu metabolického fenotypu a voľbu optimálnej terapeutickej stratégie. Súčasné poznatky umožňujú predikovať fenotyp na základe genotypu len s určitou pravdepodobnosťou a tak je tomu aj v prípade farmakologickej liečby kofaktorom. Na druhej strane, niektoré genotypy boli nespočetne krát evaluované v klinických skúškach a javia konštantné výsledky. V týchto prípadoch môže genotyp slúžiť ako užitočný prediktor v plánovaní záťažového testu kofaktorom.
Táto publikácia vznikla vďaka finančnej podpore projektov: „Diagnostika spoločensky závažných ochorení na Slovensku, založená na moderných biotechnológiách“ ITMS 26240220058, a APVV-0240-12 „Funkčná analýza novoidentifikovaných DNA variantov v génoch zodpovedných za cystickú fibrózu a fenylketonúriu“.
Došlo: 17. 7. 2015
Přijato: 23. 10. 2015
RNDr. Emil Polák, PhD.
Katedra molekulárnej biológie B2-211
Prírodovedecká fakulta Univerzity Komenského
Ilkovičova 6
842 15 Bratislava,
Slovenská republika
e-mail: polakemo@gmail.com
Sources
1. Eisensmith RC, Woo SL. Molecular basis of phenylketonuria and related hyperphenylalaninemias: mutations and polymorphisms in the human phenylalanine hydroxylase gene. Hum Mutat 1992; 1 : 13–23.
2. Thony B, Blau N. Mutations in the BH4-metabolizing genes GTP cyclohydrolase I, 6-pyruvoyl-tetrahydropterin synthase, sepiapterin reductase, carbinolamine-4a-dehydratase, and dihydropteridine reductase. Hum Mutat 2006; 27 : 870–878.
3. Blau N, van Spronsen FJ, Levy HL. Phenylketonuria. Lancet 2010; 376 : 1417–1427.
4. Zschocke J. Phenylketonuria mutations in Europe. Hum Mutat 2003; 21 : 345–356.
5. Votava F, Kožich V, Chrastina P. Výsledky rozšířeného novorozeneckého screeningu v České republice. Čes-slov Pediat 2014; 69 (2): 77–86.
6. Scriver CR. The PAH gene, phenylketonuria, and a paradigm shift. Hum Mutat 2007; 28 : 831–845.
7. Guthrie R, Susi A. A simple phenylalanine method for detecting phenylketonuria in large populations of mewborn infants. Pediatrics 1963; 32 : 338–343.
8. Chace DH, Sherwin JE, Hillman SL, et al. Use of phenylalanine-to-tyrosine ratio determined by tandem mass spectrometry to improve newborn screening for phenylketonuria of early discharge specimens collected in the first 24 hours. Clin Chem 1998; 44 : 2405–2409.
9. Blau N, Hennermann JB, Langenbeck U, et al. Diagnosis, classification, and genetics of phenylketonuria and tetrahydrobiopterin (BH4) deficiencies. Mol Genet Metab 2011; 104 (Suppl): S2–9.
10. Zerjav Tansek M, Groselj U, Angelkova N, et al. Phenylketonuria screening and management in southeastern Europe – survey results from 11 countries. Orphanet J Rare Dis 2015; 10 : 68.
11. Guldberg P, Rey F, Zschocke J, et al. A European multicenter study of phenylalanine hydroxylase deficiency: classification of 105 mutations and a general system for genotype-based prediction of metabolic phenotype. Am J Hum Genet 1998; 63 : 71–79.
12. van Spronsen FJ, van Rijn M, Dorgelo B, et al. Phenylalanine tolerance can already reliably be assessed at the age of 2 years in patients with PKU. J Inherit Metab Dis 2009; 32 : 27–31.
13. Polak E, Ficek A, Radvanszky J, et al. Phenylalanine hydroxylase deficiency in the Slovak population: genotype-phenotype correlations and genotype-based predictions of BH4-responsiveness. Gene 2013; 526 : 347–355.
14. Groselj U, Tansek MZ, Kovac J, et al. Five novel mutations and two large deletions in a population analysis of the phenylalanine hydroxylase gene. Mol Genet Metab 2012; 106 : 142–148.
15. Sterl E, Paul K, Paschke E, et al. Prevalence of tetrahydrobiopterine (BH4)-responsive alleles among Austrian patients with PAH deficiency: comprehensive results from molecular analysis in 147 patients. J Inherit Metab Dis 2013; 36 : 7–13.
16. Mitchell JJ, Trakadis YJ, Scriver CR. Phenylalanine hydroxylase deficiency. Genet Med 2011; 13 : 697–707.
17. Blau N, Bonafe L, Thony B. Tetrahydrobiopterin deficiencies without hyperphenylalaninemia: diagnosis and genetics of dopa-responsive dystonia and sepiapterin reductase deficiency. Mol Genet Metab 2001; 74 : 172–185.
18. Longo N. Disorders of biopterin metabolism. J Inherit Metab Dis 2009; 32 : 333–342.
19. Lidsky AS, Law ML, Morse HG, et al. Regional mapping of the phenyl-alanine hydroxylase gene and the phenylketonuria locus in the human genome. Proc Natl Acad Sci U S A 1985; 82 : 6221–6225.
20. Woo SL, Lidsky AS, Guttler F, et al. Cloned human phenylalanine hydroxylase gene allows prenatal diagnosis and carrier detection of classical phenylketonuria. Nature 1983; 306 : 151–155.
21. Konecki DS, Wang Y, Trefz FK, et al. Structural characterization of the 5’ regions of the human phenylalanine hydroxylase gene. Biochemistry 1992; 31 : 8363–8368.
22. Wang Y, Hahn TM, Tsai SY, et al. Functional characterization of a unique liver gene promoter. J Biol Chem 1994; 269 : 9137–9146.
23. Lichter-Konecki U, Hipke CM, Konecki DS. Human phenylalanine hydroxylase gene expression in kidney and other nonhepatic tissues. Mol Genet Metab 1999; 67 : 308–316.
24. Lei XD, Kaufman S. Identification of hepatic nuclear factor 1 binding sites in the 5’ flanking region of the human phenylalanine hydroxylase gene: implication of a dual function of phenylalanine hydroxylase stimulator in the phenylalanine hydroxylation system. Proc Natl Acad Sci U S A 1998; 95 : 1500–1504.
25. Dobrowolski SF, Andersen HS, Doktor TK, et al. The phenylalanine hydroxylase c.30C>G synonymous variation (p.G10G) creates a common exonic splicing silencer. Mol Genet Metab 2010; 100 : 316–323.
26. Scriver CR, Hurtubise M, Konecki D, et al. PAHdb 2003: what a locus-specific knowledgebase can do. Hum Mutat 2003; 21 : 333–344.
27. Kozak L, Hrabincova E, Kintr J, et al. Identification and characterization of large deletions in the phenylalanine hydroxylase (PAH) gene by MLPA: evidence for both homologous and non-homologous mechanisms of rearrangement. Mol Genet Metab 2006; 89 : 300–309.
28. Waters PJ, Parniak MA, Nowacki P, et al. In vitro expression analysis of mutations in phenylalanine hydroxylase: linking genotype to phenotype and structure to function. Hum Mutat 1998; 11 : 4–17.
29. Pey AL, Stricher F, Serrano L, et al. Predicted effects of missense mutations on native-state stability account for phenotypic outcome in phenylketonuria, a paradigm of misfolding diseases. Am J Hum Genet 2007; 81 : 1006–1024.
30. Gersting SW, Kemter KF, Staudigl M, et al. Loss of function in phenylketonuria is caused by impaired molecular motions and conformational instability. Am J Hum Genet 2008; 83 : 5–17.
31. Dobrowolski SF, Heintz C, Miller T, et al. Molecular genetics and impact of residual in vitro phenylalanine hydroxylase activity on tetrahydrobiopterin responsiveness in Turkish PKU population. Mol Genet Metab 2011; 102 : 116–121.
32. Channon S, Mockler C, Lee P. Executive functioning and speed of processing in phenylketonuria. Neuropsychology 2005; 19 : 679–686.
33. Feldmann RE Jr, Maurer MH, Hunzinger C, et al. Reduction in rat phosphatidylethanolamine binding protein-1 (PEBP1) after chronic corticosterone treatment may be paralleled by cognitive impairment: a first study. Stress 2008; 11 : 134–147.
34. Procházková D. Současné možnosti léčby hyperfenylalaninémie. Čes-slov Pediat 2010; 65 (7–8): 452–458.
35. Kure S, Hou DC, Ohura T, et al. Tetrahydrobiopterin-responsive phenyl-alanine hydroxylase deficiency. J Pediatr 1999; 135 : 375–378.
36. Blau N, Erlandsen H. The metabolic and molecular bases of tetrahydrobiopterin-responsive phenylalanine hydroxylase deficiency. Mol Genet Metab 2004; 82 : 101–111.
37. Underhaug J, Aubi O, Martinez A. Phenylalanine hydroxylase misfolding and pharmacological chaperones. Curr Top Med Chem 2012; 12 : 2534–2545.
38. Cerreto M, Cavaliere P, Carluccio C, et al. Natural phenylalanine hydroxylase variants that confer a mild phenotype affect the enzyme’s conformational stability and oligomerization equilibrium. Biochim Biophys Acta 2011; 1812 : 1435–1445.
39. Keil S, Anjema K, van Spronsen FJ, et al. Long-term follow-up and outcome of phenylketonuria patients on sapropterin: a retrospective study. Pediatrics 2013; 131: e1881–1888.
40. Muntau AC, Leandro J, Staudigl M, et al. Innovative strategies to treat protein misfolding in inborn errors of metabolism: pharmacological chaperones and proteostasis regulators. J Inherit Metab Dis 2014; 37 : 505–523.
41. Wettstein S, Underhaug J, Perez B, et al. Linking genotypes database with locus-specific database and genotype-phenotype correlation in phenylketonuria. Eur J Hum Genet 2015; 23 : 302–309.
42. Leandro J, Leandro P, Flatmark T. Heterotetrameric forms of human phenylalanine hydroxylase: co-expression of wild-type and mutant forms in a bicistronic system. Biochim Biophys Acta 2011; 1812 : 602–612.
43. Leandro J, Nascimento C, de Almeida IT, et al. Co-expression of different subunits of human phenylalanine hydroxylase: evidence of negative interallelic complementation. Biochim Biophys Acta 2006; 1762 : 544–550.
44. Zschocke J, Preusse A, Sarnavka V, et al. The molecular basis of phenylalanine hydroxylase deficiency in Croatia. Hum Mutat 2003; 21 : 399.
45. Scriver CR, Waters PJ. Monogenic traits are not simple: lessons from phenylketonuria. Trends Genet 1999; 15 : 267–272.
46. Stojiljkovic M, Zukic B, Tosic N, et al. Novel transcriptional regulatory element in the phenylalanine hydroxylase gene intron 8. Mol Genet Metab 2010; 101 : 81–83.
47. Zurfluh MR, Zschocke J, Lindner M, et al. Molecular genetics of tetrahydrobiopterin-responsive phenylalanine hydroxylase deficiency. Hum Mutat 2008; 29 : 167–175.
48. Trefz FK, Scheible D, Gotz H, et al. Significance of genotype in tetrahydrobiopterin-responsive phenylketonuria. J Inherit Metab Dis 2009; 32 : 22–26.
49. Tansek MZ, Groselj U, Murko S, et al. Assessment of tetrahydrobiopterin (BH(4))-responsiveness and spontaneous phenylalanine reduction in a phenylalanine hydroxylase deficiency population. Mol Genet Metab 2012; 107 : 37–42.
50. Quirk ME, Dobrowolski SF, Nelson BE, et al. Utility of phenylalanine hydroxylase genotype for tetrahydrobiopterin responsiveness classification in patients with phenylketonuria. Mol Genet Metab 2012; 107 : 31–36.
51. Gersting SW, Staudigl M, Truger MS, et al. Activation of phenylalanine hydroxylase induces positive cooperativity toward the natural cofactor. J Biol Chem 2010; 285 : 30686–30697.
52. Staudigl M, Gersting SW, Danecka MK, et al. The interplay between genotype, metabolic state and cofactor treatment governs phenylalanine hydroxylase function and drug response. Hum Mol Genet 2011; 20 : 2628–2641.
53. Danecka MK, Woidy M, Zschocke J, et al. Mapping the functional landscape of frequent phenylalanine hydroxylase (PAH) genotypes promotes personalised medicine in phenylketonuria. J Med Genet 2015; 52 : 175–185.
54. Kadasi L, Polakova H, Ferakova E, et al. PKU in Slovakia: mutation screening and haplotype analysis. Hum Genet 1995; 95 : 112–114.
55. Ferakova E, Ferak V, Kadasi L, et al. A unique RFLP haplotype at the phenylalanine hydroxylase locus in Czechoslovak Gypsies with phenylketonuria. Funct Dev Morphol 1992; 2 : 139–140.
56. Polák E, Ficek A, Baldovič M, et al. Komplexná mutačná analýza génu PAH u slovenských pacientov postihnutých fenylketonúriou. Čes-slov Pediat 2008; 63 (10): 528–534.
Labels
Neonatology Paediatrics General practitioner for children and adolescentsArticle was published in
Czech-Slovak Pediatrics

2015 Issue 6
- What Effect Can Be Expected from Limosilactobacillus reuteri in Mucositis and Peri-Implantitis?
- The Importance of Limosilactobacillus reuteri in Administration to Diabetics with Gingivitis
Most read in this issue
- Primární (autoimunitní) sklerozující cholangitida u pacienta s nespecifickým střevním zánětem
- Nikotin ovlivňuje vývoj mozku
- Vliv valgozity paty na pohyb nohy při chůzi u dětí ve věku 3 až 8 let
- Aspergillus fumigatus a plicní postižení u cystické fibrózy – přehled problematiky