
Trombotické mikroangiopatie – hemolyticko-uremické syndromy a trombotická trombocytopenická purpura
Thrombotic microangiopathies – hemolytic-uremic syndromes and thrombotic thrombocytopenic purpura
Thrombotic microangiopathies are a heterogeneous group of disease that causes damage of endothelial cells of small arteries in different organs that lead to thrombi formation and organ ischemia. The laboratory markers of TMA are typically a triad of non-immune hemolytic anemia, thrombocytopenia and acute kidney injury. Thrombotic microangiopathies could be differentiated according their etiology to hemolytic-uremic syndromes (HUS) – typical HUS (caused by Shiga-toxin producing E. coli) and atypical HUS (aHUS, caused mainly by dysregulation of alternative pathway of complement) and thrombotic thrombocytopenic purpura (TTP, severe deficiency of von Willebrand factor cleaving protease called ADAMTS13). Treatment of TMA depend on its etiology – only symptomatic therapy in typical HUS (dialysis, transfusion), plasmapheresis, immunosuppression or plasma infusions in TTP and monoclonal anti-C5 antibody eculizumab in aHUS.
KEY WORDS:
thrombotic microangiopathies, thrombotic thrombocytopenic purpura, hemolytic-uremic syndrome
Authors:
T. Seeman 1; Ľ. Podracká 2; Š. Štolbová 1; K. Bláhová 1
Authors‘ workplace:
Pediatrická klinika 2. LF UK a FN Motol, Praha
1; Detská klinika, Lekárska fakulta Univerzity Komenského a DFNsP, Bratislava
2
Published in:
Čes-slov Pediat 2017; 72 (2): 99-108.
Category:
Overview
Trombotické mikroangiopatie (TMA) jsou heterogenní skupina chorob různých příčin, která je charakterizována postižením endotelu drobných cév v různých orgánech, která vede k tvorbě trombů v mikrocirkulaci s následnou poruchou funkce postižených orgánů, hlavně ledvin. Laboratorně se projevují zejména triádou neimunní hemolytická anémie, trombocytopenie a zvýšené parametry ledvinných funkcí. Z etiologického hlediska se trombotické mikroangiopatie dělí na hemolyticko-uremické syndromy (HUS) – typický HUS (způsoben Shiga-toxin produkující E. coli) a atypický HUS (aHUS, způsoben nejčastěji funkční poruchou alternativní složky komplementu) a trombotickou trombocytopenickou purpuru (TTP, deficit štěpící proteázy von Willebrandova faktoru zvaného ADAMTS13). Léčba TMA závisí na příčině, terapie typického HUS je pouze symptomatická, léčba TTP zahrnuje zejména plazmaferézy, imunosupresiva nebo infuze plazmy, atypický HUS se nově léčí monoklonální protilátkou proti C5 složce komplementu eculizumabem.
KLÍČOVÁ SLOVA:
trombotické mikroangiopatie, trombotická trombocytopenická purpura, hemolyticko-uremický syndrom
DEFINICE A ROZDĚLENÍ TROMBOTICKÝCH MIKROANGIOPATIÍ
Trombotické mikroangiopatie (TMA) jsou skupina chorob různých příčin, která je charakterizována postižením endotelu drobných cév v různých orgánech (mikroangiopatie), která vede k tvorbě trombů v mikrocirkulaci s následnou poruchou funkce postižených orgánů. Laboratorně se projevuji zejména triádou hemolytická anémie, trombocytopenie a poruchou funkce ledvin.
Z klinického a etiologického hlediska se TMA dělí na:
- hemolyticko-uremické syndromy (HUS),
- trombotickou trombocytopenickou purpuru (TTP).
Do doby objevení příčin těchto chorob se často tyto diagnózy používaly pod společným názvem „HUS//TTP“, s objevem příčin těchto závažných a život ohrožujících onemocnění na začátku 21. století je nutné tyto choroby přísně oddělovat, protože kromě odlišných příčin mají i odlišnou léčbu a prognózu. Pokud se tedy u dětského pacienta objeví známky TMA, je nutné vždy odhalit její příčinu a co nejrychleji ji začít léčit podle ní.
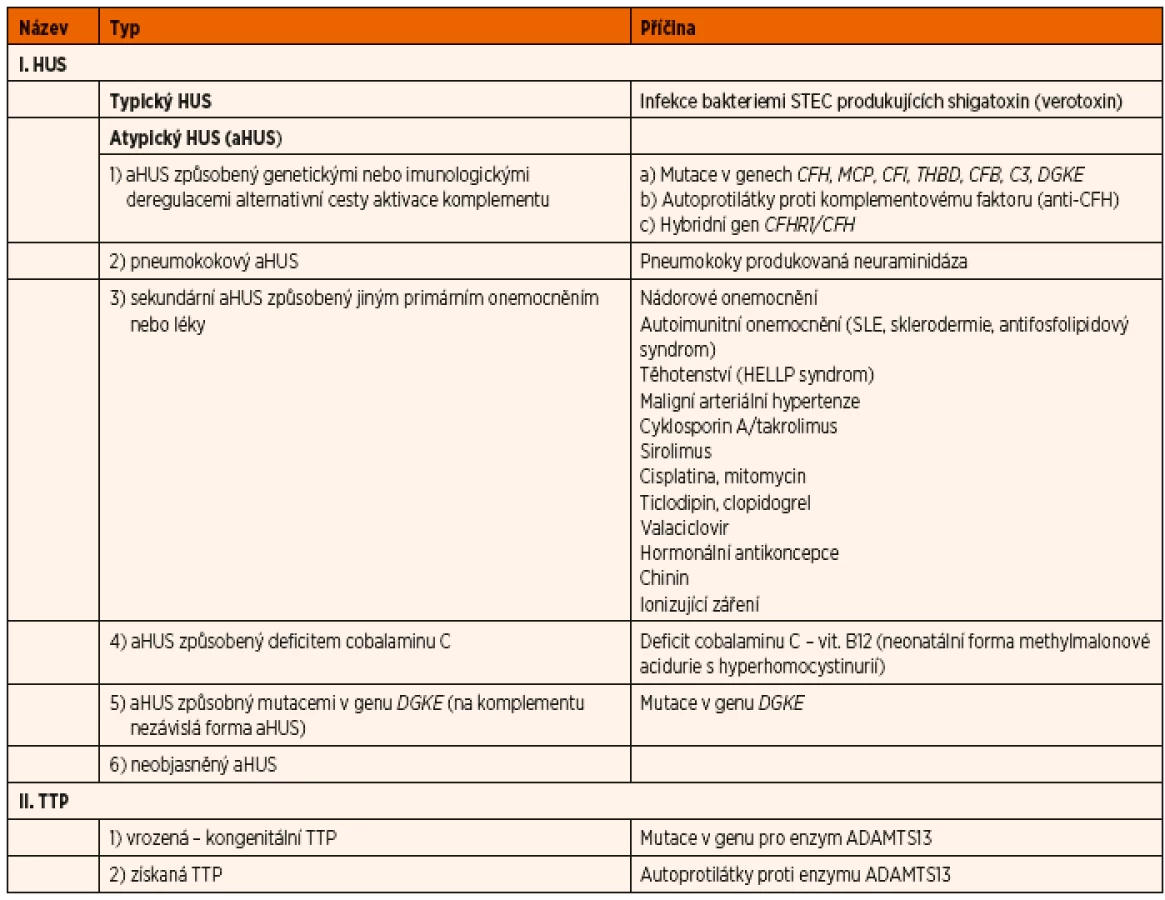
Rozděleni trombotických mikroangiopatií je uvedeno v tabulce 1.
TROMBOTICKÁ TROMBOCYTOPENICKÁ PURPURA (TTP)
V roku 1924 opísal Moschcowitz závažný klinický prípad 16-ročného dievčaťa s výraznou bledosťou, purpurou a hemiparézou, ktoré za 14 dní exitovalo na kardiálne zlyhanie [1]. Autopsia prekvapivo odhalila početné hyalinné tromby v terminálnych arteriolách a kapilárach viacerých orgánoch vrátane obličiek. Táto zaujímavá kazuistika predstavuje prvý známy prípad TMA, pravdepodobne TTP, ktorá sa tiež označuje ako TMA mediovaná deficienciou štiepiacej proteázy ADAMTS13.
- Príčina TTP
O necelých 60 rokov neskôr sa v sére pacientov s chronickou relabujúcou (hereditárnou?) TTP detegovali veľké multiméry von-Willebrandovho faktora [2]. Tento nález viedol k objaveniu štiepiacej proteázy von Willebrandovho faktora, ktorá sa nazvala ADAMTS13 [3]. ADAMTS13 štiepi multiméry von Willebrandovho faktora uvolňované z endotelových buniek ciev. Deficit proteázy ADAMTS13 vedie k hromadeniu veľkých multimérov von Willebrandovho faktora v cirkulácii a riziku trombov v malých cievach.
Hereditárna TTP (tiež označovaná ako Upshaw-Schulman syndróm) je zapríčinená homozygótnou alebo heterozygótnou mutáciou ADAMTS13 [3]. Pacienti s heterozygótnou mutáciou nemusia mať žiadne zjavné abnormality [4]. Získaná TTP je autoimunitné ochorenie spôsobené tvorbou autoprotilátok proti ADAMTS13. Incidencia získanej TTP je oveľa častejšia u dospelých (2,9 prípadov na 1 milión/rok) ako u detí (0,1 prípad/1 milión/rok) [5].
- Klinický obraz a diagnóza TTP
TTP v porovnaní s ostatnými primárnymi TMA syndrómami zriedkavejšie vyvoláva závažné akútne zlyhanie obličiek. Hereditárna TTP sa prejavuje rekurentnými epizódami mikroangiopatickej hemolytickej anémie a trombocytopénie, ktoré sú často sprevádzané neurologickými prejavmi alebo postihnutím viacerých orgánov. Diagnóza hereditárnej TTP sa opiera o dôkaz deficitu ADAMTS13, chýbanie autoprotilátok a potvrdenie mutácie ADAMTS13. Hereditárna TTP sa môže manifestovať už hneď po narodení mikroangiopatickou hemolytickou anémiou a trombocytopéniou, alebo až v dospelosti po provokujúcom inzulte napr. tehotenstve [4, 6]. Hoci závažnosť priebehu sa odvíja od typu mutácie, z heterogénneho klinického obrazu u postihnutých súrodencov sa usudzuje, že pre manifestáciu sú dôležité aj ďalšie genetické a environmentálne faktory [7].
Klinické prejavy sú pestré, a to od minimálnych abnormalít až po dramatické život ohrozujúce stavy. V úvode je často prítomná slabosť, gastrointestinálne symptómy, následne sa pridružuje purpura a tranzientné neurologické prejavy. Treba však uviesť, že až u 1/3 pacientov chýba neurologická symptomatológia. Väčšina chorých má normálny alebo iba prechodne ľahko zvýšený sérový kreatinín. Diagnostické kritériá sa opierajú o prítomnosť mikroangiopatickej hemolytickej anémie a trombocytopénie bez zjavnej vyvolávajúcej príčiny. Diagnózu TTP podporí dôkaz aktivity ADAMTS13 < ako 10 % z normálnej hodnoty. Na druhej strane, nízka koncentrácia ADAMTS13 nie je dostatočne senzitívna, aby identifikovala všetkých pacientov s TTP ani dostatočne špecifická, aby vylúčila pacientov s prebiehajúcim základným ochorením [8].
- Liečba TTP
Terapia je podporná. Pri hereditárnej TTP sa pravidelne podávajú náhradné infúzie plazmy, pri získanej forme môže byť účinná plazmaferéza. Pri alergii na plazmu sa odporúča koncentrát faktora VIII, ktorý obsahuje ADAMTS13 [9]. Hypertenzia a neurologické abnormality môžu perzistovať aj po odoznení akútnej fázy ochorenia, ale prechod do chronickej obličkovej choroby je zriedkavý [10].
Plazmaferéza signifikantne zlepšila prežívanie pacientov so získanou formou TTP. V komplikovaných prípadoch sa indikujú glukokortikoidy, rituximab a ostatné imunosupresíva. Napriek tomu pacienti so získanou TTP majú riziko relapsov, zvýšenú prevalenciu kognitívnych porúch, depresie, hypertenziu a predčasnú smrť [5].
HEMOLYTICKO-UREMICKÉ SYNDROMY (HUS)
1. Typický hemolyticko-uremický syndrom (D+HUS)
V r. 1955 Gasser se spolupracovníky popsal poprvé toto onemocnění u 5 kojenců a malých dětí [11]. Definoval je jako akutní selhání ledvin, trombocytopénii a hemolytickou anémii. Označením onemocnění jako syndrom již tehdy vystihl skutečnost, že nejde o jednu klinicko-patologickou jednotku z hlediska etiologického, klinického, léčebného i prognostického. Typická forma hemolyticko-uremického syndromu (D+HUS) je označována jako idiopatický, epidemický HUS s charakteristickým prodromálním stadiem průjmů (diarrhoe).
- Epidemiologie D+HUS
D+HUS se vyskytuje především u dětí nižších věkových kategorií, mladších než 3 roky, téměř nikdy u novorozenců. Publikované incidence na 100 000 dětí do 15 let věku v západní Evropě, Severní Americe i v České republice jsou: 0,28 % (Itálie), 0,7 (Rakousko), 1,4 (Česká republika), 1,44 (Kanada), 1,6–1,74 (USA). Vztaženo na věk do 5 let je pak uváděna incidence v těchto regionech od 2–3 dětí na 100 000 ročně. Kmeny Escherichia coli označované synonymy Shiga-toxin produkující E. coli (STEC) nebo verotoxin (verocytotoxin) produkující E. coli (VTEC) jsou známy jako původci lidského onemocnění od r. 1982, kdy kmeny patřící k tehdy neobvyklému sérotypu O157:H7 vyvolaly dvě epidemie hemoragické kolitidy v USA. Následovaly obdobné epidemie v Ontariu v r. 1982, Québecu v r. 1983, Nebrasce v r. 1984 a opět v Ontariu v r. 1985. Ve velké většině bylo zdrojem infekce požití nedovařeného hovězího masa.
Spojení mezi STEC 0157:H7 a několika dalších sérotypů (0111, 026 a dalších) bylo poprvé dokumentováno Karmalim se spolupracovníky [12]. V následujících letech klinické a epidemiologické studie z různých oblastí světa potvrdily, že STEC jsou hlavními původci D+HUS [13–16]. Dosud k nejzávažnějšímu epidemickému výskytu došlo v r. 2011 v Německu, infikovaných bylo 3816 pacientů, z nichž u 845 (22 %) se rozvinul HUS, 54 pacientů zemřelo. V souboru pacientů převažovali dospělí (medián 42 let) a ženské pohlaví (68 %). Infekční, vysoce virulentní kmen E. coli O104:H4 (produkující Stx2) byl posléze prokázán v „bio“ pokrmech připravených z tzv. řeckého sena [17].
Přirozeným rezervoárem STEC je především hovězí dobytek (jejich zažívací trakt, výkaly), E. coli O157:H7 byla však izolována i od ovcí, koz, prasat, koní, vysoké zvěře. Vylučování STEC je u zvířat asymptomatické, přechodné (většinou ne déle než 1 měsíc), s vrcholem v teplém období roku. Infekční dávka patogenu je nízká. Analýza dat z epidemií prokázala, že k nejčastějšímu přenosu na člověka dochází po požití kontaminované potravy a vody (k přenosu může dojít i kontaktem s rezervoárovými zvířaty), výjimečně dochází k přenosu interpersonálnímu (azylové domy, hospice).
- Klinický obraz, laboratorní nálezy u D+HUS
Pro prodromální stadium D+HUS je charakteristická přítomnost průjmů. Gastrointestinální symptomy předcházejí plně rozvinutému onemocnění (4–6 dní), výjimečně po nich může následovat i krátké bezpříznakové období. Hemoragická kolitida se projevuje bolestí břicha, vodnatým průjmem, často s příměsí krve, někdy s poruchami pasáže, prolapsem rekta, invaginací, v ojedinělých případech může dojít až k perforaci střeva. Plně rozvinutý HUS je charakterizován náhle vzniklou anémií s nápadnou bledostí, často dušností. Je přítomen subikterus až ikterus, tendence ke krvácení do kůže. Renální symptomy zahrnují mikro - či makrohematurii, proteinurii (občas nefrotického typu), oligurii až anurii, v další fázi onemocnění polyurii. Jsou přítomny edémy, hypertenze (zpočátku spíše volumová), která spolu s možným (spíše však ojedinělým) výskytem myokarditidy či perikarditidy může vyústit v srdeční selhání. Symptomatologie postižení centrálního nervového systému různého stupně (somnolence, apatie, zvýšená dráždivost, epileptiformní křeče, kóma) se vyskytuje až u 50 % pacientů. Závažná CNS symptomatologie (na které se podílí hyponatrémie, možný edém mozku, mikroangiopatické léze v CNS, vlastní působení toxinu, urémie) pak koreluje s tíží onemocnění a je i jedním z negativních prognostických faktorů dalšího vývoje.
Z výsledků rutinně prováděných laboratorních vyšetření stojí v popředí pokles hemoglobinu, jeho hodnota se obyčejně pohybuje v rozmezí 50–100 g/l, může však klesnout i pod hodnotu 50 g/l. Nález schistocytů (2–10 %) a helmicovitých buněk v periferní krvi svědčí pro fragmentaci erytrocytů. Počet retikulocytů bývá většinou zvýšen. Průkazným nálezem hemolýzy je zvýšená hodnota LDH a nízký či nedetekovatelný haptoglobin. Nekonjugovaná hyperbilirubinémie mírného stupně je častá. Trombocytopenie, která je odrazem intravaskulární koagulace, se obvykle pohybuje v hodnotách 30–100 x109/l. Trombocytopenie pod 20x109/l a méně může způsobit krvácení. Koagulační testy jsou obvykle normální, stejně tak i Coombsův test je negativní. Přítomná bývá leukocytóza. Při typickém průběhu D+HUS jsou dalšími laboratorními nálezy hyperazotémie (zvýšená hladina urey, kreatininu), může být přítomna hyperkalémie, ale i hodnoty kalia v mezích normy (ztráty kalia stolicí v prodromálním stadiu), diluční hyponatrémie, metabolická acidóza, hyperfosfatémie, hypokalcémie, hypertriglyceridémie, hyperurikémie, hypoalbuminémie, mírná elevace transamináz, amylázy, lipázy, hyperglykémie. Při zachovalé diuréze hematurie, proteinurie.
- Mikrobiologická diagnostika
Při podezření na infekci jde především o rychlou izolaci STEC kmenů (kultivace na MacConkeyho agaru, detekce antigenů metodou ELISA). V další fázi je to pak detekce genů odpovědných za produkci Shiga toxinů 1, 2 metodou PCR (Stx1 a Stx2). Toto vyšetření provádí většinou již specializovaná laboratoř, klinický význam má především průkaz virulentnějšího typu toxinu – Stx2 [18]. Při hromadném výskytu onemocnění či dokonce při epidemickém výskytu je vhodná i spolupráce s veterinárními lékaři pro odhalení rezervoáru infekce.
- Patogeneze
Po expozici STEC dojde k jeho kolonizaci a adhezi na enterocyty v tenkém i tlustém střevě. STEC produkují dva odlišné typy toxinu, Shiga toxin-1 (Stx1) a Shiga toxin-2 (Stx2 – více potentní). Toxin je složen z enzymaticky aktivní podjednotky A a několika vazebných podjednotek B, které zprostředkují vazbu toxinu ke specifickým receptorům. Stx přechází přes střevní sliznici do systémové cirkulace, kde dochází k vazbě na polymorfonukleáry, ale i na erytrocyty (P1 antigen erytrocytární membrány), destičky, monocyty. Z cirkulace se Stx dostává k cílovým orgánům (již za 2 hodiny po požití), kde se váže na tzv. Gb3 receptory endotelových buněk (Gb3 receptor – globotriosylceramid receptor). Již za 30 minut po vazbě k endotelovým buňkám (v ledvinách, CNS, pankreatu a jiných parenchymových orgánech) dojde k inaktivaci ribozomů, následně k inhibici proteosyntézy a apoptóze. Postižení endotelu pak spouští kaskádu dalších reakcí: je snížena produkce prostacyklinu (PGI2 – má vazodilatační účinky, inhibuje agregaci a adhezi trombocytů), zvyšuje se lokální koncentrace zánětlivých cytokinů (např. IL-1, IL-2), dále faktoru nekrotizující nádory (TNF-alfa) a transformujícího růstového faktoru-beta1 (TGF-beta1). Je snížena syntéza NO, naopak zvýšeně jsou uvolňovány protrombotické faktory, jako např. inhibitor aktivující plazminogen, trombomedulin a jiné. Dalším důsledkem dysfunkce vaskulárního endotelu je uvolnění neobvykle velkých multimérů von Willebrandova faktoru (vWF) do cirkulace. Výsledkem výše popsaných změn je trombotická mikroangiopatie (zduření endotelu s rozšířením subendoteliálního prostoru, fibrinoidní nekróza stěny arteriol, přítomnost hyalinních trombů, organizace trombů především v aneuryzmatických úsecích), která v případě ledvin vede k akutnímu selhání ledvin.
I když mutace v regulačních genech komplementu jsou spojeny s atypickou formou HUS, k výrazné dysregulaci komplementu dochází též u D+HUS (Stx váže faktor H, dochází k alternativní cestě aktivace komplementu) [19].
- Léčba
Ke standardním léčebným postupům patří:
- Úprava vnitřního prostředí: Dosažení negativní vodní a sodíkové bilance (hyperhydratace je často zvládnutelná až dialýzou), úprava případné hyperkalémie (iontoměniče, beta-2-mimetika, dialýza), hyperfosfatémie (dialýza, iontoměniče – u dětí nejnižších věkových kategorií se běžně neužívají), hypokalcémie (perorální či parenterální substituce).
- Krevní převody: Korekce anémie (většinou až při poklesu hemoglobinu pod 60 g/l), podání trombocytárního náplavu (při poklesu trombocytů pod 20, resp. 10x109/l), jsou-li přítomny projevy krvácení a především, když je v této situaci plánován invazivní výkon (zavedení centrálního žilního katetru, PD katetru). Aplikace erytropoetinu v akutní fázi je alternativní či spíše krajním řešením v případě, že rodiče z náboženských důvodů krevní převod odmítají.
- Léčba hypertenze a léčba křečí podle běžných terapeutických schémat.
- Dialyzační léčba: Kritéria zahájení dialyzační léčby odpovídají obecným požadavkům u dítěte s akutním selháním ledvin (kalium vyšší než 7 mmol/l, urea vyšší než 35 mmol/l, anurie či výrazná oligurie s diurézou pod 0,5 ml/kg/hod trvající déle než 72 hodin, pomocným kritériem je pokles pH krve pod 7,2). Bez ohledu na tato kritéria se dialyzační léčba (ev. kontinuální venovenózní hemodiafiltrace – CVVHD) zahajuje při klinických známkách edému plic a mozku. Ve srovnání s ostatními příčinami akutního selhání ledvin je u HUS tendence zahajovat dialyzační léčbu spíše dříve. Volba mezi peritoneální dialýzou (PD), hemodialýzou (HD) či kontinuální očišťovací metodou (CVVHD) je dána především věkem dítěte, průběhem onemocnění. U kojenců a batolat je preferována PD.
- Zajištění optimálního stavu výživy: Dostatečné kalorické zajištění pacienta s akutním selháním ledvin zlepšuje jeho pozdní prognózu. Vzhledem k alteraci GIT je zpočátku výhodné upřednostnit výživu parenterální cestou.
Antibiotická léčba gastroenteritidy v akutní fázi onemocnění je kontroverzní, spíše se nedoporučuje. Recentní in vitro studie prokázaly, že např. minimální inhibiční koncentrace ciprofloxacinu a trimetoprimu vedly naopak ke zvýšené produkci Stx [20].
V souvislosti s rozsáhlou epidemií D+HUS v Německu v r. 2011 a při poznatcích, že i u D+HUS dochází k alternativní aktivaci komplementu, byla u nejtěžších případů D+HUS, především s CNS symptomatologií (poruchy vědomí, edém mozku, krvácení do CNS), podána monoklonální protilátka proti C5 složce komplementu eculizumab s příznivým efektem na další průběh onemocnění). Indikace k jeho podání je v současné době kontroverzně diskutována [21, 22].
- Extrarenální postižení u pacientů s D+HUS
Při včasném zahájení léčby (především dialýzy) se v současné době minimalizovala úmrtí v souvislosti s akutním selháním ledvin. Extrarenální postižení je v časném stadiu HUS nejčastější příčinou úmrtí a zhoršuje i dlouhodobou prognózu. Jde o postižení gastrointestinálního traktu (kolitida, invaginace, prolaps rekta, perforace střeva), přítomná může být akutní pankreatitida, endokrinní i exokrinní insuficience pankreatu, hepatomegalie s elevací transamináz. 30–50 % pacientů má projevy postižení CNS různého stupně (při krvácení do CNS bývá nejčastěji postižena oblast bazálních ganglií). Následkem trombotické mikroangiopatie může dojít i ke změnám v myokardu (kardiomyopatie), ke kožním projevům (petechie, purpury), postižení svalů (rhabdomyolýza), očí, parotické žlázy, ovarií, nadledvin. Postižení plic je spíše vzácné [23, 24].
- Prognóza pacientů s D+HUS
Ještě v 70. letech minulého století byla mortalita v akutní fázi D+HUS zhruba 20%, nyní se pohybuje kolem 5 %, nikoliv však z renálních příčin. Na druhé straně vysoká prevalence vzniku renálního postižení s odstupem několika let (10–15 let) po úspěšné léčbě akutní fáze onemocnění přiměla řadu autorů určit potenciální prognostické ukazatele v akutní fázi, které by mohly s pozdější alterací renálních funkcí souviset. Jsou to: věk pacientů (nižší věk je spojen s lepší prognózou), délka anurie (více jak 8 dní), délka trvání trombocytopenie, délka dialyzační léčby (více jak 4 týdny), závažná CNS symptomatologie, perzistující hypertenze. Někteří autoři uvádějí i leukocytózu (více než 20x109/l).
V souboru českých pacientů s D+HUS jsme po více než 10 letech prokázali, že pouze 25 % pacientů je zcela zdrávo, 40 % má tzv. renální reziduální symptomatologii (hematurie a/nebo proteinurie a/nebo hypertenze s normální či mírně sníženou hodnotou glomerulární filtrace – ne méně než 60 ml/min/1,73 m2) a 35 % má renální nedostatečnost či chronické renální selhání [25]. Meta-analytická studie kanadských autorů z r. 2003 porovnala výsledky 49 studií z 18 zemí z let 1950–2001, což představovalo soubor 3476 pacientů po D+HUS. 25 % pacientů vykazovalo při dlouhodobém přežití závažnou renální symptomatologii, negativním prognostickým ukazatelem byla jednoznačně CNS symptomatologie [26]. Z těchto důvodů je nutné pacienty i po klasické formě HUS dlouhodobě dispenzarizovat [27]. Z hlediska prevence je žádoucí vyloučit případné rezervoáry infekcí patogenních E. coli, do budoucnosti jsou určité možnosti spatřovány v pasivní imunoterapii v podobě např. monoklonálních protilátek proti Shiga toxinu.
2. Atypický hemolyticko-uremický syndrom (aHUS)
Atypický hemolyticko-uremický syndrom (aHUS) je definován jako jakákoliv forma HUS, která není typickým HUS, tzn. není způsobená infekcí Shiga-toxin produkující E. coli (STEC).
Atypický HUS tvoří cca 10 % případů dětí s HUS. Jedná se o velmi vzácnou chorobu s incidencí ve světě kolem 0,6 případů ročně na milion dětské populace [28], tj. ročně onemocní nově touto chorobou v ČR jen asi jedno dítě.
- Příčiny aHUS
Podle etiologie se aHUS dělí na:
- aHUS způsobený genetickými nebo imunologickými dysregulacemi alternativní cesty aktivace komplementu
- aHUS způsobený pneumokokovou infekcí, resp. enzymem neuraminidázou tvořenou pneumokoky (tzv. pneumokokový nebo neuraminidázový HUS)
- sekundární aHUS způsobený jiným primárním onemocněním (např. nádorovými onemocněními, SLE) nebo léky (např. cyklosporin, cisplatina)
- aHUS způsobený deficitem cobalaminu C
- aHUS způsobný mutacemi v genu DGKE (diacylglycerol kináza epsilon)
- neobjasněný aHUS
Přehled jednotlivých příčin aHUS je uveden v tabulce 1.
- Atypický HUS způsobený genetickými nebo imunologickými abnormalitami v systému komplementu (komplementopatie)
Snížené hladiny C3 složky komplementu zjišťované u pacientů s atypickým průběhem HUS již v 70. letech 20. století svědčily pro to, že tyto atypické formy HUS jsou způsobeny patologicky zvýšenou aktivací alternativní cesty komplementu [29]. I histologické nálezy v ledvinách pacientů s aHUS prokazovaly lokální aktivaci komplementu až k jeho lytické složce (depozice C3 v arteriolách a glomerulech a zvýšené množství finální lytické složky C5b-9 v místech intravaskulárních trombů). Avšak až v roce 1998 bylo poprvé prokázáno, že mutace v genu pro komplementový faktor H (CFH) způsobují aHUS [30]. V dalších 18 letech bylo objeveno celkem 8 genů, jejichž mutace způsobují nebo jsou predisponujícím faktorem pro vznik aHUS (geny pro komplementové faktory H, I a B, gen pro MCP/CD46, C3 složku komplementu, thrombomodulin a komplement faktor H příbuzné proteiny 1 a 3 – CFHR1 a CFHR3, hybridní CFHR1/CFH gen). Mutace v těchto genech vedou k nekontrolovatelné nadměrné aktivaci alternativní cesty komplementu až k terminálnímu komplementovému lytickému komplexu (TCC), který poškozuje cílové buňky, zejména endotelové. Obnažená subendoteliální matrix pak aktivuje koagulaci a dochází ke vzniku intravaskulárních trombů a tím k trombotické mikroangiopatii se všemi jejími důsledky, jako jsou tkáňová ischémie, nekróza a selhání funkce orgánů, zejména ledvin.
Dalším typem komplementopatického aHUS je aHUS způsobený inhibičními autoprotilátkami proti komplementovému faktoru H. Autoprotilátky proti komplementovému faktoru H (CFH) byly poprvé popsány ve spojení s aHUS v roce 2005 u tří dětských pacientů. aHUS asociovaný s protilátkami proti faktoru H tvoří ve světových registrech 6–25 % všech případů aHUS [31, 32]. Protilátky proti faktoru H se váží na C-terminální část CFH, tím snižují jeho regulační funkci, jejímž výsledkem je aktivace alternativní cesty komplementu [32]. Tyto autoprotilátky pravděpodobně vznikají u geneticky predisponovaných jedinců (u více než 90 % pacientů s tímto podtypem aHUS byla popsána homozygotní delece genů pro proteiny příbuzné s CFH - CFHR1 a CFHR3, způsobené nonalelickou homologní rekombinací na chromozomu 1q32, která způsobuje deficienci těchto proteinů. aHUS vzniklý jako kombinace HUS s tvorbou IgG protilátek proti CFH a deficience CFHR proteinů se označuje jako DEAP-HUS (Deficient for CFHR plasma proteins and factor H autoantibody positive HUS) [31]. Jde vlastně o autoimunitní onemocnění, které vede ke snížené inhibiční funkci komplementového faktoru H a tím opět k hyperaktivaci alternativní cesty komplementu a poškození endotelu. Tento typ aHUS je v ČR nejčastější a tvoří cca 50 % všech forem aHUS u českých dětí. I přes relativně nízkou pravděpodobnost výskytu jiných mutací spojených s aHUS u toho podtypu aHUS [32] se podle mezinárodních doporučení i přes nález protilátek proti faktoru H doporučuje provést u všech pacientů také molekulárně genetické vyšetření [33]. Věk manifestace onemocnění se pohybuje mezi 4.–17. rokem života. Zhruba 50 % pacientů s tímto podtypem aHUS se manifestuje průjmy stejnými jako u STEC-HUS, u 84 % pacientů se může objevit výrazná bolest břicha a zvracení, 23 % má příznaky pankreatitidy (zvýšenou sérovou amylázu a/nebo lipázu) a 50 % pacientů může mít elevované jaterní testy. Vzácně byly na začátku onemocnění popsány křeče, Malloryho-Weissův syndrom, urtika a přechodný edém obličeje. Kvůli akumulaci autoprotilátek je zde na rozdíl od ostatních forem aHUS vyšší počet relapsů onemocnění (58 %) a s každým relapsem roste riziko chronického onemocnění ledvin a chronického renálního selhání [31].
- Atypický HUS způsobený pneumokokovou infekcí, resp. neuraminidázou produkovanou pneumokoky
Tento typ aHUS je způsobený pneumokokovou infekcí (nejčastěji pneumokokovou pneumonií, ale i pneumokokovou sepsí nebo meningitidou), resp. enzymem neuraminidázou, který pneumokoky produkují. Tento enzym odštěpuje neuraminidovou kyselinu z glykoproteinů a glykolipidů membrány lidských erytrocytů, čímž dochází k odkrytí tzv. Thomsenova-Friedenreichova kryptantigenu (TF - nebo T-antigen). Tím dochází k hemolýze erytrocytů a hemolytické anémii s trombotickou mikroangiopatií. Diagnostickým kritériem pneumokokového HUS je kromě triády HUS (hemolytická anémie, akutní selhání ledvin a trombocytopenie) také průkaz infekce Streptococcus pneumoniae a pozitivní Coombsův test.
- Klinický obraz
Při aHUS onemocnění většinou nepředchází prodromální stadium kolitidy s průjmy (aHUS nesouvisí s infekcí E. coli, resp. Shiga-like toxinem). V případě pneumokokového aHUS je v předchorobí pneumonie, často komplikovaná pleurálním výpotkem, může však být přítomna i pneumokoková meningitida, sinusitida, otitida nebo sepse.
Onemocnění má výrazně těžší klinický průběh než typický HUS. V akutním stadiu (před zavedením nové léčby eculizumabem) byla uváděna mortalita u aHUS způsobeného poruchami komplementu 20–30 %. Mortalita pneumokokového aHUS byla v minulosti kolem 50 %, nyní se pohybuje „jen“ kolem 10 %.
Klinické projevy jsou až na chybění průjmu (ale až 30 % pacientů s aHUS může mít gastrointestinální příznaky včetně průjmu) neodlišitelné od typického HUS (bledost, únava, ikterus, petechie, makrohematurie, anurie).
Kolem 30 % pacientů má též extrarenální trombotické projevy choroby v CNS (křeče, poruchy vědomí), cévách končetin, GIT (játra, pankreas) či srdci.
- Diagnostika
a) Základní klinická a laboratorní diagnostika
Všichni pacienti s klinickým podezřením na aHUS musí být podrobně vyšetřeni – kromě základních vyšetření (krevní obraz (KO) s trombocyty, parametry renálních funkcí, moč chemicky a sediment) musí mít vyšetřeny také schistocyty v KO, Coombsův test, laktátdehydrogenázu, haptoglobin, C3 a C4 složku komplementu a celkovou komplementovou hemolytickou aktivitu (CH50 nebo CH100). Pacienti by měli mít základní neurologické vyšetření a EEG. Děti s podezřením na pneumokokový aHUS musí mít vyšetření mikrobiologické na přítomnost Streptococcus pneumoniae a proveden přímý Coombsův test.
Doporučený diagnostický a diferenciálně diagnostický algoritmus u pacientů s TMA (HUS/TTP) je uveden na schématu 1.

b) Molekulárně genetická diagnostika
DNA analýza genů způsobujících komplementopatické formy aHUS se provádí až po stanovení klinické diagnózy aHUS a po zahájení léčby. Mutace v některých genech způsobují velmi závažné formy HUS s velmi špatnou prognózou a časnou manifestací, jiné mutace, např. v genu MCP, mají lepší výhled s minimem relapsů po transplantaci ledviny (tab. 2). V současné době se daří prokázat genetickou anomálii v asociovaných genech u 70–80 % pacientů s aHUS. Většina mutací je heterozygotních a penetrance se zdá být relativně nízká, kolem 50 %. Často se najde mutace u jednoho z rodičů či sourozence, který je kompletně zdravý. Tato nízká penetrance nemoci ji vyčleňuje ze standardního konceptu monogenní choroby. Zdá se, že mutace je pouze predisponujícím faktorem a ke spuštění nemoci je třeba více faktorů, zejména zevních (např. infekce, stres apod.). Kombinace mutací ve dvou genech pro aHUS je velmi vzácná (3–6 %). Pro nízkou penetranci je i eticky obtížné určit indikaci k prenatální diagnostice. Kromě tradiční Sangerovy sekvenace je nutné i vyšetření MLPA (multiplex ligation-dependent probe amplification) k odhalení případné přítomnosti hybridního CFHR1/CFH genu, který neodhalí tradiční sekvenace.

Z klinického průběhu a základních laboratorních vyšetření nelze bohužel určit, o jakou genetickou formu D-HUS se jedná. V současné době však existuje několik rychlých screeningových vyšetření pro předběžné určení etiologie aHUS (např. vyšetření protilátek proti komplementovému faktoru H nebo exprese membránového kofaktorového proteinu (MCP) na leukocytech).
Znalost genetického pozadí onemocnění (odhalení mutací v genech pro aHUS – viz výše) a vyšetření protilátek proti faktoru H je tedy nezbytné pro přesné určení příčiny a odhad prognózy onemocnění. Od doby zavedení léčby aHUS eculizumabem však není z terapeutického hlediska zásadní, jakou formou aHUS pacient onemocní, protože příznivá odpověď na tuto novou léčbu je stejná u všech forem způsobených hyperaktivací komplementu. Molekulárně genetické vyšetření však má zásadní význam u pacientů, kteří jsou již dialyzováni a plánuje se zařazení do transplantačního programu, neboť různé formy aHUS mají různě vysoké riziko rekurence v transplantované ledvině (10–90 %) (tab. 2).
- Léčba
Možnosti léčby aHUS byly až donedávna omezené (dialýza, transfuze). Dlouhá léta se vědělo o příznivém efektu plazmaferézy s doplněním čerstvé plazmy. Léčbou první volby u aHUS byla až do nedávné doby plazmaferéza. Plazmaferézu bylo doporučováno zahájit v akutní fázi co nejdříve v průběhu nemoci a ideální je pokračovat v opakovaných plazmaferézách s cílem dosáhnout klinické i laboratorní remise. Plazmaferéza je ovšem i přes svou nespornou účinnost extrémně zatěžující a u některých forem neefektivní (např. mutace v MCP – anomálie membránového kofaktorového proteinu, nikoliv plazmatického faktoru).
V roce 2009 byly publikovány první práce s úspěšnou léčbou aHUS monoklonální protilátkou proti C5 složce komplementu (eculizumab) [34]. Tento lék cíleně míří na hyperaktivovanou alternativní cestu komplementu blokádou na úrovni faktoru C5 a přitom netlumí odstraňování imunokomplexů ani opsonizaci, ale pouze lytickou schopnost komplementu, který v případě hyperaktivace poškozuje buňky zejména endotelové. Cílená léčba touto monoklonální protilátkou umožňuje zvládnout akutní ataky aHUS a je vhodná i pro chronickou blokaci komplementu u pacientů, kteří byli rezistentní na plazmaferézu (efektivita léčby u plazmaferéza-dependentních i plazmaferéza-rezistentních forem aHUS je velmi vysoká, přes 90 % pacientů dosáhne remise), prevenci relapsů, včetně rekurencí po transplantaci ledviny [35]. Od roku 2015 je eculizumab mezinárodními experty konsensuálně doporučován jako léčba první volby u aHUS ještě před plazmaferézou [33]. Jeho nevýhodou je, že preparát je nutno podávat dlouhodobě (u některých podtypů komplementopatických aHUS zřejmě celoživotně) vždy po 14 dnech v intravenózní infuzi a jeho velmi vysoká cena. Naopak u některých podtypů aHUS (např. MCP-aHUS) je možné zvážit vysazení eculizumabu po dosažení remise onemocnění.
U pacientů s aHUS asociovaného s protilátkami proti faktoru H (DEAP-HUS) současný konsensus doporučuje zahájit včasnou léčbu eculizumabem, přestože eculizumab nesnižuje produkci protilátek proti faktoru H. Pouze u závažného průběhu onemocnění je vhodné zvážit zahájení plazmaferéz v kombinaci s imunosupresivní terapií (kortikosteroidy, cyklofosfamid, rituximab). Během terapie je nutná pravidelná kontrola hladin protilátek, při snížení jejich titru pod 1000 AU/ml je možné eculizumab vysadit bez další léčby nebo předtím zahájit imunosupresivní léčbu v kombinaci mykofenolát mofetil a kortikosteroidy ve snaze snížit titr protilátek. Pokud po jednom roce této terapie zůstávání hladina protilátek proti faktoru H pod 1000 AU/ml a C3 složka komplementů je normální, je možné terapii úplně vysadit [33].
U pacientů s pneumokokovým HUS je léčba většinou jen symptomatická, některá kazuistická sdělení ukazují na možný pozitivní vliv plazmaferéz na osud pacientů (uvažuje se o pozitivním vlivu odstraňování anti-TF protilátek a snižování neuraminidázové aktivity plazmy pacientů), což však nikdy nebylo dokázáno v kontrolované studii.
- Prevence
Prevence komplementopatických forem aHUS není možná vzhledem ke genetickému podkladu většiny typů. Naproti tomu u pneumokokového aHUS lze prevencí infekcí Streptococcus pneumoniae, zejména očkováním, snížit výskyt pneumokokových infekcí a tím i jejich komplikací jako je pneumokokový aHUS. Bohužel však očkovací látky nepokrývají celé spektrum serotypů Streptococcus pneumoniae, které může pneumokokový aHUS vyvolat.
- Prognóza
Prognózu pacientů s komplementopatickými formami aHUS revolučně změnilo zavedení léčby eculizumabem do klinické praxe v roce 2009. Proto je v současné době nutné prognózu pacientů rozdělit na éru před eculimabem a po jeho zavedení. Před érou eculizumabu byla v akutním stadiu mortalita pacientů 20–30%, navíc až 50 % dětí, které přežily akutní stadium, končilo v dalším průběhu v chronickém selhání ledvin – tedy dlouhodobá prognóza aHUS byla před érou eculizumabu významně horší než u typického HUS. Jednotlivé genetické formy se od sebe značně lišily svou prognózou. Mutace v některých genech způsobují velmi závažné vrozené formy HUS s velmi špatnou prognózou (geny pro faktor H, faktor B, C3), jiné mutace, např. v genu MCP měly lepší prognózou v akutním stadiu, navíc s minimem rekurencí po transplantaci ledviny.
Po zavedení léčby eculizumabu do klinické praxe se prognóza velmi výrazně zlepšila – přes 90 % léčených dětí se dostane do remise, navíc mortalita je téměř nulová. Navíc tato velmi příznivá odpověď na léčbu eculizumabem není závislá na typu mutovaného genu (pokud mutace zasahuje geny regulující komplement – tj. všechny kromě MCP), což je další výhoda této léčby, protože léčebný efekt nezávisí na specifické příčině a můžeme tedy zahájit léčbu hned po stanovení diagnózy hus, aniž bychom museli čekat na výsledek molekulárně genetického vyšetření.
Nejhorší prognózu mají v současnosti pacienti s pneumokokovým aHUS, u kterých zůstává mortalita kolem 10 %, navíc většina přeživších dětí končí po určité době v chronickém selhání ledvin a vyžaduje léčbu dialýzou a transplantací. Jednou z mála současných výhod této formy aHUS je fakt, že onemocnění nerekuruje po transplantaci ledviny.
Atypický HUS je život a ledviny ohrožující onemocnění, jehož prognóza závisí na jeho příčině. Díky nejmodernější biologické léčbě je prognóza komplementopatických forem aHUS velmi výrazně zlepšená oproti minulosti. Proto má v současné době nejhorší prognózu pneumokokový aHUS, jehož mortalita zůstává kolem 10 % a většina přeživších dětí dospěje do chronického selhání ledvin.
Podporováno grantem AZV MZ ČR reg. č. 15-31586A.
Prof. MUDr. Tomáš Seeman, CSc.
Pediatrická klinika 2. LF UK a FN Motol
V Úvalu 84
150 06 Praha 5
e-mail: tomas.seeman@lfmotol.cuni.cz
Sources
1. Moschcowitz E. Hyaline thrombosis of the terminal arterioles and capillaries: a hitherto undescribed disease. Proc N Y Pathol Soc 1924; 24 : 21–24.
2. Moake JL, Rudy CK, Troll JH, et al. Unusually large plasma factor VIII: von Willebrand factor multimers in chronic relapsing thrombotic thrombocytopenic purpura. N Engl J Med 1982; 307 : 1432–1435.
3. Levy GG, Nichols WC, Lian EC, et al. Mutations in a member of the ADAMTS gene family cause thrombotic thrombocytopenic purpura. Nature 2001; 413 : 488–494.
4. Fujimura Y, Matsumoto M, Isonishi A, et al. Natural history of Upshaw-Schulman syndrome based on ADAMTS13 gene analysis in Japan. J Thromb Haemost 2011; 9 (Suppl 1): 283–301.
5. Deford CC, Reese JA, Schwartz LH, et al. Multiple major morbidities and increased mortality during long-term follow-up after recovery from thrombotic thrombocytopenic purpura. Blood 2013; 122 : 2023–2029.
6. Moatti-Cohen M, Garrec C, Wolf M, et al. Unexpected frequency of UpshawSchulman syndrome in pregnancy-onset thrombotic thrombocytopenic purpura. Blood 2012; 119 : 5888–5897.
7. Lotta LA, Wu HM, Mackie IJ, et al. Residual plasmatic activity of ADAMTS13 is correlated with phenotype severity in congenital thrombotic thrombocytopenic purpura. Blood 2012; 120 : 440–448.
8. Kremer Hovinga JA, Vesely SK, Terrell DR, et al. Survival and relapse in patients with thrombotic thrombocytopenic purpura. Blood 2010; 115 : 1500–1511.
9. Naik S, Mahoney DH. Successful treatment of congenital TTP with a novel approach using plasma-derived factor VIII. J Pediatr Hematol Oncol 2013; 35 : 551–553.
10. Rosales A, Hofer J, Zimmerhackl L-B, et al. Need for long-term follow-up in enterohemorrhagic Escherichia coli-associated hemolytic uremic syndrome due to late emerging sequelae. Clin Infect Dis 2012; 54 : 1413–1421.
11. Gasser C, Gautier E, Steed A, et al. Hemolytisch-uremische syndrome. Bilaterale nierenrindendekrosen by akuten erworbenchen hamolytischen anemiam. Schweiz Med Wochenschr 1955; 85 : 905–909.
12. Karmali MA, Patric M, Lim C, et al. The association between idiopathic hemolytic-uremic syndrome and infection by verotoxin-producing Escherichia coli. J Infect Dis 1985; 151 : 775–782.
13. Bielazsewská M, Janda J, Bláhová K. Human Escherichia coli O157:H7 infection associated with the consumption of unpasteurized goat milk. Epidem Inf 1997; 119 : 299–305.
14. Cummings KC, Mohle-Boetani JC, Werner SB, et al. Population-based trends in pediatric hemolytic uremic syndrome in California, 1994-1999. Substantial underreporting and public health implication. Am J Epidemiol 2002; 15 : 941–948.
15. Yoshioka K, Yagi K, Moriguchi N. Clinical features and treatment of children with hemolytic uremic syndrome caused by enterohemorrhagic Escherichia coli O 15:H7 infection: experience of an outbreak in Sakai City, 1966. Pediatr Inf 1999; 41 : 223–227.
16. Bielazsewska M, Mellmann A, Zhang W, et al. Characterisation of the Escherichia coli strain associated with an outbreak of hemolytic uremic syndorme in Germany 2011: a microbiological study. Lancet Infect Dis 2011; 11 : 671–676.
17. Franc C, Werber D, Cramer JP, et al. Epidemic profile of Shiga-toxi-producing Escherichia coli 0104:H4 Outbreak in Germany. N Engl J Med 2011; 365 : 1771–1780.
18. Bielazsewska M, Mellmann A, Bletz S, et al. Enterohemorrhagic escherichia coli O26:H11/H-: A new virulent clone emerges in Europe. Clin Infect Dis 2013; 56 : 1373–1381.
19. Morigi M, Galbusera M, Gastoldi S, et al. Alternative pathway activation of complement by Shiga toxin promotes exuberant C3a formation that triggers microvascular thrombosis. J Immunol 2011; 187 : 172–180.
20. McGannon CM, Fuller Ca, Weiss A. Different classes of antibiotics differentially influence Shiga toxin production. Antimicrob Agents Chemother 2010; 54 : 3790–3798.
21. Lapeyraque AL, Malina M, Fremeaux-Bacchi V, et al. Eculizumab in severe shiga-toxin-associated HUS. N Engl J Med 2011; 364 : 2561–2563.
22. Kielstein JT, Beutel G, Fleig S, et al. Collaborators of the DGIN STEC--HUS registry. Best supportive care and therapeutic plasma exchange with or without eculizumab in Shiga-toxin-producing E. coli O104:H4 induced haemolytic-uraemic syndrome: an analysis of the German STEC--HUS registry. Nephrol Dial Transplant 2012; 27 : 3807–3815.
23. Siegler RL. Spectrum of extrarenal involvement in postdiarrheal hemolytic-uremic syndrome. J Pediatr 1994; 125 : 511–518.
24. Theobald I, Kuwertz-Broking E, Schiborr M, et al. Central nervous system involvement in hemolytic-uremic syndrome. Nephron 2002; 92 : 363–368.
25. Bláhová K, Janda J, Kreisinger J, et al. Long-term follow-up of Czech children with D+hemolytic-uremic syndrome. Pediatr Nephrol 2002; 17 : 400–403.
26. Garg AX, Suri RS, Barrowman N, et al. Long-term renal prognosis of diarrhea-associated hemolytic uremic syndrome. A systematic review, meta-analysis, and meta-regresion. JAMA 2003; 290 : 1360–1370.
27. Spinale JM, Ruebner RL, Copelovitch L, et al. Long-term outcomes of Shiga toxin hemolytic uremic syndrome. Pediatr Nephrol 2013; 28 : 2097–2105.
28. Ardissino G, Salardi S, Colombo E, et al. Epidemiology of haemolytic uremic syndrome in children. Data from the North Italian HUS network. Eur J Pediatr 2016 Apr; 175 (4): 465–473.
29. Stühlinger W, Kourilsky O, Kanfer A, Sraer JD. Haemolytic-uraemic syndrome: evidence for intravascular C3 activation. Lancet 1974 Sep 28; 2 (7883): 788–789.
30. Warwicker P, Goodship TH, Donne RL, et al. Genetic studies into inherited and sporadic hemolytic uremic syndrome. Kidney Int 1998 Apr; 53 (4): 836–844.
31. Noone D, Waters A, Pluthero FG, et al. Successful treatment of DEAP--HUS with eculizumab. Pediatr Nephrol 2014; 29 (5): 841–851.
32. Dragon-Durey MA, Sethi SK, Bagga A, et al. Clinical features of anti-factor H autoantibody-associated hemolytic uremic syndrome. J Am Soc Nephrol 2010; 21 (12): 2180–2187.
33. Loirat C, Fakhouri F, Ariceta G, et al.; HUS International. An international consensus approach to the management of atypical hemolytic uremic syndrome in children. Pediatr Nephrol 2016 Jan; 31 (1): 15–39.
34. Nürnberger J, Philipp T, Witzke O, et al. Eculizumab for atypical hemolytic-uremic syndrome. N Engl J Med 2009 Jan 29; 360 (5): 542–544.
35. Legendre CM, Licht C, Muus P, et al. Terminal complement inhibitor eculizumab in atypical hemolytic-uremic syndrome. N Engl J Med 2013 Jun 6; 368 (23): 2169–2181.
Labels
Neonatology Paediatrics General practitioner for children and adolescentsArticle was published in
Czech-Slovak Pediatrics

2017 Issue 2
- What Effect Can Be Expected from Limosilactobacillus reuteri in Mucositis and Peri-Implantitis?
- The Importance of Limosilactobacillus reuteri in Administration to Diabetics with Gingivitis
Most read in this issue
- Trombotické mikroangiopatie – hemolyticko-uremické syndromy a trombotická trombocytopenická purpura
- Nefrotický syndrom v dětském věku
- Doporučení Pracovní skupiny dětské nefrologie České pediatrické společnosti pro diagnostiku a léčbu infekcí močových cest u dětí a dorostu
- Neurofibromatóza u 5letého dítěte – diagnóza na základě huhňavosti