
Osteogenesis imperfecta – současný pohled na problematiku
Osteogenesis imperfecta – state of art
Osteogenesis imperfecta presents a group of genetically determined disorders, resulting in brittle bones. This condition is mostly caused by mutation in the genes encoding type I. collagen. Data about different types of illness, its hereditability, and treatment possibilities significantly increased during recent years. This review should bring state of art information about this disease in childhood.
Key words:
osteogenesis imperfecta, children, symptoms, diagnosis, therapy
Authors:
M. Bayer
Authors‘ workplace:
Osteocentrum, Oddělení revmatologie a rehabilitace, Thomayerova nemocnice, Praha
; Klinika dětí a dorostu 3. LF UK a FN Královské Vinohrady, Praha
Published in:
Čes-slov Pediat 2017; 72 (4): 212-222.
Category:
Overview
Jako osteogenesis imperfecta je označována skupina onemocnění na genetickém podkladě, vedoucí ke zvýšené lomivosti kostí. Stav je většinou způsoben mutací genů kódujících kolagen I. typu. V posledních letech významně narůstá množství poznatků o různých formách choroby, její dědičnosti a možnostech terapie. Článek by měl přinést současné informace o této nemoci u dětí.
KLÍČOVÁ SLOVA:
osteogenesis imperfecta, děti, příznaky, diagnóza, léčba
ÚVOD
Názvem osteogenesis imperfecta (OI) se označuje skupina geneticky determinovaných onemocnění. Jedná se o dědičná postižení pojivové tkáně s autozomálně dominantním či recesivním přenosem, popsány jsou již také genetické poruchy s fenotypem odpovídajícím OI vázané na chromozom X. V rodinách bez předchozí zátěže může jít o spontánní nové mutace. Převážná většina nemocných má mutaci jednoho ze dvou genů, COL1A1 nebo COL1A2, jež kódují kolagen I. typu. V genech COL1A1 a COL1A2, umístěných na chromozomech 7 a 17, bylo dosud nalezeno více než 1300 různých mutací. Choroba se vyskytuje jednou na 10 000–30 000 porodů. Příslušnost k etnické skupině ani pohlaví nemá na její incidenci vliv. Kolagen nemocných je horší kvality, což má za následek i poruchu orientace krystalů hydroxyapatitu při mineralizaci novotvořené kostní tkáně. Postižen je jak skelet, tak ostatní tkáně, jež kolagen obsahují.
PATOFYZIOLOGIE ONEMOCNĚNÍ A GENETICKÉ ASPEKTY
Kostní hmota dětí s OI při vyšetření v elektronovém mikroskopu nebo počítačovou tomografií s vysokým rozlišením a mechanickými testy vykazuje více resorpčních kavit, větší hustotu osteocytárních lakun a nižší pružnost (Youngův modulus). Je tedy více náchylná k fraktuře [1]. Jedním z rizikových faktorů je zřejmě nedostatečná maturace osteoblastů. K narušení vývoje nových osteoblastů mohou vést patologické změny v některých transkripčních faktorech, signálním glykoproteinu Wnt-1 (tato kaskáda je jedním z klíčových regulačních systémů pro správnou funkci osteoblastů), intracelulárním kationovém kanálu typu b a jiných substancích [2]. I nezralé osteoblasty nicméně stimulují formaci a diferenciaci osteoklastů, což vede ke zvýšené resorpci s úbytkem kostní hmoty [3].
Nejde však vždy jen o kvantitativní poruchu a/nebo deficitní mineralizaci. Patologie může též spočívat v abnormální organizaci minerální matrix s menšími, nevhodně orientovanými krystaly apatitu, jež postrádají konektivitu. Výsledkem je zhoršení mechanických vlastností kosti jako materiálu [4].
Nejčastějšími příčinami vzniku OI bývají dominantní mutace genů, kódujících řetězce kolagenu I. typu (tab. 1). Kolagen I. typu je tvořen trojitou spirálou dvou řetězců alfa 1 a jednoho řetězce alfa 2. Za ně zodpovídají geny COL1A1 a COL1A2. Po translaci probíhají následné modifikace pro-alfa řetězců v hrubém endoplazmatickém retikulu a Golgiho aparátu osteoblastů, kde vznikají vlastní trojité spirály. Tyto procesy řídí (kromě jiných) geny CRTAP, LEPRE1, PPIB a FKBP10. Prokolagen se posléze exocytózou dostává z buňky a po odštěpení propeptidu N a C se formují molekuly kolagenu, jež spřaženy příčnými můstky tvoří kolagenní vlákna. Ta jsou základem kostní hmoty, která je následně mineralizována.

Gen CRTAP kóduje tzv. „cartilage-associated protein“, který je potřebným kofaktorem posttranslační modifikace řetězců kolagenu. Při jeho chybění dochází k těžké osteochondrodystrofii, zlomeninám, deformitám končetin a poruše růstu. Stoupá množství vysoce mineralizované primární kosti s poruchou jejího dalšího vývoje [5]. Gen LEPRE1 kóduje prolyl 3-hydroxylázu (P3H1). Jeho mutace způsobí těžkou recesivní formu OI. Pokusná zvířata s inaktivovaným genem mají podle nálezů v elektronovém mikroskopu také abnormity ultrastruktury kolagenních vláken šlach a kůže [6]. Gen PPIB kóduje cyklofilin B (CyPB). CyPB spolu s výše uvedenými látkami tvoří v hrubém endoplazmatickém retikulu osteoblastu komplex, který řídí hydroxylaci prolinu v alfa řetězci prokolagenu I. typu. CRTAP a P3H1 se v tomto komplexu vzájemně stabilizují, zatímco CyPB jejich mutace neovlivňují. Dysfunkce celého komplexu P3H1/CRTAP//CyPB jsou zřejmě příčinou řady recesivních forem OI [7]. CyPB navíc kromě tvorby šroubovnice kolagenních řetězců hraje roli i při prostorové formaci C-terminálního propeptidu prokolagenu I. typu a jeho deficit se projeví na závažnosti manifestace OI [8]. V nedávné studii byly u 598 jedinců s OI nalezeny mutace ve 12 různých genech, v 11 % recesivní. Převaha dominantních mutací se týká COL1A1/COL1A2, malá část byla v genu IFITM5 nebo P4HB. Z recesivních genů se nejčastěji objevily mutace v SERPINF1 a CRTAP [9]. V poslední době genů spojených s autozomálně recesivní formou OI přibývá. Jejich výčet nyní zahrnuje také například geny SP7/Osterix nebo FKBP10 [10]. Recesivní mutace v FKBP10 způsobuje závažnou formu OI nebo Bruckův syndrom (OI s kongenitálními kontrakturami) [11]. Zde a též u genů PLOD2 a SERPINF1 je patofyziologickým mechanismem opožděná sekrece prokolagenu I. typu se současně porušenou schopností molekulární stability a posttranslační modifikace vedoucí ke snížení mechanické odolnosti kostní hmoty i ke vzniku kontraktur [12].
Ojediněle se jako příčina onemocnění objevují mutace zcela jiné, např. na dlouhém raménku 11. chromozomu [13]. Velmi vzácné recesivní formy OI mohou být vázané i na X chromozom [14].
Pokud jeden z rodičů trpí klasickou OI s autozomální dominantní dědičností, je riziko onemocnění pro každé jeho dítě 50%. Rodiče dítěte s recesivním typem OI mají stálé 25% riziko, že bude postiženo i další dítě. Narodí-li se zdravé, pak je ve ⅔ případů heterozygotem a mutovanou alelu dále přenáší.
Fenotyp nemocného nemá přímý vztah ke genotypu. Při téže mutaci může míra závažnosti klinických projevů u jednotlivých členů rodiny výrazně kolísat. Průkaz identické mutace tedy nelze použít k prognóze stavu postiženého jedince. U všech typů OI také může nastat situace, kdy se choroba v rodině u nikoho jiného nevyskytuje. Pak bývá stav způsoben spontánní novou mutací. Rizikové faktory pro jejich vznik během gravidity dosud objasněny nebyly. Je-li na vině spontánní výskyt nové mutace, dosahuje riziko OI v další graviditě pouze 2–5 %.
Identifikace mutace tak nabývá na důležitosti zejména pro určení rizika v další graviditě a plánování prenatální péče. Genetické testování asymptomatického rodiče nemocného dítěte by mohlo prokázat, zda rodič má dosud nediagnostikovanou OI; mozaiku mutace pro dominantní typ OI, nebo zda rodiče či sourozenci dítěte jsou přenašeči recesivní formy OI.
KLINICKÉ PROJEVY A KOMPLIKACE
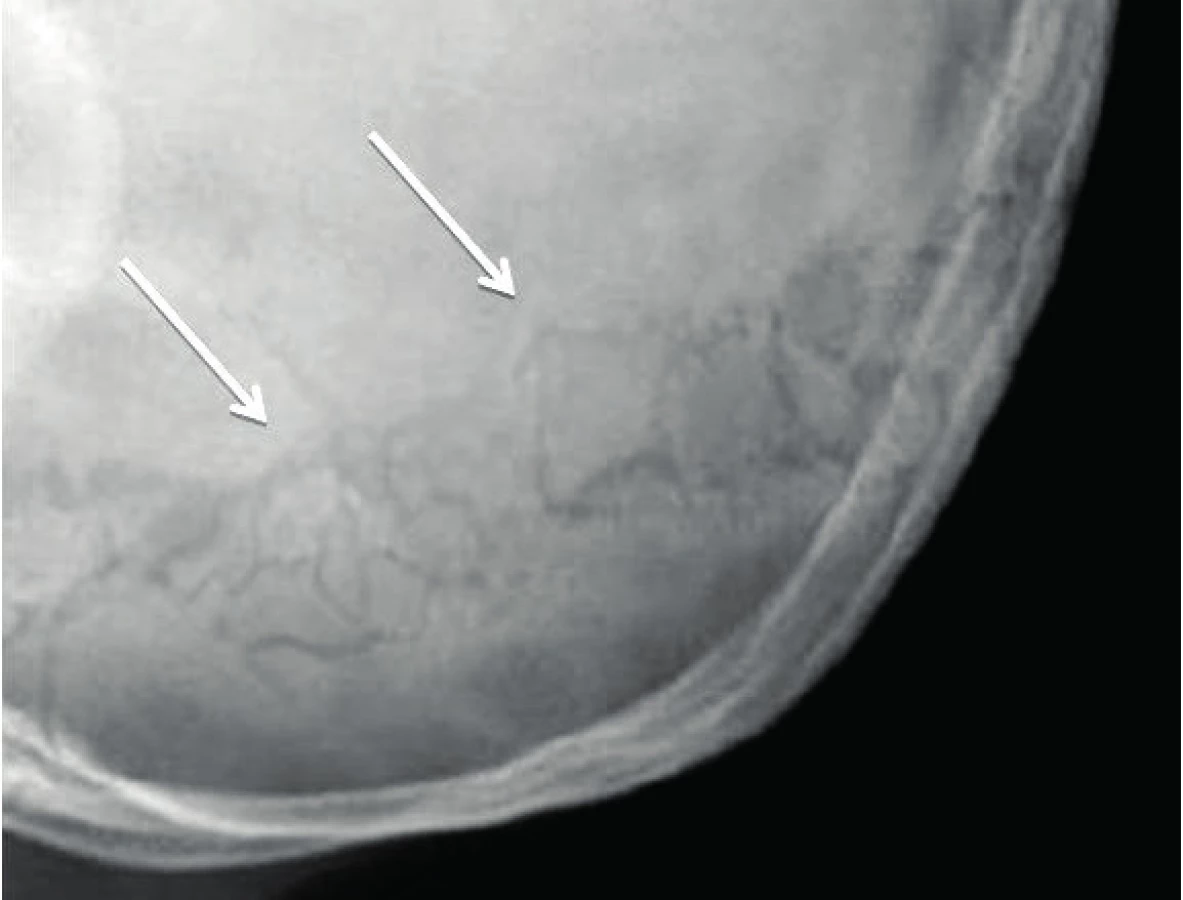
Manifestace OI může být různorodá. Typickým nálezem jsou zlomeniny, které mohou postihnout kteroukoli kost (obr. 1). Kompresivní fraktura obratle u dítěte při jeho běžných aktivitách je vždy známkou poklesu mechanické odolnosti kosti. Podezření by dále měly budit dvě zlomeniny dlouhých kostí do desátého roku věku nebo tři a více fraktur během celého dětství. Zlomeniny prstů či nosu se nepočítají, stejně jako fraktury způsobené úrazu přiměřeným mechanismem (pády z výšek, kolo, lyže, kontaktní sporty apod.). Jedinci s těžšími formami choroby mívají velmi malou postavu. Na růstové faktory však typ OI významný vliv nemá, u nemocných s různě závažnou manifestací choroby jsou IGF-I a IGFBP-3 nacházeny v dolní části normálního rozmezí [15]. U kojenců je možné pozorovat velké rozměry fontanel, které se také později uzavírají. Část nemocných mívá na lebce tzv. wormiánské kůstky (obr. 2). Jde o nadpočetné kůstky lebního krytu kompletně oddělené linií švu. Vznikají z nadpočetných osifikačních center na lebce. Jejich častou lokalizací je lambdový či koronární šev. Na vzniku wormiánských kůstek se zřejmě podílí napětí dura mater a rozšíření lebečních švů. Může se tak stát při omezené osifikaci lebky, k níž dochází při různých metabolických osteopatiích [16]. Jejich nález tedy není pro diagnózu OI určující, podezření na chorobu však nepochybně zvyšuje. Za významný počet se považuje nález deseti a více wormiánských kůstek na nativním rentgenovém snímku lbi. Takřka ve třech čtvrtinách případů jsou zachytitelné v prvém roce života. Prevalence výskytu významného počtu wormiánských kůstek je u nemocných s OI nepřímo úměrná Z-skóre výšky postavy. Ve skupině 195 dětí s OI byl zjištěn jejich významný počet u 35 % nemocných s OI I. typu, v 96 % při OI III. typu a 78 % OI IV. typu [17].

S OI jsou typicky spojovány modré skléry (obr. 3). Jsou tmavší a mohou mít šedý nebo modravý nádech. Tento projev má ovšem ve skutečnosti pouze asi polovina nemocných. Lehce namodralé skléry mohou mít i zcela zdravé děti ve věku do 18 měsíců. U všech typů OI bývá také ztenčená rohovka [18]. Postižení častěji než zdravá populace trpí myopií.

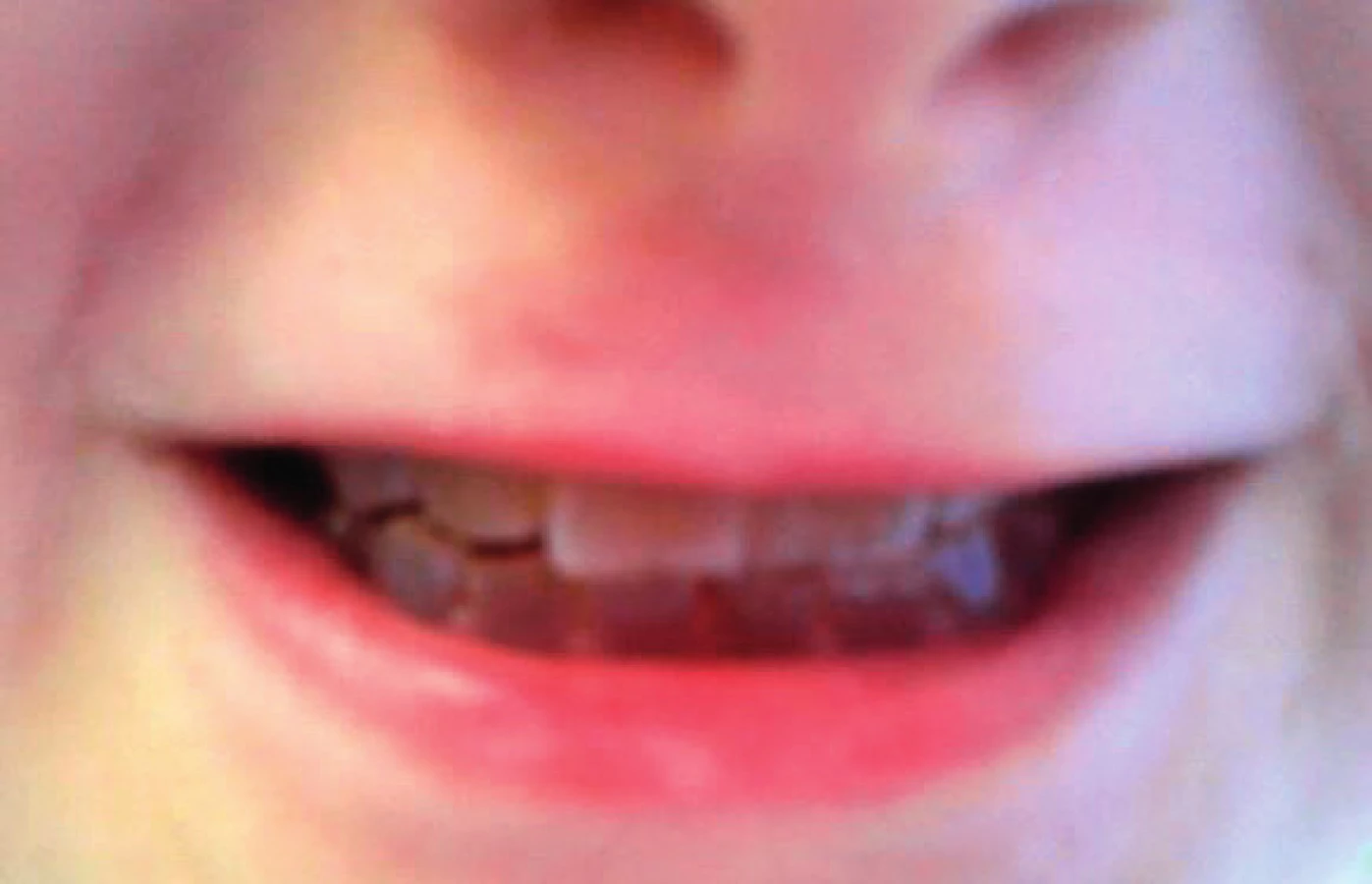
Další nálezy se týkají chrupu. Zuby u jedinců s neléčenou OI prořezávají o něco dříve a také dochází k rychlejší obměně primární dentice. Terapie bisfosfonáty tyto děje mírně zpomaluje a vývoj chrupu vůči věku je pak stejný jako u zdravých dětí [19]. Jedinci s OI způsobenou kvalitativní poruchou kolagenu I. typu mívají často oligodoncii. Prevalence ageneze zubů je u nemocných s OI vysoká, okolo 17 % [20]. Přibližně u 50 % nemocných lze zastihnout dentinogenesis imperfecta, dysplazii dentinu s našedlými a opalescentními zuby, které jsou nápadně křehké (obr. 4). Postižena bývá již první dentice. I jedinci z různých rodin mají za předpokladu téže mutace shodný výskyt dentinogenesis imperfecta v 90 % (a modrých sklér v 75 %). Do určité míry je tedy možné na základě prokázané mutace alespoň některé fenotypické projevy nemocného dítěte předpovědět [21].
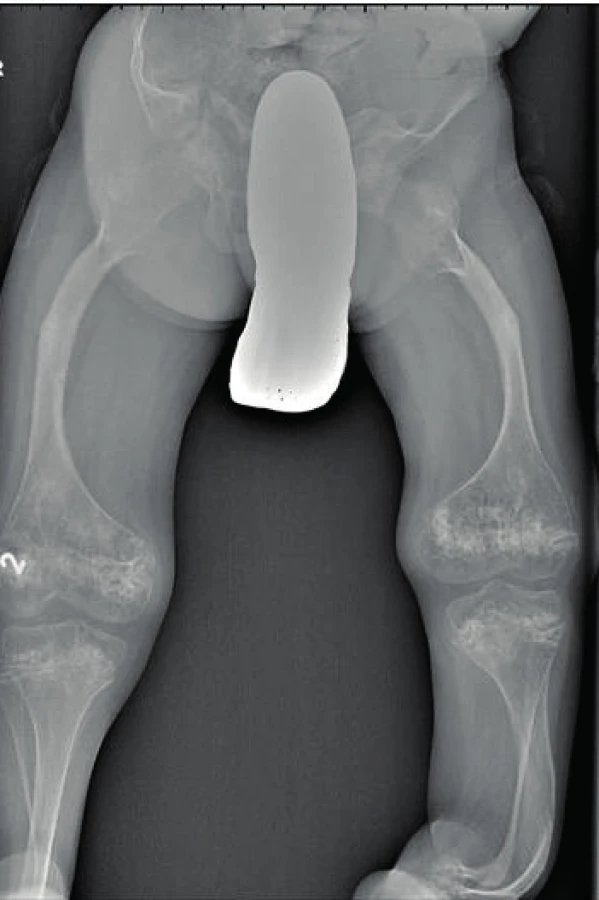
Opakované fraktury vedou k zakřivení dlouhých kostí (obr. 5, 6). Časté jsou také deformity hrudníku (pectus carinatum, pectus excavatum, abnormity žeber) a páteře (skolióza, kyfóza). S těmito změnami souvisí bolesti zad a poruchy chůze, je-li jí pacient schopen. Ve vysokém procentu mívají skoliózu nemocní s typem OI III a IV, u typu I je výskyt řidší. Bylo pozorováno, že farmakoterapie (bisfosfonáty) zahájená do věku šesti let může u OI III. typu manifestaci skoliózy příznivě ovlivnit [22]. V důsledku abnormit žeber, obratlů i páteře, snížené síly mezižeberních svalů a nedostatečného objemu hrudníku pacienti s OI trpí častěji infekty dýchacích cest a poruchami plicních funkcí.

Kvalitativní a/nebo kvantitativní porucha kolagenu nepostihuje pouze kostní tkáň. Děti mají uvolněné vazy, což vede k hypermobilitě a nestabilitě kloubů, častěji se objevují hernie. Dochází ke snadné tvorbě hematomů. Nemocní mívají ploché nohy a jejich svalová síla je snížena, což lze doložit aplikací funkčních hodnotících škál [23]. Některé děti s mírnou formou OI mohou jevit i rysy Ehlersova-Danlosova syndromu s výraznou kloubní hyperlaxicitou, volnou kůží se špatným hojením poranění a nápadnou fragilitou cév. V takových případech je pečlivá biochemická analýza kolagenu potřebná k upřesnění diagnózy [24].
Mezi nejčastější kardiovaskulární projevy OI patří dilatace kořene aorty, vedoucí k aortální či mitrální regurgitaci [25, 26]. U dospělých je diagnóza OI v porovnání se zdravými jedinci nezávislým faktorem snížené funkce pravé komory srdeční [27]. Výskyt nedomykavosti aortální a mitrální chlopně, fibrilace síní, aneuryzmat cév a kardiálního selhání u dospělých pacientů s OI a u zdravé populace v nedávné dánské studii dokládá, že nemocní s OI mají vyšší riziko kardiovaskulárních onemocnění, a to i po korekci na známé faktory přispívající k rozvoji těchto chorob [28].
Ve třetí či čtvrté dekádě života, méně často i dříve, jsou nemocní s OI ohroženi poruchou sluchu. Ta může být způsobena kochleární otosklerózou, zlomeninami či atrofií kůstek vnitřního ucha nebo kochleární degenerací [29]. Porucha sluchu je velmi variabilní a jako u ostatních klnických projevů OI zde chybí korelace mezi genotypem a fenotypem [30]. Sluch je proto nutné u dětí s OI opakovaně vyšetřovat, dysfunkce středního ucha ve spojitosti s otitidami ohrožují nemocné mladší dvaceti let mnohem častěji než jejich zdravé vrstevníky [31].
Nezbytná je také zmínka o abnormitách baze lební. Nějaká kraniocervikální anomálie byla zastižena u 37 % nemocných s OI. Může se vyvinout i navzdory léčbě bisfosfonáty. Nutno to mít na paměti zejména u těžších forem onemocnění [32, 33]. Nezávislým faktorem predispozice k takové abnormitě se při OI ukazuje hodnota Z-skóre výšky postavy. Při Z-skóre pod -3,48 mají abnormitu baze lební prakticky všichni pacienti bez ohledu na věk, pohlaví, barvu sklér, denzitu kostního minerálu v oblasti páteře či anamnézu terapie bisfosfonáty [34]. U závažnějších průběhů OI může nestabilita obratlů C1-C2 způsobit životu nebezpečnou komplikaci, bazilární invaginaci a kompresi kmene [35]. Nelze ji předvídat. Projeví se bolestmi hlavy, nystagmem, spazmy v obličeji, slabostí končetin, případně pádem. Může při ní dojít k náhlému úmrtí. Diagnózu atlanto-axiální subluxace stanoví počítačová tomografie. U některých nemocných s těžší OI se rozvíjí hydrocefalus. Většinou však bývá komunikující a nevyžaduje vždy řešení zkratem.
Přestože je u řady dětí s OI možno pozorovat opoždění vývoje hrubé motoriky, zejména v důsledku hypotonie a opakovaných zlomenin, jejich mentální vývoj je většinou normální.
KLASIFIKACE JEDNOTLIVÝCH TYPŮ OSTEOGENESIS IMPERFECTA
V roce 1979 navrhl David Sillence klasifikaci OI na typy I, II, III a IV [36]. Vycházel přitom ze způsobu dědičnosti, klinického obrazu a nálezu na rtg. Na přelomu století pak postupně přibyly další typy OI s odlišnými klinickými projevy nebo jinou genetickou příčinou. Spolu s bouřlivým rozvojem poznatků o genetickém podkladě jednotlivých poruch vedoucích ke klinické manifestaci patologických stavů s fenotypem OI, provázených zlomeninami, vznikla potřeba stávající klasifikaci revidovat. V současné době je ve spojitosti s OI známo více než 1500 různých mutací [37]. V převážně většině se týkají poruchy tvorby, prostorového uspořádání a maturace kolagenu I. typu. Nepoměrně vzácnější jsou poruchy signálních systémů a jiných funkcí osteoblastů. Pro běžnou klinickou praxi nicméně postačí původní základní dělení, doplněné o OI V. typu [38].
OI I. typu
Nejčastější (asi polovina všech nemocných) a nejmírnější forma. Závažné kostní deformity nevznikají. Modré skléry jsou časté. Postižení mají jen mírně sníženou výšku postavy. U části pacientů lze nalézt dentinogenesis imperfecta. Počty zlomenin jsou různé, po ukončení růstu jich významně ubývá. Tento typ OI se dědí autozomálně dominantně, velmi časté jsou i nové spontánní mutace.
OI II. typu
Je formou nejzávažnější. Příčinou stavu bývá nová dominantní mutace nebo parentální mozaicismus. Deformity a zlomeniny dlouhých kostí, obratlů a žeber jsou patrné již intrauterinně. Dítě může mít makrocefalii a rodí se většinou s těžkou poruchou mineralizace lebního krytu. Skléry jsou šedé nebo tmavě modré. Deformity hrudníku vedou k hypoplazii plic, která je limitující pro prognózu. K úmrtí většinou dochází během několika týdnů.
OI III. typu
Nejtěžší forma, která přežívá. Také u těchto dětí nalézáme fraktury již před narozením. Novorozené děti mají měkkou lebku, šedomodré skléry, deformity hrudníku a končetin. Spolu s dalšími zlomeninami deformity progredují. Častá je těžká skolióza. Výsledná výška postavy je v dospělosti nezřídka jen kolem jednoho metru. Postižení jsou většinou upoutáni na vozík. Onemocnění je nejčastěji způsobeno spontánní novou mutací, dědičnost však může být i dominantní či recesivní.
OI IV. typu
Na rozdíl od prvého typu mívají tito nemocní významně nižší postavu. Komprese obratlových těl, deformity páteře a zlomeniny dlouhých kostí se zde vyskytují v různé míře. Namodralá barva sklér není nápadná a u některých dětí postupně vymizí. Tento typ se dědí autozomálně dominantně, v mnoha případech jde o novou mutaci.
OI V. typu
Typickým znakem je tvorba nápadně hypertrofického svalku po fraktuře nebo operačním zákroku na kosti [39]. Na dlouhých kostech bývají vícečetné osifikace, nezřídka též dochází ke kalcifikaci interoseální membrány na horních i dolních končetinách. Pokud se vytvoří mezi radiem a ulnou, významně omezuje rotační pohyb předloktí. Může dojít k dislokaci hlavičky radia. Nemocní si bývají podobní, s krátkým vzhůru natočeným nosem, malými ústy, prominující bradou a šedomodrými sklérami [40]. Radiologický obraz na dlouhých kostech se během prvého roku života vyvíjí od nálezu podobného rachitidě k typickým denzním pruhům v metafýzách. Zřejmě se patofyziologický proces týká kosti i chrupavky [41]. Onemocnění se dědí autozomálně dominantně.
DIAGNÓZA A DIFERENCIÁLNÍ DIAGNOSTIKA
Základem diagnostiky OI je pečlivé fyzikální vyšetření dítěte spolu s podrobnou osobní i rodinnou anamnézou. Zachycení klinických příznaků je pro stanovení diagnózy velmi důležité a v řadě případů je pro ni určující. Nutno však mít na paměti, že třeba nálezy modrých sklér nebo malé postavy sice do obrazu OI patří, ale nejsou pro ni zcela patognomické. Nativní rentgenové snímky skeletu zdokumentují nové i starší fraktury, kompresivní deformity obratlů, zakřivení dlouhých kostí a wormiánské kůstky na lebce. Denzitometrické vyšetření (nejčastěji dvouenergiovou rtg absorpciometrii bederní páteře, případně i celotělovou) je možné na různých typech přístrojů provést od dvou, tří nebo pěti let věku. Výsledek Z-skóre, korigovaný na aktuální velikost postavy dítěte, má prognostický význam predispozice ke zlomeninám. Denzita kostního minerálu (bone mineral density, BMD) může být snížena u všech typů OI, ale existují také varianty onemocnění, při nichž může být i zvýšena, např. mutace genu pro kostní morfogenetický protein 1 (BMP1 – viz tab. 1). Moderní denzitometrické přístroje již umožňují i hodnocení deformity obratlů (tzv. vertebral fracture assessment, VFA). Po počátečních nejistotách se tato metoda, jež je spojena jen s malou dávkou záření, zdá u dětí s OI vůči konvenční radiografii bezpečnější a praktickou alternativou [42]. Pouze na samotnou hodnotu BMD ostatně spoléhat nelze. Kompresi obratle má poměrně vysoké procento dětí s OI a nález normální BMD ji nevylučuje.
Laboratorní vyšetření v případě OI není v diagnostice klíčové. Většina nemocných má laboratorní nálezy zcela normální. U části z nich lze zachytit různý stupeň hyperkalciurie, která však většinou nevede k rozvoji nefrolitiázy.
Laboratorní ukazatele kostního obratu se během terapie OI mohou významně měnit, ale se změnami BMD, hybnosti, výskytem bolestí, ani s přítomností kompresivních zlomenin obratlů nekorelují [43]. Molekulárně genetickým vyšetřením lze dnes odhalit převážnou většinu mutací genů pro řetězce alfa 1 nebo alfa 2 kolagenu I. typu. V případě nejasností připadají v úvahu další geny zodpovědné za proteiny, které se uplatňují při tvorbě a maturaci kolagenu nebo v signálních kaskádách činnosti osteoblastů. Průkaz konkrétní genetické příčiny zejména u těžkých forem OI je důležitý pro prenatální diagnostiku a určení rizika v dané rodině. Nutno však mít na paměti, že negativní nález při molekulárně genetickém vyšetření OI nijak nevylučuje. Kostní biopsie je při diagnostice OI další možnou, ne však rutinní metodou. Vzorek kostní tkáně je možné získat během ortopedické operace nebo lze (u dětí s hmotností nad deset kilogramů) provést izolovanou biopsii v celkové anestezii, nejčastěji z lopaty kosti kyčelní. Vyšetření vzorku a jeho interpretace vyžaduje zkušeného hodnotitele. Zatím nejsou k dispozici dostatečné referenční databáze histomorfometrických parametrů u zdravých dětí různého věku.
Diferenciálně diagnosticky je třeba u dětí s opakovanými frakturami uvažovat také o možném syndromu týraného dítěte. Mírnější formy OI (občasné zlomeniny s ne zcela objasněným mechanismem, časté hematomy na kůži, bez dalších projevů) mohou zejména při negativní rodinné anamnéze k takovému podezření vést. Následně se může rodina ocitnout ve velmi svízelné situaci, kdy případně hrozí i odejmutí dítěte [44]. Podrobný rozbor vzniku jednotlivých úrazů a pečlivé zhodnocení celkového stavu dítěte lékařem v problematice OI zkušeným spolu s denzitometrií skeletu a genetickým vyšetřením může do případu vnést více světla. Nicméně rutinní testování molekulárně genetickými metodami se při podezření na syndrom týraného dítěte jako dostatečně výtěžné neukázalo [45].
Mezi další vzácná onemocnění s vysokou lomivostí kostí, která mají fenotyp OI, patří například:
- osteoporóza s pseudogliomem, provázená slepotou, způsobená mutacemi genu pro LRP5 – „low density lipoprotein receptor-related protein 5“ [46];
- syndrom Cole-Carpenterův s kraniosynostózou, proptózou bulbů, hydrocefalem a poruchou růstu [47];
- syndrom Bruckův s kontrakturami kloubů, způsobený mutacemi genu pro kostní specifickou lysyl-hydroxylázu PLOD2, ovlivňující vznik vazeb mezi řetězci kolagenu [48, 49], nebo mutací genu FKBP10;
- syndrom osteogenesis imperfecta/Ehlersův-Danlosův syndrom. Onemocnění se kromě zvýšené lomivosti kostí vyznačuje tendencí k rychle progredujícímu patologickému zakřivení páteře [50].
TERAPEUTICKÉ POSTUPY
Ke každému pacientovi je nutno přistupovat s ohledem na jeho individuální stav, možnosti a rodinné prostředí. Cílem je jeho začlenění mezi vrstevníky a postupně do společnosti spolu s dosažením co možná nejvyšší míry samostatnosti a sebeobslužnosti. Je k tomu potřeba týmové práce více odborníků a samozřejmě úzká spolupráce celé rodiny. Od okamžiku stanovení diagnózy je na místě úprava životního stylu, rehabilitační péče, v případě potřeby terapie farmaky a podle indikace ortopeda léčba chirurgická.
Životní styl
U novorozence nebo kojence s OI se musí matka pod dohledem odborného personálu naučit, jak ho správně ošetřovat. Při manipulaci s dítětem je třeba se vyvarovat nadměrných tahů, rotací či tlaků na dlouhé kosti. Je vhodné relativně často měnit polohu dítěte v postýlce. Rozmrzelost a plačtivost bez jasné příčiny a zejména omezení spontánní hybnosti některé končetiny mohou být známkou nové fraktury. Nicméně postupně se zvyšující pohybové aktivitě dítěte a stimulům z okolí by se s ohledem na potřebu řádného psychomotorického vývoje nemělo bránit. Umožní-li to jejich celkový zdravotní stav, měly by tyto děti docházet už do mateřské školy. Záleží samozřejmě na dohodě rodičů a personálu, protože zvýšený dohled je nutný. Základní školní docházku není třeba odkládat. Řada školních budov je dnes již zařízena bezbariérově, takže i vozíčkáři se mohou běžně účastnit výuky. Učitelé by měli vědět, že OI nebývá spojena s mentální poruchou. Dítě s lehčí formou OI není na místě paušálně uvolňovat ani z tělesné výchovy. Po dohodě s ošetřujícím lékařem je jenom nutné stanovit konkrétní činnosti, kterých by se účastnit nemělo (cvičení na nářadí, skoky z výšky, kontaktní sporty apod.). Rodiče i učitelé by měli pochopit, že různé činnosti ve skupině dětí jistě riziko zlomeniny zvyšují, ale že za úspěšné zařazení do kolektivu je to většinou cena přijatelná. Možnost začlenit se mezi vrstevníky přináší nemocnému potřebné sebevědomí, což ve starším školním a posléze dorostovém věku může být důležitější než úzkostlivá ochrana před frakturou.
Děti s OI mohou a mají být očkovány podle platného schématu bez omezení.
Rehabilitace
Přiměřená fyzická aktivita prospívá všem pacientům s OI. Pozitivní působení svalové práce na kostní hmotu je u nemocných s OI stejné jako u zdravých jedinců. Případná svalová slabost u nich nepochybně přispívá k prohloubení již snížené mechanické odolnosti kostí [51]. Už od kojeneckého věku lze vhodným polohováním předcházet kontrakturám a cvičením posilovat trend postupného vzpřimování postavy. Nácviky správných technik sezení, vstávání a stoje jsou prevencí poruch páteře. Cvičení pod dohledem zvyšuje svalovou sílu, aerobní kapacitu a zmírňuje pocity únavy [52]. Pro děti s OI je zvláště vhodný pohyb ve vodě. V posledních letech se na pokusných zvířatech i u lidí zkoumá vliv vibrační terapie [53, 54]. Rekreační sportovní aktivity, individuálně přizpůsobené závažnosti choroby, jsou pro děti s OI jednoznačně přínosem fyzickým i psychickým.
Farmakoterapie
Příčinná léčba OI v současné době k dispozici není. Z farmak se dosud nejvíce osvědčily bisfosfonáty. Snižují výskyt zlomenin dlouhých kostí a uplatňují se v prevenci kompresivních fraktur obratlů, takže nepřímo zmírňují deficit výšky postavy [55]. U OI III. typu může terapie bisfosfonáty omezit progresi skoliózy, zejména u předškolních dětí. V adolescenci už skolióza léčbou ani typem OI významněji ovlivněna není [56]. Účinek bisfosfonátů je obecně výraznější u dětí s těžším průběhem choroby [57] a během prvých tří či čtyř let terapie [58]. Oficiální doporučení pro výběr bisfosfonátu, dávkování a dobu, po kterou by měla terapie trvat, neexistuje. Nejdéle se u dětí s OI používá intravenózní pamidronát. Poměrně spolehlivě zvyšuje BMD, snižuje počty zlomenin a zlepšuje stavy po kompresi obratlů, a to u všech sledovaných typů OI [59]. U dětí mladších dvou let se obecně doporučuje dávka pamidronátu 0,5 mg/kg/den po tři dny každé dva měsíce. V praxi je u nich vhodné při zahájení léčby podat prvou dávku nižší (0,25 mg/kg/den) a pokud se s terapií začíná u novorozence či velmi malého kojence, existují schémata ještě šetrnější. U dětí starších dvou let již přichází v úvahu standardní podávání pamidronátu v dávce 1 mg/kg po tři následující dny se čtvrtletní pauzou. Podle některých autorů je toto možné nahradit jednou infuzí s dvojnásobnou dávkou účinné látky třikrát ročně. Zatím však takový postup nebyl ověřen na větším počtu nemocných a nejsou zprávy, zda je stejně účinný v prevenci zlomenin [60]. Před léčbou je vhodné ověřit, že dítě má normální funkce ledvin, vyšetřujeme mineralogram, jaterní testy a krevní obraz. Na případnou „flu-like“ reakci postačí běžná antipyretika. Podle literárních údajů lze během terapie i.v. pamidronátem v kostní tkáni zastihnout zvětšené osteoklasty s vyšším počtem jader. Příčina jevu známa není, uvažované teorie o toxickém působení pamidronátu se nepotvrdily [61]. V některých státech se k léčbě OI u dětí používá i.v. neridronát [62], podáván byl také i.v. ibandronát v dávce 2 mg každé tři měsíce [63]. Vzestupu kostní hmoty a poklesu výskytu kostních deformit bylo dosaženo i perorálním risedronátem, u něhož se optimální dávkou zdají být 2 mg/kg jednou týdně [64]. Pomalu též narůstají zkušenosti s i.v. zoledronátem. Jeho dobrá účinnost byla popsána při použití dávky 0,05 mg/kg i.v. každých šest měsíců [65]; někteří autoři doporučují dávku zoledronátu 0,1 mg/kg i.v. v témže intervalu [66]. Objevují se i zprávy o jeho možném používání u dětí mladších dvou let v dávce 0,025 mg/kg každé tři měsíce. Aplikace zoledronátu má zřejmě s pamidronátem srovnatelnou účinnost i bezpečnostní profil [67]. Dlouhodobé vedlejší účinky zaznamenány nebyly [68], po podání zoledronátu dítěti ale také existuje popsaný případ těžké, život ohrožující reakce vyžadující intenzivní péči [69].
Pro indikaci léčby bisfosfonáty u mírných forem OI nemáme jednoznačné důkazy. Děti se závažnějším průběhem choroby s opakovanými zlomeninami, následnými deformitami dlouhých kostí a nízkou BMD však z této terapie nepochybně profitují. Frekvence fraktur se snižuje a pohybové schopnosti léčených bývají významně zlepšeny [70]. Dolní věková hranice pro zahájení terapie bisfosfonáty při OI stanovena není [71]. Někteří autoři zkoušejí po aktivní terapii přejít na snížené dávky, které po další dva roky udrží zlepšenou BMD a nízkou frekvenci fraktur [72], druhou možností je přestávka v léčbě, tzv. „drug holiday“. Při poklesu BMD, nových frakturách a bolestech v končetinách je samozřejmě možné se k terapii opět vrátit. Přerušovaná léčba umožní částečnou restituci kostního obratu u rostoucího organismu. Zastánci trvalé terapie však dokladují, že v oblasti metafýz v nově vzniklé kostní tkáni může po vysazení léčby riziko zlomeniny stoupat [73, 74].
Přestože u dětské OI existují teorie o srovnatelné účinnosti perorálních a intravenózních bisfosfonátů, studie s perorálním alendronátem přes příznivý účinek přípravku na BMD v oblasti bederní páteře průkaz o poklesu incidence zlomenin u nemocných dětí nepřinesla [75].
Dlouhý poločas bisfosfonátu v kostní tkáni znamená také nejistotu ve vztahu k možné budoucí graviditě léčených dívek [76]. Nicméně prozatím publikované zprávy o podávání bisfosfonátů ženám bezprostředně před anebo během gravidity ani jednou poškození plodu neuvádějí. Jednoznačný závěr však ještě učinit nelze [77, 78].
Podobně jako u dospělých, mezi nejčastější vedlejší účinky i.v. podaných bisfosfonátů patří i u dětí „flu-like“ reakce akutní fáze, zejména při prvé aplikaci. Podle některých prací též vlivem této léčby dochází k prodlouženému hojení operačních osteotomií, ovšem kvalita hojení dotčena není [79]. U dospělých se stále intenzivně studují vztahy mezi antiresorpční terapií, stomatologickými zákroky a výskytem osteonekrózy čelisti. U dětí tato komplikace zatím popsána nebyla. Je však vždy vhodné provést plánované invazivní stomatologické výkony ještě před zahájením léčby, případně ji přerušit, dokud není rána řádně zhojena. U dětí léčených dlouhodobě bisfosfonáty jsou rovněž popsány neobvyklé únavové zlomeniny v subtrochanterické oblasti nebo diafýze femuru, a to i přes přítomnost nitrodřeňových hřebů. Byla zaznamenána podobnost s atypickými frakturami femuru u dospělých [80]. Průkaz klasické atypické fraktury femuru ve vztahu k této léčbě však v současné době u dětí s OI neexistuje.
Recentní pohled na terapii bisfosfonáty při OI přinesla nedávno publikovaná práce, která vyhodnotila randomizované studie, porovnávající bisfosfonáty vůči placebu, žádné léčbě nebo komparátoru u všech typů OI. Bylo posouzeno 14 studií s celkem 819 účastníky. Ze závěru vyplývá, že léčba dětí i dospělých s OI intravenózními či perorálními bisfosfonáty vede ke zvýšení BMD, přičemž účinky jednotlivých bisfosfonátů se mezi sebou významně nelišily. Není zcela jasné, jestli se vzestupem BMD vždy souhlasně klesá riziko zlomenin, ale nikdy nebyl při léčbě zaznamenán jejich vzestup [81].
Ne vždy je odpověď na terapii bisfosfonáty uspokojivá. Je to známo například u vzácných forem OI na podkladě mutace v genu SERPINF1. V takových případech již byla v léčbě úspěšně použita monoklonální protilátka proti RANKL (ligand receptoru aktivátoru nukleárního faktoru kappa B) – denosumab [82].
Kromě antiresorpčních přípravků se v terapii osteoporotických změn u dospělých používá anabolická léčba parathormonem. Ta již byla také úspěšně podávána u dospělých nemocných s OI [83, 84]. V období rostoucího skeletu je však terapie parathormonem kontraindikována pro možnost onkologických komplikací. Určitou možností k farmakologickému posílení kostní formace by do budoucna mohla být léčba protilátkou proti sklerostinu, která u myšího modelu OI zaznamenala nadějné výsledky [85].
Velmi častým nálezem u OI je malá postava. Kombinovaná léčba bisfosfonáty a rekombinantním růstovým hormonem měla na růst postavy příznivý vliv, zejména při kvantitativním defektu syntézy kolagenu [86]. V současné době však zatím růstový hormon do rutinní praxe terapie OI u dětí nepatří.
Děti léčené pro OI by měly mít zajištěn řádný přívod minerálů a měly by být dostatečně saturovány vitaminem D.
Operační léčebné postupy
Spolupráce s ortopedem znalým problematiky je při komplexní péči o děti s OI nezbytná. Indikace operačního řešení zlomenin dlouhých kostí, jejich následných deformit nebo progredující patologie páteře patří zcela do jeho rukou. Některá pracoviště stále preferují při osteosyntéze dlouhých kostí standardní nitrodřeňové výztuže (typu Kirschnerových drátů) s poměrně dobrými výsledky. Nehledě na jejich jednoznačně nižší cenu jsou vhodné zejména při zvýšeném riziku poškození kloubního povrchu. Je však třeba je sledovat a u rostoucí kosti včas vyměňovat [87]. V poslední dekádě nachází u operatérů pečujících o děti s OI oblibu nitrodřeňové hřebování s použitím teleskopických výztuží (např. podle Fassier-Duvala), zejména v oblasti femuru. Oproti solidním hřebům je nutné mít na paměti zřídkavou možnost jejich funkčního selhání a včas ji podchytit. Prvým signálem bývá ohnutí hřebu, které může předznamenat následné uvolnění proximální a distální části. Při ohnutí hřebu je nutné nemocného sledovat a podle potřeby indikovat případnou výměnu [88]. Snížit výskyt možných komplikací – rozlomení, rozpojení či ohnutí – se podle některých autorů může dařit kombinací této metody s podpůrnou zevní fixací [89]. Nitrodřeňové teleskopické hřebování se začíná u dětských pacientů více používat také při léčbě deformit humeru [90] a dalších dlouhých kostí končetin. Pokud je u dítěte plánována osteotomie, není třeba předchozí podání bisfosfonátu omezovat, postačí alespoň týdenní odstup. Následující dávka se dříve po operaci oddalovala o řadu měsíců pro možné opožděné hojení. Současné kvalitnější operační postupy umožňují začít s následující léčbou bisfosfonáty dříve [91]. Postup je ale nutné zvážit u každého konkrétního případu zvlášť.
Další chirurgické zákroky mohou být potřebné u těžkých deformit hrudníku omezujících dýchání, které provázejí nejzávažnější přežívající formu – OI III. typu [92]. Při patologické fixaci středoušních kůstek je přiměřenou a bezpečnou metodou volby stapedotomie, která umožní zlepšení převodní nedoslýchavosti [93].
Transplantace kmenových buněk
Lidské fetální kmenové buňky se v kostní dřeni myších plodů s geneticky navozenou OI diferencovaly na zralé osteoblasty produkující osteokalcin a proteiny řetězců kolagenu, které vlastní organismus netvořil. Počty zlomenin byly významně redukovány a mechanická odolnost kosti stoupala na molekulární, tkáňové i orgánové úrovni [94, 95]. Kmenové buňky lidského choria, podané intraperitoneálně novorozeným myším s geneticky navozenou OI, snižují počty zlomenin a navozují vzestup počtu hypertrofických chondrocytů i lepší enchondrální a intramembranózní osifikaci. Tyto exogenní buňky se převážně usidlují v epifýzách dlouhých kostí, stimulují diferenciaci osteoblastů a tvorbu kolagenu. Placenta se tak zdá být dobrým zdrojem kmenových buněk k léčbě OI [96]. Podle některých recentních prací je však účinek transplantace kmenových buněk v léčbě OI pouze dočasný [97].
Přestože jsou nové možnosti léčebných postupů předmětem intenzivního výzkumu, na jejich ověření a uplatnění v klinické praxi bude třeba ještě čekat.
ZÁVĚR
Dítě s OI vždy vyžaduje komplexní péči řady odborníků, koordinovanou praktickým lékařem pro děti a dorost. Mírné formy OI nedělají zvláštní problémy. U těžších forem se objevují závažné deformity dlouhých kostí a hrudníku, které vedou ke komplikacím kardiovaskulárního a respiračního systému. Počty utrpěných fraktur jsou velmi individuální. Pravidlem však bývá jejich pokles po ukončení růstu.
Genetická heterogenita OI zmiňovaná již v r. 1979 Davidem Sillencem se molekulárně genetickými studiemi v posledních dvou dekádách potvrzuje. Počet objasněných genetických poruch vedoucích k manifestaci OI narůstá. Protože jedinci s toutéž mutací mohou mít zcela odlišnou míru závažnosti klinických projevů, nabývá na významu fenotyp nemocného. Jeho pečlivé zhodnocení umožní volit vhodné postupy (operační či neinvazivní) tak, aby se podařilo dosáhnout co nejlepší kvality života.
Poradenství a pomoc rodinám s dětmi postiženými OI poskytuje několik nadnárodních sdružení. Ve Spojených státech je to OIF (Osteogenesis Imperfecta Foundation; http://www.oif.org/), nověji se otázkami OI zabývá skupina odborníků sdružená v Brittle Bone Disorders Consortium (https://www.rarediseasesnetwork.org/cms//bbd). V Evropě pracuje OIFE (Osteoporosis Imperfecta Federation Europe; http://www.oife.org/en), sdružující nyní 24 států z celého světa. Česká republika bohužel zatím členem této federace není.
Prof. MUDr. Milan Bayer, CSc.
Klinika dětí a dorostu 3. LF UK
a FN Královské Vinohrady
Šrobárova 50
100 34 Praha 10
e-mail: milan.bayer@fnkv.cz; milan.bayer@ftn.cz
Sources
1. Imbert L, Aurégan JC, Pernelle K, Hoc T. Microstructure and compressive mechanical properties of cortical bone in children with osteogenesis imperfecta treated with bisphosphonates compared with healthy children. J Mech Behav Biomed Mater 2015; 46 : 261–270.
2. Marini JC, Reich A, Smith SM. Osteogenesis imperfecta due to mutations in non-collagenous genes: lessons in the biology of bone formation. Curr Opin Pediatr 2014 Aug; 26 (4): 500–507.
3. Li H, Jiang X, Delaney J, et al. Immature osteoblast lineage cells increase osteoclastogenesis in osteogenesis imperfecta murine. Am J Pathol 2010; 176 (5): 2405–2413.
4. Vanleene M, Porter A, Guillot PV, et al. Ultra-structural defects cause low bone matrix stiffness despite high mineralization in osteogenesis imperfecta mice. Bone 2012; 50 (6): 1317–1323.
5. Fratzl-Zelman N, Morello R, Lee B, et al. CRTAP deficiency leads to abnormally high bone matrix mineralization in a murine model and in children with osteogenesis imperfecta type VII. Bone 2010; 46 (3): 820–826.
6. Vranka JA, Pokidysheva E, Hayashi L, et al. Prolyl 3-hydroxylase 1 null mice display abnormalities in fibrillar collagen-rich tissues such as tendons, skin and bones. J Biol Chem 2010; 285 (22): 17253–17262.
7. Van Dijk FS, Nesbitt IM, Zwikstra EH. PPIB mutations cause severe osteogenesis imperfecta. Am J Hum Genet 2009; 85 (4): 521–527.
8. Pyott SM, Schwarze U, Christiansen HE, et al. Mutations in PPIB (cyclophilin B) delay type I procollagen chain association and result in perinatal lethal to moderate osteogenesis imperfecta phenotypes. Hum Mol Genet 2011; 20 (8): 1595–1609.
9. Bardai G, Moffatt P, Glorieux FH, Rauch F. DNA sequence analysis in 598 individuals with a clinical diagnosis of osteogenesis imperfecta: diagnostic yield and mutation spectrum. Osteoporos Int 2016; 27 (12): 3607–3613.
10. Parker MJ, Deshpande C, Rankin J, et al. Type 1 collagenopathy presenting with a Russell-Silver phenotype. Am J Med Genet A 2011; 155 (6): 1414–1418.
11. Barnes AM, Duncan G, Weis M, et al. Kuskokwim syndrome, a recessive congenital contracture disorder, Extends the phenotype of FKBP10 mutations. Hum Mutat 2013; 34 (9): 1279–1288.
12. Schwarze U, Cundy T, Pyott SM, et al. Mutations in FKBP10, which result in Bruck syndrome and recessive forms of osteogenesis imperfecta, inhibit the hydroxylation of telopeptide lysines in bone collagen. Hum Mol Genet 2013; 22 (1): 1–17.
13. Kamoun-Goldrat A, Pannier S, Huber C, et al. A new osteogenesis imperfecta with improvement over time maps to 11q. Am J Med Genet A 2008; 146A (14): 1807–1814.
14. Lindert U, Cabral WA, Ausavarat S, et al. MBTPS2 mutations cause defective regulated intramembrane proteolysis in X-linked osteogenesis imperfecta. Nat Commun 2016; 7 : 11920.
15. Hoyer-Kuhn H, Höbing L, Cassens J, et al. Children with severe osteogenesis imperfecta and short stature present on average with normal IGF-I and IGFBP-3 levels. J Pediatr Endocrinol Metab 2016; 29 (7): 813–818.
16. Bellary SS, Steinberg A, Mirzayan N, et al. Wormian bones: A review. Clin Anat 2013; 26 (8): 922–927.
17. Semler O, Cheung MS, Glorieux, FH, Rauch F. Wormian bones in osteogenesis imperfecta: Correlation to clinical findings and genotype. Am J Med Genet A 2010; 152A (7): 1681–1687.
18. Evereklioglu C, Madenci E, Bayazit YA, et al. Central corneal thickness is lower in osteogenesis imperfecta and negatively correlates with the presence of blue sclera. Ophthalmic Physiol Opt 2002; 22 (6): 511–515.
19. Kamoun-Goldrat A, Ginisty D, Le Merrer M. Effects of bisphosphonates on tooth eruption in children with osteogenesis imperfecta. Eur J Oral Sci 2008; 116 (3): 195–198.
20. Malmgren B, Andersson K, Lindahl K, et al. Tooth agenesis in osteogenesis imperfecta related to mutations in the collagen type I genes. Oral Dis 2017; 23 (1): 42–49.
21. Rauch F, Lalic L, Roughley P, Glorieux FH. Genotype-phenotype correlations in nonlethal osteogenesis imperfecta caused by mutations in the helical domain of collagen type I. Eur J Hum Genet 2010; 18 (6): 642–647.
22. Anissipour AK, Hammerberg KW, Caudill A, et al. Behavior of scoliosis during growth in children with osteogenesis imperfecta. J Bone Joint Surg Am 2014; 96 (3): 237–243.
23. Caudill A, Flanagan A, Hassani S, et al. Ankle strength and functional limitations in children and adolescents with type I osteogenesis imperfecta. Pediatr Phys Ther 2010; 22 (3): 288–295.
24. Malfait F, Symoens S, Goemans N, et al. Helical mutations in type I collagen that affect the processing of the amino-propeptide result in an Osteogenesis Imperfecta/Ehlers-Danlos syndrome overlap syndrome. Orphanet J Rare Dis 2013; 8 (1): 78.
25. Bonita RE, Cohen IS, Berko BA. Valvular heart disease in osteogenesis imperfecta: presentation of a case and review of the literature. Echocardiography 2010; 27 (1): 69–73.
26. Ashournia H, Johansen FT, Folkestad L, et al. Heart disease in patients with osteogenesis imperfecta - A systematic review. Int J Cardiol 2015; 196 : 149–157.
27. Radunovic Z, Steine K. Prevalence of cardiovascular disease and cardiac symptoms: left and right ventricular function in adults with osteogenesis imperfecta. Can J Cardiol 2015; 31 (11): 1386–1392.
28. Folkestad L, Hald JD, Gram J, et al. Cardiovascular disease in patients with osteogenesis imperfecta - a nationwide, register-based cohort study. Int J Cardiol 2016; 225 : 250–257.
29. Santos F, McCall AA, Chien W, Merchant S. Otopathology in osteogenesis imperfecta. Otol Neurotol 2012; 33 (9): 1562–1566.
30. Swinnen FK, Coucke PJ, De Paepe AM, et al. Osteogenesis imperfecta: the audiological phenotype lacks correlation with the genotype. Orphanet J Rare Dis 2011; 6 : 88.
31. Pillion JP, Shapiro I. Audiological findings in osteogenesis imperfecta. J Am Acad Audiol 2008; 19 (8): 595–601.
32. Arponen H, Mäkitie O, Haukka J, et al. Prevalence and natural course of craniocervical junction anomalies during growth in patients with osteogenesis imperfecta. J Bone Miner Res 2012; 27 (5): 1142–1149.
33. Arponen H, Vuorimies I, Haukka J, et al. Cranial base pathology in pediatric osteogenesis imperfecta patients treated with bisphosphonates. J Neurosurg Pediatr 2015; 15 (3): 313–320.
34. Cheung MS, Arponen H, Roughley P, et al. Cranial base abnormalities in osteogenesis imperfecta: Phenotypic and genotypic determinants. J Bone Miner Res 2011; 26 (2): 405–413.
35. Ibrahim AG, Crockard HA. Basilar impression and osteogenesis imperfecta: a 21-year retrospective review of outcomes in 20 patients. J Neurosurg Spine 2007; 7 (6): 594–600.
36. Silence DO, Senn A, Danks DM. Genetic heterogeneity in osteogenesis imperfecta. J Med Genet 1979; 16 (2): 101–116.
37. Trejo P, Rauch F. Osteogenesis imperfecta in children and adolescents – new developments in diagnosis and treatment. Osteoporos Int 2016; 27 : 3427–3437.
38. Bregou Bourgeois A, Aubry-Rozier B, Bonafé L, et al. Osteogenesis imperfecta: from diagnosis and multidisciplinary treatment to future perspectives. Swiss Med Wkly 2016; 146: w14322.
39. Glorieux FH, Rauch F, Plotkin H, et al. Type V osteogenesis imperfecta: a new form of brittle bone disease. J Bone Miner Res 2000; 15 (9): 1650–1658.
40. Balasubramanian M, Parker MJ, Dalton A, et al. Genotype-phenotype study in type V osteogenesis imperfecta. Clin Dysmorphol 2013; 22 (3): 93–101.
41. Arundel P, Offiah A, Bishop NJ. Evolution of the radiographic appearance of the metaphyses over the first year of life in type V osteogenesis imperfecta: clues to pathogenesis. J Bone Miner Res 2011; 26 (4): 894–898.
42. Diacinti D, Pisani D, D‘Avanzo M, et al. Reliability of vertebral fractures assessment (VFA) in children with osteogenesis imperfecta. Calcif Tissue Int 2015; 96 (4): 307–312.
43. Aström E, Magnusson P, Eksborg S, Söderhäll S. Biochemical bone markers in the assessment and pamidronate treatment of children and adolescents with osteogenesis imperfekta. Acta Paediatr 2010; 99 (12): 1834–1840.
44. Singh Kocher M, Dichtel L. Osteogenesis imperfecta misdiagnosed as child abuse. J Pediatr Orthop B 2011; 20 (6): 440–443.
45. Zarate YA, Clingenpeel R, Sellars EA, et al. COL1A1 and COL1A2 sequencing results in cohort of patients undergoing evaluation for potential child abuse. Am J Med Genet A 2016; 170 (7): 1858–1862.
46. Pyott SM, Tran TT, Leistritz DF, et al. WNT1 mutations in families affected by moderately severe and progressive recessive osteogenesis imperfecta. Am J Hum Genet 2013; 92 (4): 590–597.
47. Rauch F, Fahiminiya S, Majewski J, et al. Cole-Carpenter syndrome is caused by a heterozygous missense mutation in P4HB. Am J Hum Genet 2015; 96 (3): 425–431.
48. Ha-Vinh R, Alanay Y, Bank RA, et al. Phenotypic and molecular characterization of Bruck syndrome (osteogenesis imperfecta with contractures of the large joints) caused by a recessive mutation in PLOD2. Am J Med Genet 2004; 131 (2): 115–120.
49. Berg C, Geipel A, Noack F, et al. Prenatal diagnosis of Bruck syndrome. Prenat Diagn 2005; 25 (7): 535–538.
50. Mackenroth L, Fischer-Zirnsak B, Egerer J, et al. An overlapping phenotype of osteogenesis imperfecta and Ehlers-Danlos syndrome due to a heterozygous mutation in COL1A1 and biallelic missense variants in TNXB identified by whole exome sequencing. Am J Med Genet A 2016; 170A (4): 1080–1085.
51. Veilleux LN, Pouliot-Laforte A, Lemay M, et al. The functional muscle-bone unit in patients with osteogenesis imperfecta type I. Bone 2015; 79 : 52–57.
52. van Brussel M, Takken T, Uiterwall CS, et al. Physical training in children with osteogenesis imperfecta. J Pediatr 2008; 152 (1): 111–116.
53. Semler O, Fricke O, Vezyroglou K, et al. Results of a prospective pilot trial on mobility after whole body vibration in children and adolescents with osteogenesis imperfecta. Clin Rehabil 2008; 22 (5): 387–394.
54. Vanleene M, Shefelbine SJ. Therapeutic impact of low amplitude high frequency whole body vibrations on the osteogenesis imperfecta mouse bone. Bone 2013; 53 (2): 507–514.
55. Hasegawa K, Inoue M, Seino Y, et al. Growth of infants with osteogenesis imperfecta treated with bisphosphonate. Pediatr Int 2009; 51 (1): 54–58.
56. Sato A, Ouellet J, Muneta T, et al. Scoliosis in osteogenesis imperfecta caused by COL1A1/COL1A2 mutations – genotype-phenotype correlations and effect of bisphosphonate treatment. Bone 2016; 86 : 53–57.
57. Munns CF, Rauch F, Travers R, Glorieux FH. Effects of intravenous pamidronate treatment in infants with osteogenesis imperfecta: clinical and histomorphometric outcome. J Bone Miner Res 2005; 20 (7): 1235–1243.
58. Rauch F, Travers R, Glorieux FH. Pamidronate in children with osteogenesis imperfecta: histomorphometric effects of long-term therapy. J Clin Endocrinol Metab 2006; 91 (2): 511–516.
59. Lindahl K, Kindmark A, Rubin CJ, et al. Decreased fracture rate, pharmacogenetics and BMD response in 79 Swedish children with osteogenesis imperfecta types I, III and IV treated with Pamidronate. Bone 2016; 87 : 11–18.
60. Palomo T, Andrade MC, Peters BS, et al. Evaluation of a modified pamidronate protocol for the treatment of osteogenesis imperfecta. Calcif Tissue Int 2016; 98 (1): 42–48.
61. Cheung MS, Glorieux FH, Rauch F. Large osteoclasts in pediatric osteogenesis imperfecta patients receiving intravenous pamidronate. J Bone Miner Res 2009; 24 (4): 669–674.
62. Gatti D, Antoniazzi F, Prizzi R, et al. Intravenous neridronate in children with osteogenesis imperfecta: a randomized controlled study. J Bone Miner Res 2005; 20 (5): 758–763.
63. Li M, Xia W, Xing X, et al. Benefit of infusions with ibandronate treatment in children with osteogenesis imperfecta. Chin Med J 2011; 124 (19): 3049–3053.
64. Bishop N,Harrison R, Ahmed F, et al. A randomised controlled dose-ranging study of risedronate in children with moderate and severe osteogenesis imperfecta. J Bone Miner Res 2010; 25 (1): 32–40.
65. Vuorimies I, Toiviainen-Salo S, Hero M, Mäkitie O. Zoledronic acid treatment in children with osteogenesis imperfecta. Horm Res Paediatr 2011; 75 (5): 346–353.
66. Otaify GA, Aglan MS, Ibrahim MM, et al. Zoledronic acid in children with osteogenesis imperfecta and Bruck syndrome: a 2-year prospective observational study. Osteoporos Int 2016; 27 (1): 81–92.
67. Brown JJ, Zacharin MR. Safety and efficacy of intravenous zoledronic acid in paediatric osteoporosis. J Pediatr Endocrinol Metab 2009; 22 (1): 55–63.
68. Kumar C, Panigrahi I, Somasekhara Aradhya A, et al. Zoledronate for osteogenesis imperfecta: evaluation of safety profile in children. J Pediatr Endocrinol Metab 2016; 29 (8): 947–952.
69. Trivedi S, Al-Nofal A, Kumar S, et al. Severe non-infective systemic inflammatory response syndrome, shock, and end-organ dysfunction after zoledronic acid administration in a child. Osteoporos Int 2016; 27 (7): 2379–2382.
70. Sousa T, Bompadre V, White KK. Musculoskeletal functional outcomes in children with osteogenesis imperfecta: associations with disease severity and pamidronate therapy. J Pediatr Orthop 2014; 34 (1): 118–122.
71. Antoniazzi F, Zamboni G, Lauriola S, et al. Early bisphosphonate treatment in infants with severe osteogenesis imperfecta. J Pediatr 2006; 149 (2): 174–179.
72. Biggin A, Zheng L, Briody JN, et al. The long-term effects of switching from active intravenous bisphosphonate treatment to low-dose maintenance therapy in children with osteogenesis imperfecta. Horm Res Paediatr 2015; 83 (3): 183–189.
73. Rauch F, Munns C, Land C, Glorieux FH. Pamidronate in children and adolescents with osteogenesis imperfecta: effect of treatment discontinuation. J Clin Endocrinol Metab 2006; 91 (4): 1268–1274.
74. Rauch F, Cornibert S, Cheung M, Glorieux FH. Long-bone changes after pamidronate discontinuation in children and adolescents with osteogenesis imperfecta. Bone 2007; 40 (4): 821–827.
75. Ward LM, Rauch F, Whyte MP, et al. Alendronate for the treatment of pediatric osteogenesis imperfecta: a randomized placebo-controlled study. J Clin Endocrinol Metab 2011; 96 (2): 355–364.
76. Chan B, Zacharin M. Maternal and infant outcome following pamidronate treatment of polyostotic fibrous dysplasia and osteogenesis imperfecta before conception: a report of four cases. J Clin Endocrinol Metab 2006; 91 (6): 2017–2020.
77. Djokanovic N, Kliger-Grossmann C, Koren G. Does treatment with bisphosphonates endanger the human pregnancy? J Obstet Gynaecol Can 2008; 30 (12): 1146–1148.
78. Green SB, Pappas AL. Effects of maternal bisphosphonate use on fetal and neonatal outcomes. Am J Health Syst Pharm 2014; 71 (23): 2029–2036.
79. Munns CF, Rauch F, Zeitlin L, et al. Delayed osteotomy but not fracture healing in pediatric osteogenesis imperfecta patients receiving pamidronate. J Bone Miner Res 2004; 19 (11): 1779–1786.
80. Hegazy A, Kenawey M, Sochett E, et al. Unusual femur stress fractures in children with osteogenesis imperfecta and intramedullary rods on long-term intravenous pamidronate therapy. J Pediatr Orthop 2016; 36 (7): 757–761.
81. Dwan K, Phillipi CA, Steiner RD, Basel D. Bisphosphonate therapy for osteogenesis imperfecta. Cochrane Database Syst Rev 2016; 10: CD005088.
82. Hoyer-Kuhn H, Semler O, Schoenau E. Effect of denosumab on the growing skeleton in osteogenesis imperfecta. J Clin Endocrinol Metab 2014; 99 (11): 3954–3955.
83. Gatti D, Rossini M, Viapiana O, et al. Teriparatide treatment in adult patients with osteogenesis imperfecta type I. Calcif Tissue Int 2013; 93 (5): 448–452.
84. Orwoll ES, Shapiro J, Veith S, et al. Evaluation of teriparatide treatment in adults with osteogenesis imperfecta. J Clin Invest 2014; 124 (2): 491–498.
85. Sinder BP, White LE, Salemi JD, et al. Adult Brtl/+ mouse model of osteogenesis imperfecta demonstrates anabolic response to sclerostin antibody treatment with increased bone mass and strength. Osteoporos Int 2014; 25 (8): 2097–2107.
86. Antoniazzi F, Monti E, Venturi G, et al. Growth hormone in combination with bisphosphonate treatment in osteogenesis imperfecta. Eur J Endocrinol 2010;163 (3): 479–487.
87. Imajima Y, Kitano M, Ueda T. Intramedullary fixation using Kirschner wires in children with osteogenesis imperfecta. J Pediatr Orthop 2015; 35 (4): 431–434.
88. Lee RJ, Paloski MD, Sponseller PD, Leet AI. Bent telescopic rods in patients with osteogenesis imperfecta. J Pediatr Orthop 2016; 36 (6): 656–660.
89. Franzone JM, Kruse RW. Intramedullary nailing with supplemental plate and screw fixation of long bones of patients with osteogenesis imperfecta: operative technique and preliminary results. J Pediatr Orthop B 2016 Nov 9. [Epub ahead of print].
90. Grossman LS, Price AL, Rush ET, et al. Initial experience with percutaneous IM rodding of the humeri in children with osteogenesis imperfecta. J Pediatr Orthop 2016 Sep 22. [Epub ahead of print].
91. Anam EA, Rauch F, Glorieux FH, et al. Osteotomy healing in children with osteogenesis imperfecta receiving bisphosphonate treatment. J Bone Miner Res 2015; 30 (8): 1362–1368.
92. Kaplan L, Barzilay Y, Hashroni A, et al. Thoracic elongation in type III osteogenesis imperfecta patients with thoracic insufficiency syndrome. Spine (Phila Pa 1976) 2013; 38 (2): E94–100.
93. Vincent R, Wegner I, Stegeman I, Grolman W. Stapedotomy in osteogenesis imperfecta: a prospective study of 32 consecutive cases. Otol Neurotol 2014; 35 (10): 1785–1789.
94. Guillot PV, Abass O, Bassett JH, et al. Intrauterine transplantation of human fetal mesenchymal stem cells from first trimester blood repairs bone and reduces fractures in osteogenesis imperfecta mice. Blood 2008; 111 (3): 1717–1725.
95. Vanleene M, Saldanha Z, Cloyd KL, et al. Transplantation of human fetal blood stem cells in the osteogenesis imperfecta mouse leads to improvement in multi-scale tissue properties. Blood 2011; 117 (3): 1053–1060.
96. Jones GN, Moschidou D, Abdulrazzak H, et al. Potential of human fetal chorionic stem cells for the treatment of osteogenesis imperfecta. Cells Dev 2014; 23 (3): 262–276.
97. Westgren M, Götherström C. Stem cell transplantation before birth – a realistic option for treatment of osteogenesis imperfecta? Prenat Diagn 2015; 35 (9): 827–832.
Labels
Neonatology Paediatrics General practitioner for children and adolescentsArticle was published in
Czech-Slovak Pediatrics

2017 Issue 4
- What Effect Can Be Expected from Limosilactobacillus reuteri in Mucositis and Peri-Implantitis?
- The Importance of Limosilactobacillus reuteri in Administration to Diabetics with Gingivitis
Most read in this issue
- Osteogenesis imperfecta – současný pohled na problematiku
-
Infantilní hemangiomy.
Současné léčebné postupy - Hypofosfatázie – onemocnění skeletu, na které musíme myslet
- Tolerogenní dendritické buňky a jejich využití v léčbě imunopatologických stavů