
Talasemické syndromy
Thalassemias
Thalassemias represent heterogenic group of inhereted red blood cells disorders. It is caused by imbalance of globin chains in hemoglobin molecule, which leads to ineffective erythropoiesis in a bone marrow and shortening of erythrocyte´s life span. Clinical and laboratory presentation is heterogenic and depends on number of affected genes. Thalassemia carriers are asymptomatic with microcytic erythrocytes and hemoglobin level can be decreased in some cases. Severe forms of thalassemia are associated with severe microcytic hypochromic anemia, an increased hemolysis and with a related complications. Diagnosis of thalassemia consists of analysis of hemoglobin spectrum and detection of the causal mutations by molecular genetic methods.. Carriers of thlassemia allele require no therapy, patients with severe form need a regular transfusion regime. As a curative procedures, hematopoetic stem cell transplantation and gene therapy can be used. Prenatal testing is also available.
Keywords:
α-thalassemia, β-thalassemia, diagnostics, molecular genetic methods
Authors:
L. Sulovská 1; M. Divoká 2; D. Pospíšilová 1
Authors‘ workplace:
Dětská klinika, Fakultní nemocnice a Lékařská fakulta Univerzity Palackého, Olomouc
1; Hemato-onkologická klinika, Fakultní nemocnice a Lékařská fakulta Univerzity Palackého, Olomouc
2
Published in:
Čes-slov Pediat 2017; 72 (8): 457-463.
Věnováno panu profesorovi Hrodkovi, zakladateli moderní dětské hematologie v České republice
Overview
Talasémie tvoří heterogenní skupinu vrozených poruch červené krevní řady. Příčinou je nerovnováha globinových řetězců v molekule hemoglobinu, která vede k neefektivní erytropoéze v kostní dřeni a ke zkrácení životního cyklu erytrocytů. Klinická a laboratorní manifestace je značně rozdílná a závisí na počtu postižených globinových genů. Jedinci s nosičstvím talasemické alely jsou asymptomatičtí a nacházíme u nich jen mikrocytózu erytrocytů a u některých snížení hladiny hemoglobinu. Těžší formy talasémií jsou provázeny závažnou mikrocytární anémií s vystupňovanou hemolýzou a s tím souvisejícími komplikacemi. Diagnostika talasémií spočívá v analýze hemoglobinového spektra a v detekci kauzální mutace molekulárně genetickými metodami. Léčba nositelů talasemické alely většinou není nutná, u těžších forem talasémie spočívá v pravidelné substituci erytrocytárními koncentráty a léčbě přidružených komplikací. Kauzální terapií je transplantace kmenových buněk krvetvorby a genová terapie. U vybraných párů je dostupná prenatální diagnostika.
Klíčová slova:
α-talasémie, β-talasémie, diagnostika, molekulárně genetické metody
ÚVOD
Talasémie tvoří heterogenní skupinu geneticky podmíněných onemocnění červené krevní řady vedoucích k mikrocytární anémii. Společným rysem pro všechny talasemické syndromy je defektní či chybějící syntéza jednoho nebo více globinových řetězců v molekule hemoglobinu. Jedná se o kvantitativní poruchu syntézy globinu, při které nedochází ke změně pořadí aminokyselin v řetězci.
U zdravých lidí je molekula hemoglobinu (Hb) tvořená čtyřmi podjednotkami, z nichž každá je složená z hemové skupiny (stejné pro všechny typy hemoglobinů), a z globinového řetězce, který zastoupením jednotlivých aminokyselin určuje typ hemoglobinu. Existuje celkem šest různých globinových řetězců: alfa (α), beta (β), gamma (γ), delta (δ), epsilon (ε), zeta (ζ). Více než 95 % Hb dospělého člověka tvoří hemoglobin A (HbA), který je složený ze dvou α a dvou β řetězců (αα/ββ). Minoritně jsou pak u dospělého člověka zastoupeny HbA2 (αα/δδ) a fetální HbF (αα/γγ). V průběhu ontogeneze se vyskytují embryonální Hb Gower–1 (ζζ/εε), Hb Gower–2 (αα/εε) a Hb Portland (ζζ/γγ), které jsou postupně ve druhém trimestru nahrazovány fetálním HbF. Vyšší afinita embryonálních Hb a fetálního Hb ke kyslíku zaručuje dostatečnou oxygenaci tkání plodu.
Syntéza jednotlivých globinových řetězců je přísně regulovaná. Geny pro šest různých globinových řetězců jsou lokalizované na chromozomu 11 v HBB lokusu („β-like“ řetězce: ε, γ, δ a β) a na chromozomu 16 v HBA lokusu („α-like“ řetězce: ζ a α) a jsou seřazeny v pořadí, v jakém jsou během ontogeneze exprimované. Pokud není syntéza globinů narušená, pak poměr α - a β-řetězců v molekule hemoglobinu je roven 1,00 ± 0,05. V případě pacientů s talasémií je tento poměr narušený sníženou (α+ - nebo β+-talasémie) nebo úplnou absencí jednoho z řetězců (α0 - nebo β0-talasémie), což vede k akumulaci nadbytečných nepostižených globinových řetězců. Tyto řetězce nemohou být spárované s odpovídajícím „partnerem“, dochází k jejich spontánní aglutinaci a tvorbě inkluzí, které narušují fyziologii hematopoézy [1]. Typ postiženého globinového řetězce určuje i typ talasémie – nejčastěji α-talasémie a β-talasémie. Vzácně se vyskytují jiné typy např. δβ-talasémie, které nejčastěji vznikají v důsledku delece příslušných úseků DNA s následnou fúzí a vznikem hybridních δ/β genů (např. hemoglobinová varianta Hb Lepore s talasemickým fenotypem).
EPIDEMIOLOGIE
Talasémie postihuje přibližně 7 % celosvětové populace, ale její nejvyšší výskyt je koncentrovaný do oblasti Středomoří, zemí Blízkého východu, Indie, jihovýchodní Asie a severní Afriky (obr. 1) [2]. Mluvíme o takzvaném talasemickém pásu. Mezi evropské oblasti s nejvyšší prevalencí β-talasémie patří Řecko, Kypr a Sardinie (6–19 %). Vysoká koncentrace mutovaných alel v oblasti talasemického pásu vede proto k častějšímu výskytu dvojitých heterozygotních nosičů talasemické alely nebo ke kombinaci talasemického nosičství s jiným typem hemoglobinopatie u jednoho pacienta. V důsledku globalizace a migrace dochází také v posledních letech k rozšíření talasemických alel i do oblastí dříve postižených minimálně, mezi které můžeme zařadit i Českou republiku [3, 4]. V zemích s vysokým výskytem talasémie je zároveň vysoká prevalence malárie a uvádí se, že přítomnost talasemické alely jedincům poskytuje určitou selektivní výhodu [5]. Například genotyp αα/α - je spojený s o 40 % nižším rizikem úmrtí na malárii při srovnání se zdravou populací, u genotypu α-/α - je toto riziko nižší až o 60 % [6]. Přesný mechanismus této rezistence k onemocnění malárií dosud není známý, pravděpodobně se podílí snížení replikační schopnosti plazmodia v postižených erytrocytech a zvýšená destrukce infikovaných erytrocytů [7, 8].
![Světová distribuce talasémie. Nejvíce postižené oblasti tvoří tzv. talasemický pás, který zahrnuje oblasti Středomoří, zemí Blízkého východu, Indie, jihovýchodní Asie a severní Afriky. Převzato a upraveno [2].
Fig. 1. World distribution of thalassemia. The most affected areas form the so-called thalassemic belt, which includes the Mediterranean, the Middle East, India, Southeast Asia and North Africa. Adapted by [2].](https://www.prelekara.sk/media/cache/resolve/media_object_image_large/media/image/3bf12aad05cb0f4d5cc5ef925b71e722.jpg)
α-TALASÉMIE
V lidském haploidním genomu se nachází dva α-globinové geny (HBA2, HBA1), normální diploidní genotyp je tedy αα/αα. Tato sada čtyř funkčních genů může být v důsledku mutace snížena o 1, 2, 3 nebo 4 kopie genu (obr. 2). Nejčastěji se jedná o deleci funkčního genu, méně často poruchu syntézy globinového řetězce způsobuje bodová mutace.
![Genotypové varianty delečních α-talasémií. Nahoře vlevo: normální diploidní genotyp. Nahoře vpravo: heterozygot pro α⁺-talasémii, tzv. němé (nebo tiché) nosičství. Uprostřed: u homozygotů pro α⁺-talasémii (nejčastěji delece dvou genů, –α/–α) nebo u heterozygotů pro α⁰-talasémii (– –/αα) hovoříme o nosičství α-talasémie. Dole vlevo: ztráta tří α-globinových genů (dvojití heterozygoti pro α⁺-talasémii a α⁰-talasémii, nejčastěji – – /–α), vede k chorobě HbH se středně těžkou anémií a s produkcí HbH (β4). Při inkubaci s briliant-kresylovou modří nacházíme v erytrocytech precipitovaný HbH. Dole vpravo: chybění čtyř α-globinových genů, tj. homozygotní stav pro α⁰-talasémii (– –/– –), není slučitelné s životem a vede ke vzniku fetálního hydropsu nebo syndromu Hb Bart’s (γ4). Upraveno podle [9].](https://www.prelekara.sk/media/cache/resolve/media_object_image_large/media/image/fe7db077f6dc38f939e6a4ebced658de.jpg)
α-talasémie minima nebo také tiché nosičství α-talasémie je způsobena delecí jednoho α-globinového genu. Většinou se neprojeví ani při klinickém vyšetření, ani při vyšetření krevního obrazu. Elektroforéza Hb je normální. Jedinou možností identifikace těchto jedinců je analýza DNA. Předpokládá se, že výskyt v České republice je častý [9].
α-talasémie minor (nosičství α-talasémie) je způsobeno delecí dvou genů, která může postihnout jeden chromozom (αα/--, cis forma), nebo jeden gen na každém chromozomu (α-/α, trans forma). Nosičství α-talasémie se může manifestovat mírnou anémií, mikrocytózou a hypochromázií erytrocytů (tab. 1). Na rozdíl od pacien-tů s β-talasémií minor nedetekujeme při vyšetření hemoglobinového spektra zvýšenou hladinu HbA2 [10].
Ztráta tří funkčních genů pro α-globinový řetězec způsobuje nadbytek β-globinových řetězců, které spontánně agregují do tetramerů (β4). Výsledkem je HbH, který je nestabilní a má vysokou afinitu ke kyslíku. Pacienti s HbH bývají symptomatičtí již při narození, protože α-globin je součástí i HbF. Po narození proto bývá přítomná anémie a ikterus. Intrauterinně volné γ řetězce agregují do tetramerů (Hb Bart‘s), při elektroforéze Hb v prvních týdnech života je jeho frakce detekovatelná v koncentraci 20–40 %. Pacienti jsou postiženi mírnou až středně těžkou anémií (70–100 g/l), v dospělosti se manifestují i důsledky chronické hemolýzy – hepatosplenomegalie, ikterus, cholecystolitiáza. Dysregulace metabolismu železa (Fe) u těchto pacientů vede v pozdějším věku k hemosideróze jater a myokardu i přesto, že většina pacientů nevyžaduje transfuze erytrocytárních koncentrátů [10, 11, 12].
α-talasémie major je důsledkem delece všech čtyř genů pro α-globinový řetězec. Je neslučitelná s extrauterinním životem. Již intrauterinně totiž dochází k tvorbě Hb Bart‘s, který má desetinásobně vyšší afinitu ke kyslíku. Chronická hypoxie způsobuje závažné poruchy vývoje plodu a v důsledku srdečního selhávání vede k prosáknutí všech tkání (fetální hydrops) [9]. Většina plodů umírá intrauterinně. V literatuře bylo popsáno několik případů živě narozených dětí, které však umírají během prvních hodin života [13]. Současně dochází k ohrožení života matky toxémií, polyhydramnionem, akutním hemoragickým šokem aj. [10, 14].
β-TALASÉMIE
Gen pro β-globinový řetězec (HBB) je lokalizovaný na chromozomu 11 v lokusu HBB pouze v jedné kopii (na rozdíl od genů pro α-globin), normální diploidní genotyp je tedy označen β/β. Na rozdíl od α-talasémií jsou β-globinové geny nejčastěji postiženy bodovými mutacemi, malými inzercemi, méně často pak rozsáhlými delecemi. Historicky byly β-talasémie děleny podle klinického průběhu na formu minor, intermedia a major. Zavedení DNA sekvenování identifikuje i pacienty s tzv. tichým nosičstvím β-talasémie. Tito pacienti mají v jednom genu pro β-globinový řetězec mutaci, která vede jen k velmi diskrétnímu snížení produkce β-řetězců, což se při vyšetření krevního obrazu neprojeví [10, 15].
Heterozygotní mutace postihující gen pro β-globinový řetězec vede k β-talasémii minor. Tito pacienti mají obvykle výraznou mikrocytózu, hypochromii erytrocytů, kompenzatorní erytrocytózu a mohou mít mírnou anémii. V nátěru periferní krve se vyskytují různé formy patologických tvarů erytrocytů (poikilocyty, eliptocyty, terčovité erytrocyty) a může být přítomné bazofilní tečkování. Většina nosičů β-talasemické alely má zvýšenou hladinu HbA2 (tab. 1). U části pacientů je přítomná hepatomegalie a splenomegalie. Slezina je hmatná u méně než 20 % jedinců, při ultrazvukovém vyšetření mají tito pacienti slezinu větší o 29–67 % než zdravé kontroly [16, 17]. Naprostá většina pacientů během svého života nevyžaduje podání transfuze.
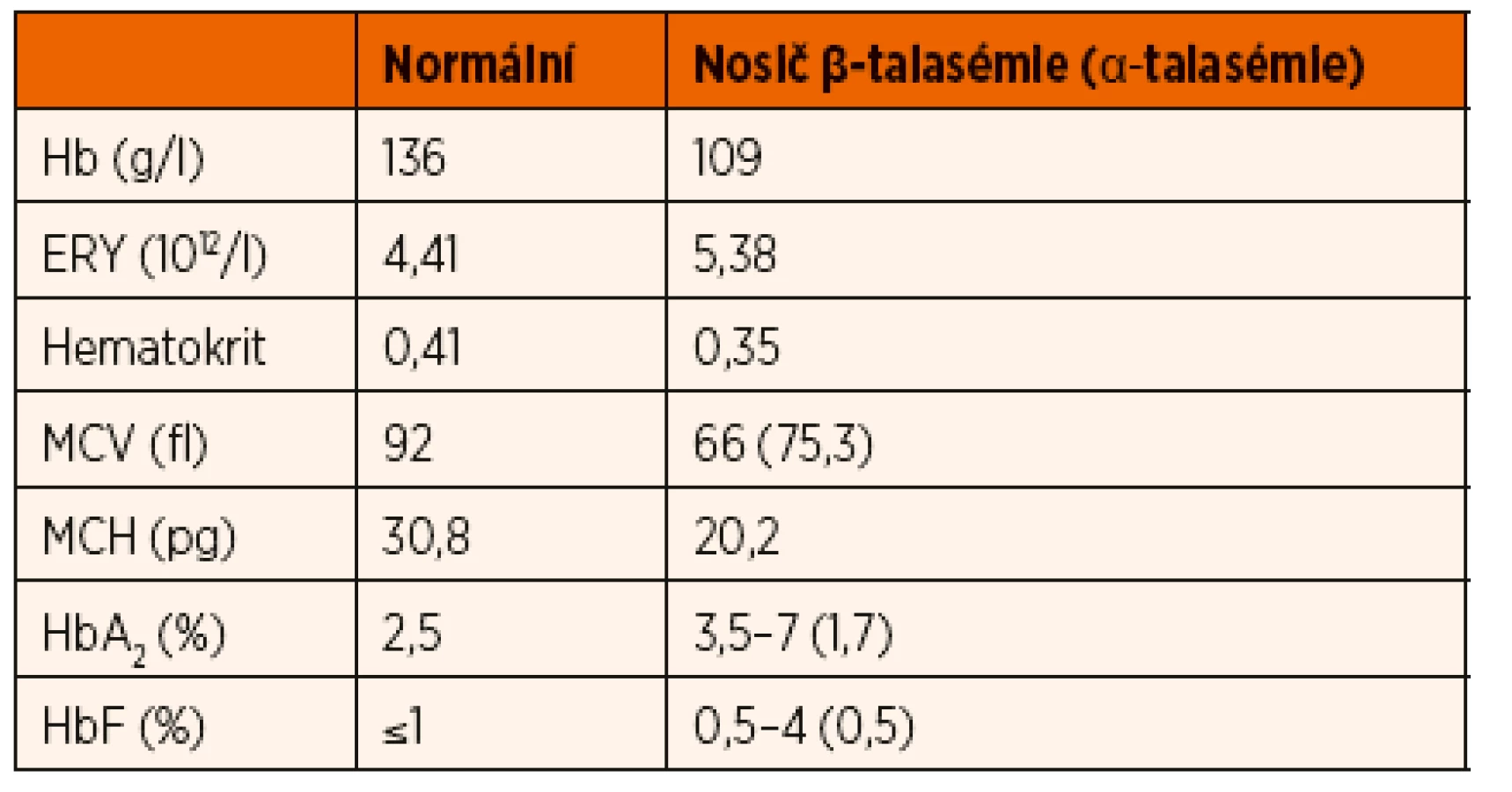
β-talasémie intermedia je definovaná závažnější anémií, než jakou nacházíme u pacientů s talasémií minor. K udržení stabilní hladiny hemoglobinu a kvality života pacienti nevyžadují pravidelnou substituci erytrocytárními koncentráty, na rozdíl od pacientů postižených talasémií major. U většiny pacientů s β-talasémií intermedia se hladina hemoglobinu pohybuje nad 70 g/l. Genotyp pacientů je značně heterogenní, od homozygotní formy mutací vedoucích k mírnému snížení syntézy řetězců až po dvojité heterozygotní mutace s různým dopadem na syntézu globinu [18, 19].
Klinická manifestace β-talasémie intermedia je velmi rozdílná – různě závažná anémie s charakteristickými talasemickými rysy (mikrocytóza, hypochromie, erytrocytóza, patologické morfologické odchylky erytrocytů v nátěru periferní krve). Pacienti s tímto typem talasémie i přes absenci chronické erytrocytární substituce trpí komplikacemi z přetížení Fe. Akumulace Fe je pravděpodobně způsobena alterací regulace příjmu Fe ze stravy. Klíčový význam má v tomto případě neefektivní erytropoéza, která přes dosud neobjasněnou signální dráhu snižuje hladinu hepcidinu s následným zvýšením příjmu Fe ze stravy [20, 21]. Přetížení Fe se u těchto pacientů objevuje o 10–20 let později než u pacientů s pravidelným podáváním transfuzí. V následujících letech lze očekávat, že intenzivní výzkum v oblasti terapeutických zásahů do regulace metabolismu Fe povede ke zlepšení prognózy pacientů.
U nosičů β-talasemické alely se můžeme setkat s klinickým obrazem talasémie intermedia také v případě, kdy současné postižení α-globinových genů (triplikace) prohloubí vzájemný nepoměr α - a β-globinových řetězců [22].
β-talasémie major je nejzávažnějším talasemickým syndromem, který je slučitelný s postnatálním životem. Absence nebo výrazné snížení produkce β-globinových řetězců je důsledkem přítomnosti homozygotní nebo dvojitě heterozygotní formy mutace HBB genu. Volné, nespárované α-řetězce jsou nestabilní, spontánně agregují do tetramerů a vytváří nerozpustná inkluzní tělíska, která poškozují erytroidní progenitory v kostní dřeni. Zralé erytrocyty uvolněné do cirkulace mají zkrácený životní cyklus a podléhají předčasnému rozpadu v monocyto-makrofágovém systému sleziny a jater [23]. Intrauterinně a během prvních měsíců života jsou homozygotní jedinci asymptomatičtí, protože dominantním hemoglobinem je HbF a jeho syntéza není ovlivněna. Při následném postnatálním fyziologickém snižování syntézy HbF a neadekvátním zvyšování HbA se objevuje závažná mikrocytární, hypochromní anémie. Naprostá většina pacientů je diagnostikovaná od 6. měsíce do 2 let věku.
Klinický obraz a laboratorní nálezy u pacientů s β-talasémií major jsou kombinací čtyř patologických jevů – anémie, chronické hemolýzy, expanze erytropoézy a zvýšeného obratu Fe. Mikrocytární anémie je závažná, při diagnóze se u pacientů hladiny Hb pohybují nezřídka mezi 20–30 g/l, v nátěru periferní krve pozorujeme kromě mikrocytů (střední objem erytrocytů pod 65 fl) i výraznou anizocytózu a četné morfologické odchylky tvaru erytrocytů – terčovité erytrocyty, eliptocyty, jaderné erytrocyty (obr. 3). Bývá také přítomno bazofilní tečkování. Osmotická rezistence erytrocytů je zvýšená [24]. Chronická hypoxie způsobuje extramedulární expanzi erytropoézy – v játrech, slezině, ledvinách a také tumoriformních masách v mediastinu nebo retroperitoneu [25]. Kostní abnormality jsou dalším důsledkem expanze erytropoézy. Nejznatelnější jsou v obličeji – prominence a zvětšení maxily, relativně malý nos a vystupující horní řezáky (facies talasemica). V plochých kostech lebky se rozšiřuje dřeňová dutina a ztenčuje se kompakta kosti. Při vyšetření kostní dřeně talasemických jedinců nacházíme typickou erytroidní hyperplazii (poměr myeloidní a erytroidní řady může dosahovat poměru 1 : 20, zatímco u zdravých lidí se tento poměr pohybuje 1,5–3 : 1) [11]. Chronicky probíhající hemolýza se projevuje ikterickým zbarvením kůže a sklér, splenomegalií s hepatomegalií a tvorbou žlučových kamenů.
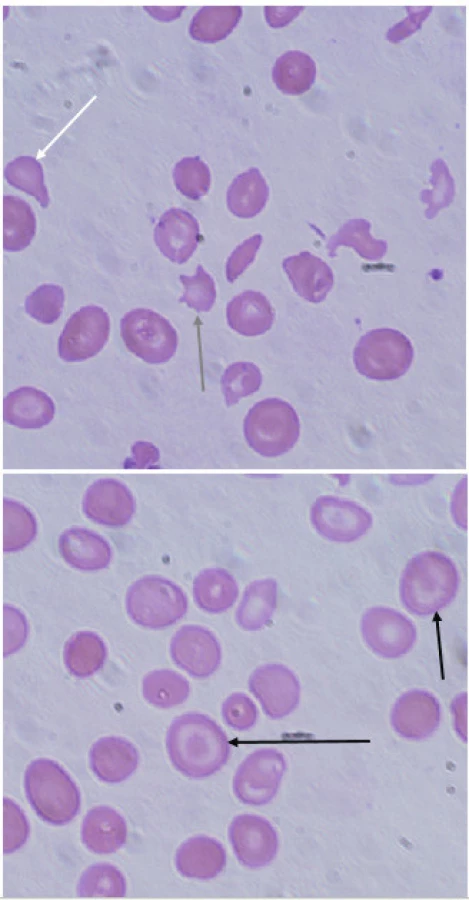
Zvýšený obrat Fe při expanzi erytropoézy a exogenní přívod Fe transfuzemi mají za následek poruchu funkce životně důležitých orgánů. Sekundární hemosideróza postihuje zejména játra, slezinu, myokard a endokrinní žlázy. U části pacientů zejména v rozvojových zemích je hepatopatie dále zhoršovaná koincidencí talasémie s infekční virovou hepatitidou B nebo C [10, 11]. Kardiální postižení se projevuje poruchami srdečního rytmu, ischemickou chorobou srdeční, dilatací levé komory a srdečním selháváním, které je nejčastější příčinou úmrtí pacientů [26]. Endokrinopatie zahrnují hypogonadismus (40–55 %), růstovou retardaci (33 %), diabetes mellitus (6–13 %), hypothyreózu a hypoparathyreózu (10–11 %). Časně zahájená chelatační léčba zamezí progresi postižení, není však dosud jasné, jestli již vzniklé změny jsou reverzibilní [27, 28]. Kvalitu života pacientů s talasémií major výrazně ovlivňuje bolest. Multicentrická prospektivní studie s účastí 258 subjektů dokumentuje negativní vliv bolesti na kvalitu života jednotlivce. 81 % pacientů pociťovalo v posledním roce bolesti. Nejčastěji se jednalo o bolesti hlavy, střední a dolní části zad a bolesti nohou. Při regresní analýze výskyt bolesti koreloval s věkem, ne však s tíží anémie, typem talasémie, počtem transfuzí, chelatační léčbou ani kostní denzitou [29].
DIAGNOSTIKA TALASÉMIÍ V ČESKÉ REPUBLICE
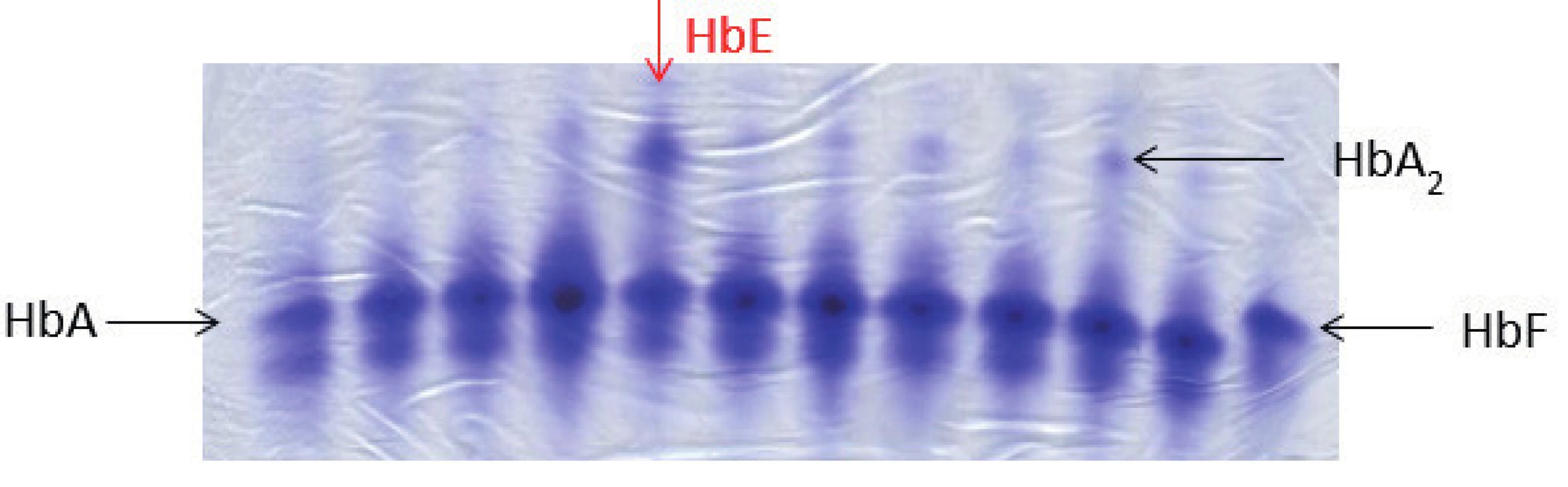
Typické nálezy v krevním obraze a nátěru periferní krve, jak byly popsány výše, jsou diagnosticky cenné, je však třeba je doplnit o další speciální hematologické a molekulárně genetické metody k potvrzení diagnózy. Mezi nejrozšířenější metody patří elektroforéza Hb, pomocí které lze identifikovat pacienty s abnormálním hemoglobinovým spektrem (obr. 4). Další používanou metodou je chromatografické stanovení hladiny HbA2, která může být až dvojnásobně zvýšená u pacientů s β-talasémií minor, někteří z těchto jedinců mají zároveň zvýšenou frakci HbF, kterou lze poměrně přesně kvantifikovat pomocí metody alkalické denaturace. U homozygotů pro β0-talasémii detekujeme pouze HbF a případně normální nebo jen mírně zvýšený HbA2. Na molekulární úrovni lze prokázat bodové mutace v rámci HBB genu pomocí Sangerova sekvenování. Deleční formy β-talasémií analyzujeme pomocí metody MLPA (Multiplex Ligation Probe Amplification). Dosud již bylo identifikováno přes 300 delečních a bodových mutací vedoucích k β-talasémii.
U pacientů s inaktivací tří α-globinových řetězců lze prokázat precipitovaný HbH v erytrocytech inkubací s briliant-kresylovou modří, po obarvení můžeme pozorovat v erytrocytech inkluze tvořené shlukem volných β-globinových řetězců. Kauzální mutace způsobující α-talasémie se detekují pomocí molekulárně genetických metod. Většina mutací má charakter delecí (ztráta určitého úseku DNA), k jejich potvrzení se používá nejčastěji metoda MLPA nebo multiplexní polymerázová řetězová reakce. Nedeleční formy α-talasémií způsobené bodovou mutací v HBA genu lze detekovat podobně jako u β-talasémií pomocí Sangerova sekvenování.
LÉČBA
Pacienti s talasémií minor většinou nevyžadují žádnou léčbu. Zejména v zemích, kde se nosičství talasémie vyskytuje minoritně (včetně České republiky), jsou často tito pacienti mylně léčeni preparáty Fe. Mikrocytární a hypochromní anémie je v tomto případě považována za anémii sideropenickou. Nosičství talasémie je pak diagnostikované až při neadekvátní odpovědi na tuto léčbu. Základem terapie těžších forem β-talasémie je substituce erytrocytárními koncentráty. Pacienti s β-talasémií intermedia vyžadují léčbu transfuzemi erytrocytů v dětství a v době dospívání jen ojediněle a k pravidelné substituční terapii, pokud vůbec musí být zahájena, dospějí až po ukončení druhé dekády života. Nemocní s β-talasémií major zahajují pravidelnou transfuzní léčbu většinou ihned po stanovení diagnózy, tedy v časném dětství. Časové rozmezí podávání jednotlivých erytrocytárních koncentrátů je 2–4 týdny.
Pravidelný přívod Fe transfuzemi a neefektivní erytropoézou zprostředkovaná hyperabsorpce Fe u pacientů s talasémií intermedia a talasémií major způsobují sekundární hemosiderózu [30, 31]. Klíčovou roli v tomto procesu sehrává molekula hepcidinu, hlavní regulační protein metabolismu Fe v lidském těle. Suprese syntézy hepcidinu erytropoetickou aktivitou kostní dřeně je pravděpodobně nadřazená signálům o stavu zásob Fe v organismu [32]. Toto snížení hladiny hepcidinu je zodpovědné za přetížení Fe u pacientů s talasémií intermedia, kteří nejsou závislí na transfuzní terapii [21]. Na druhé straně u pacientů s β-talasémií major je primární příčinou přetížení Fe pravidelné podávání erytrocytárních koncentrátů. Hladiny hepcidinu u těchto pacientů jsou vyšší než u pacientů s talasémií intermedia, což je pravděpodobně způsobeno supresivním efektem podávaných transfuzí na erytropoetické signály kostní dřeně účastnících se regulace syntézy hepcidinu [33]. Alterace metabolismu Fe i přes absenci klinických příznaků byla popsána i u nositelů talasemické alely [34, 35]. Pacienti s laboratorními nebo klinickými známkami přetížení Fe proto vyžadují podávání chelatačních látek k vyvázání nadbytečného Fe. V praxi jsou používány 3 typy chelatačních látek: deferoxamin, deferiprone, deferasirox. Indikace k podávání jednotlivých preparátů závisí zejména na věku pacienta a toleranci jednotlivých léků.
Léčba přidružených komplikací u pacientů s talasémií vyžaduje multidisciplinární přístup zahrnující péči kardiologickou, endokrinologickou, ortopedickou a chirurgickou.
Kurativní léčbou talasémie jsou transplantace kmenových buněk krvetvorby (HSCT) a genová terapie. První HSCT byla provedena již v roce 1982 a od té doby byla využita k léčbě talasémie u více než 3000 pacientů [36, 37]. Overall event-free survival je 80–97 % v závislosti na pokročilosti onemocnění. Nejvhodnějším dárcem je zdravý HLA shodný sourozenec [38]. Genová terapie talasémie je pak jedinou možností kurativní léčby pro pacienty, kteří nenajdou vhodného dárce k transplantaci. S úspěchem byla poprvé použita u pacienta s nosičstvím β-talasémie v kombinaci s hemoglobinopatií HbE v roce 2010. Celkem nyní probíhají tři klinické studie zahrnující 7 pacientů, u všech byl použitý lentivirový vektor se schopností začlenit funkční gen pro β-globinový řetězec do genomu pacienta. Tato léčba je zatím experimentální, protože chybí dostupné informace o vlivu zásahu do genetické informace pacientů na mutagenezi [39].
ZÁVĚR
V České republice je onemocnění talasémií relativně vzácnou příčinou mikrocytární anémie v dětské populaci. S rozšiřující se migrační vlnou lze však očekávat nárůst diagnostikovaných pacientů – zejména pacientů s nosičstvím talasemické alely. Pacienti s mikrocytární anémií, jejíž příčinou není sideropenie a která nevykazuje adekvátní odpověď na léčbu preparáty železa, by proto měli být podrobněji hematologicky vyšetřeni ve specializovaném centru. S nárůstem migrace v Evropě dále také souvisí potřeba prenatálního vyšetření, pokud existuje podezření na výskyt hemoglobinopatie v rodině.
MUDr. Lucie Sulovská, Ph.D.
Dětská klinika
FN a LF UP
I. P. Pavlova 6
775 20 Olomouc
e-mail: luciesulovska@email.cz
Sources
1. Schechter AN. Hemoglobin research and the origins of molecular medicine. Blood 2008; 112 (10): 3927–3938.
2. Williams TN, Weatherall DJ. World distribution, population genetics, and health burden of the hemoglobinopathies. Review. Cold Spring Harb Perspect Med 2012; 2 (9): a011692.
3. Angastiniotis M, Modell B. Global epidemiology of hemoglobin disorders. Ann N Y Acad Sci 1998; 850 : 251–269.
4. Weatherall DJ, Clegg, JB. Inherited haemoglobin disorders: an increasing global health problem. Bull World Health Org 2001; 79 : 704–712.
5. Mockenhaupt FP, Ehrhardt S, Gellert S, et al. Alpha(+)-thalassemia protects African children from severe malaria. Blood 2004; 104 : 2003–2006.
6. Williams TN, Wambua S, Uyoga S, et al. Both heterozygous and homozygous α + thalassemia protect against severe and fatal Plasmodium falciparum malaria on the coast of Kenya. Blood 2005; 106 : 368–371.
7. Pattanapanyasat K, Yongvanitchit K, Tongtawe P, et al. Impairment of Plasmodium falciparum growth in thalassemic red blood cells: further evidence by using biotin labeling and flow cytometry. Blood 1999; 93 : 3116–3119.
8. Ayi K, Turrini F, Piga A, et al. Enhanced phagocytosis of ring-parasitized in sickle trait and beta-thalassemia trait. Blood 2004; 104 : 3364–3371.
9. Divoký V, Indrák K, Mojzíková R. Hemoglobinopatie: talasémie a strukturní Hb varianty. In: Pospíšilová Š, Dvořáková D, Mayer J (Eds). Molekulární hematologie. Praha: Galén, 2013 : 270–283.
10. Mehta RP, Keohane EM. Thalassemias. In: Rodak BF, Fritsma GA, Keohane OA. Hematology: Clinical Principles and Appplications. 4th ed. Missouri: Elsevier, 2012 : 408–425.
11. Benz EJ, Schrier SL Landaw SA. Clinical manifestation and diagnosis of the talassemias. www.uptodate.com. Last updated: Dec 17, 2014.
12. Lorey F, Charoenkwan P, Witkowska HE, et al. Hb H hydrops foetalis syndrome: a case report and review of literature. Br J Haematol 2001; 115 (1): 72-78.
13. Carr S, Rubin L, Dixon D et al. Intrauterine therapy for homozygous alpha-thalassemia. Obstet Gynecol 1995; 85 (5 Pt 2): 876–879.
14. Chui DH, Waye JS. Hydrops fetalis caused by alpha-thalassemia: an emerging health care problém. Blood 1998; 91 (7): 2213–2222.
15. Thein SL. Pathophysiology of beta thalassemia – a guide to molecular therapies. Hematology Am Soc Hematol Educ Program 2005 : 31–37.
16. Tassiopoulos T, Rombos Y, Konstantopoulos K, et al. Spleen size in beta-thalassaemia heterozygotes. Haematologia (Budap) 1995; 26 (4): 205–209.
17. Karimi M, Bagheri MH, Tahmtan M, et al. Prevalence of hepatosplenomegaly in beta thalassemia minor subjects in Iran. Eur J Radiol 2009; 69 (1): 120–122.
18. Galanello R, Cao A. Relationship between genotype and phenotype: thalassemia intermedia. Ann N Y Acad Sci 1998; 850 : 325–333.
19. Taher AT, Musallam KM, Karimi M, et al. Overview on practices in thalassemia intermedia management aiming for lowering complication rates across a region of endemicity: the OPTIMAL CARE study. Blood 2010; 115 (10): 1886–1892.
20. Tanno T, Bhanu NV, Oneal PA, et al. High levels of GDF15 in thalassemia suppress expression of the iron regulatory protein hepcidin. Nat Med 2007 Sep; 13 (9): 1096–1101.
21. Origa R, Galanello R, Ganz T, et al. Liver iron concentrations and urinary hepcidin in beta-thalassemia. Haematologica 2007; 92 (5): 583–588.
22. Oron V, Filon D, Oppenheim A, et al. Severe thalassaemia intermedia caused by interaction of homozygosity for alpha-globin gene triplication with heterozygosity for beta zero-thalassaemia. Br J Haematol 1994; 86 (2): 377–379.
23. Olivieri NF. The beta-thalassemias. Review. N Engl J Med 1999 Jul 8; 341 (2): 99–109. Erratum in: N Engl J Med 1999; 341 (18): 1407.
24. Sirichotiyakul S, Tantipalakorn C, Sanguansermsri T, et al. Erythrocyte osmotic fragility test for screening of alpha-thalassemia-1 and beta-thalassemia trait in pregnancy. Int J Gynaecol Obstet 2004; 86 : 347–350.
25. Dragean CA, Duquesne L, Theate I, et al. Extramedullary haemopoiesis and spinal cord compression. Lancet 2011; 377 (9761): 251.
26. Pepe A, Meloni A, Rossi G, et al. Cardiac complications and diabetes in thalassaemia major: a large historical multicentre study. Br J Haematol 2013; 163 (4): 520–527.
27. Fung EB, Harmatz PR, Lee PD, et al. Multi-Centre Study of Iron Overload Research Group. Increased prevalence of iron-overload associated endocrinopathy in thalassaemia versus sickle-cell disease. Br J Haematol 2006; 135 (4): 574–582.
28. Vogiatzi MG, Macklin EA, Trachtenberg FL, et al. Thalassemia Clinical Research Network. Differences in the prevalence of growth, endocrine and vitamin D abnormalities among the various thalassaemia syndromes in North America. Br J Haematol 2009; 146 (5): 546–556.
29. Haines D, Martin M, Carson S, et al. Thalassemia Clinical Research Network. Pain in thalassaemia: the effects of age on pain frequency and severity. Br J Haematol 2013; 160 (5): 680–687.
30. Rachmilewitz EA, Giardina PJ. How I treat thalassemia. Blood 2011; 118 (13): 3479–3488.
31. Goss C, Giardina P, Degtyaryova D, et al. Red blood cell transfusions for thalassemia: results of a survey assessing current practice and proposal of evidence-based guidelines. Transfusion 2014; 54 (7): 1773–1781.
32. Ganz T, Nemeth E. The hepcidin-ferroportin system as a therapeutic target in anemias and iron overload disorders. Hematology Am Soc Hematol Educ Program 2011; 2011 : 538–542.
33. Pasricha SR, Frazer DM, Bowden DK, et al. Transfusion suppresses erythropoiesis and increases hepcidin in adult patients with β-thalassemia major: a longitudinal study. Blood 2013; 122 (1): 124–133.
34. Guimarães JS, Cominal JG, Silva-Pinto AC, et al. Altered erythropoiesis and iron metabolism in carriers of thalassemia. Eur J Haematol 2015; 94 (6): 511–518.
35. Jones E, Pasricha SR, Allen A, et al. Hepcidin is suppressed by erythropoiesis in hemoglobin E β-thalassemia and β-thalassemia trait. Blood 2015; 125 (5): 873–880.
36. Thomas ED, Buckner CD, Sanders JE, et al. Marrow transplantation for thalassaemia. Lancet 1982; 2 (8292): 227–229.
37. Angelucci E, Baronciani D. Allogeneic stem cell transplantation for thalassemia major. Haematologica 2008; 93 (12): 1780–1784.
38. Lucarelli G, Gaziev J. Advances in the allogeneic transplantation for thalassemia. Blood Rev 2008; 22 (2): 53–63.
39. Finotti A, Breda L, Lederer CW, et al. Recent trends in the gene therapy of β-thalassemia. J Blood Med 2015; 6 : 69–85.
Labels
Neonatology Paediatrics General practitioner for children and adolescentsArticle was published in
Czech-Slovak Pediatrics

2017 Issue 8
- What Effect Can Be Expected from Limosilactobacillus reuteri in Mucositis and Peri-Implantitis?
- The Importance of Limosilactobacillus reuteri in Administration to Diabetics with Gingivitis
Most read in this issue
- Talasemické syndromy
- Chronický zánět středního ucha v dětském věku
- Mozgový absces – zriedkavá, ale závažná infekcia v detskom veku
- Talasémie a hemoglobinové varianty u dětí