
C3 glomerulopatie – nově definovaná klinická jednotka
C3 glomerulopathy – a new clinical entity
C3 glomerulopathy is a differentially, diagnostically newly defined rare clinical entity with poor prognosis. Symptomatology is variable, the diagnosis is based on the results of immunofluorescent evaluation of renal biopsy, and the disease is confirmed by genetic testing and complete examination of the complement system. The accumulation of isolated C3 deposits without antibodies is a key histological finding.
We report two female patients with the same diagnosis and a relatively different course of disease. One patient is a heterozygous carrier of rare (p.A353V) and common (MCPggaac haplotype) mutation.
Key words:
C3 glomerulopathy, alternate complement pathway, complements, genetics, renal biopsy
Authors:
E. Sládková 1; K. Pivovarčíková 2; J. Sýkora 1
Authors‘ workplace:
Dětská klinika Lékařské fakulty v Plzni, Univerzity Karlovy v Praze a Fakultní nemocnice Plzeň
1; Šiklův ústav patologie Lékařské fakulty v Plzni, Univerzity Karlovy v Praze a Fakultní nemocnice Plzeň
2
Published in:
Čes-slov Pediat 2018; 73 (3): 139-145.
Category:
Original Papers
Overview
C3 glomerulopatie představuje nově definovanou, vzácnou klinickou jednotku se závažnou prognózou. Symptomatologie je variabilní, diagnostika je založena na výsledku imunofluorescenčního vyšetření z renální biopsie, komplexní vyšetření komplementového systému a genetické vyšetření chorobu potvrdí. Hlavní renální histologický nález je izolovaná akumulace depozit C3 bez protilátek.
Prezentujeme kazuistiky dvou pacientek se stejnou diagnózou a relativně odlišným průběhem onemocnění, jedna pacientka je heterozygotním nosičem vzácné (p.A353V) a běžné (varianta MCPggaac haplotyp) mutace.
Klíčová slova:
C3 glomerulopatie, alternativní cesta, složky komplementu, genetika, renální biopsie>
ÚVOD
V poslední dekádě došlo k rozpoznání spektra chorob, u kterých hrají primární roli v patogenezi abnormality aktivace komplementu. Membranoproliferativní glomerulonefritida (GN) je nově reklasifikována na imunoglobuliny mediovanou, kdy dochází k aktivaci klasické složky komplementu, a non-imunoglobuliny mediovanou, kdy je patologie v alternativní složce komplementu [1, 2]. Tato skupina onemocnění pak byla označena souhrnným názvem C3 glomerulopatie, pod kterým je zahrnuta nemoc denzních depozit (dense deposit disease) a C3 glomerulonefritida. Patofyziologicky se podobá atypickému hemolyticko-uremickému syndromu [3].
Jedná se o velmi vzácné onemocnění postihující stejnou měrou obě pohlaví. Vyskytuje se u dětí i dospělých, prevalence se odhaduje na 2–3/1 milion [4], výskyt této choroby v naší dětské populaci však není známý. Prognóza onemocnění je závažná a progrese do terminálního selhání ledvin se odhaduje až u 50 % pacientů do 10 až 15 let. Klinická manifestace může být od asymptomatických močových nálezů charakteru mikroskopické hematurie nebo proteinurie, přes makroskopickou hematurii, nefrotický syndrom až po renální selhání.
Cílem práce je prezentovat velmi vzácnou nově definovanou pediatrickou formu C3 glomerulárního onemocnění.
SOUBOR PACIENTŮ
V letech 2015 až 2017 bylo v nefrologické ordinaci Dětské kliniky FN a LF UK v Plzni diagnostikováno 16 dětí a dospívajících s patologickými cytochemickými močovými nálezy (hematurie, proteinurie), sníženou C3 složkou komplementu a zvýšenou hodnotou ASLO. Klinicky se jednalo o asymptomatické náhodné patologické nálezy v moči, akutní nefritický syndrom s přechodnou poruchou renálních funkcí a nefrotický syndrom.
U většiny pacientů bylo podle klinického průběhu vysloveno podezření na poststreptokokovou GN s dalším ambulantním sledováním. U 2 pacientů (dívky) byl však klinický průběh a laboratorní parametry nápadně odlišné a diagnóza byla následně přehodnocena na C3 glomerulopatii.
KAZUISTIKA 1
Šestnáctiletá dívka narozená z fyziologické gravidity, rodinná a perinatální anamnéza jsou bez pozoruhodností. V 8 letech prodělala akutní pyelonefritidu. Pro opakované tonzilitidy byla ORL lékařem doporučena tonzilektomie, která nebyla provedena. Při preventivní prohlídce u praktického pediatra v 15 letech byla zjištěna mikroskopická hematurie a proteinurie, empiricky doporučená antibiotická terapie, avšak bez provedení bakteriologického vyšetření moči. Dívka pokračovala v běžných životních aktivitách včetně sportu, pro přetrvávající mikroskopickou hematurii a proteinurii byla odeslána do nefrologické ordinace naší kliniky. Laboratorně normální renální funkce, mineralogram, celková bílkovina, albumin a hypercholesterolémie. V moči poměr proteinů a kreatininu Uprot/Ukreat 231 mg/mmol. Imunologicky normální hodnoty imunoglobulinů, cirkulujících imunokomplexů a negativní autoprotilátky. Byla snížená C3 složka komplementu (0,19 g/l), C4 v normě, zvýšené hodnoty ASLO. Mikrobiologickým vyšetřením byl zjištěn Streptococcus agalactiae v koncentraci 107 v moči a Streptococcus dysgalactiae v krku.
Kromě chronické tonzilitidy byla diagnostikována vulvovaginitida. Byla zahájena terapie G-penicilinem a lokálně vaginálními čípky, byly provedeny tonzilektomie a adenotomie. Močový nález se znormalizoval, renální funkce byly trvale v normě, dívka byla v dobrém klinickém stavu. Pro přetrvávající snížení C3 složky komplementu byla provedena renální biopsie. Pro váhání rodiny se souhlasem s vyšetřením byla provedena s odstupem 2 měsíců. V té době došlo opět k významnému zhoršení proteinurie (celkový odpad bílkoviny 7 g/m2/den, Uprot/Ukreat 700 mg/mmol). V klinickém obraze byly nápadné otoky vyžadující diuretickou terapii.
Histologické vyšetření:
Histologické vyšetření ve světelném mikroskopu prokázalo lobulární uspořádání glomerulů se zvýšenou mesangiální celularitou, rozštěpy glomerulární bazální membrány (GBM) a nekrózy kapilárních kliček s extrakapilární proliferací ve formě celulárních (čerstvých) srpků, což svědčilo pro rychle progredující GN (RPGN). Tubulointersticiální kompartment a vaskulární aparát byly relativně přiměřeného vzhledu, bez významnějších patologických změn. Imunofluorescenční vyšetření ukázalo silnou (3+) granulární pozitivitu v oblasti glomerulární bazální membrány (GBM) v C3 u všech glomerulů. Pouze zcela necharakteristická tečkovitá pozitivita fokálně segmentálně byla zachycena v IgA, IgG, IgM a C1q. Ultrastrukturálním vyšetřením v elektronovém mikroskopu byla zastižena objemná splývající elektrondenzní depozita subendoteliálně a subepiteliálně (odpovídající imunofluorescenčnímu nálezu). Po klinicko-patologické korelaci byl nález označen jako GN s dominantní C3 depozicí.
Po provedení renální biopsie byla zahájena pulzní terapie kortikoidy, následovaná perorálním podáváním, pro bioptický nález rychle progredující GN (RPGN) byl podáván též cyklofosfamid. Klinický stav pacientky se komplikoval rozvojem významného asymetrického otoku vulvy, který postupně vymizel (obr. 1).
Fig. 1. Vulvar swelling – local complications in patient 1.

V současné době není v České republice dostupné kompletní vyšetření komplementu, oslovili jsme proto po schválení revizním lékařem spolupracující pracoviště v Budapešti.
Kompletní vyšetření komplementu:
Byly odeslány 2 vzorky, první odebraný v akutní fázi při kombinované imunosupresivní terapii, druhý s odstupem 4 měsíců, kdy došlo po přechodné normalizaci opět ke snížení C3 složky komplementu. V 1. vzorku byl zastižen normální profil komplementu s negativními autoprotilátkami a nízkými aktivačními produkty komplementu.

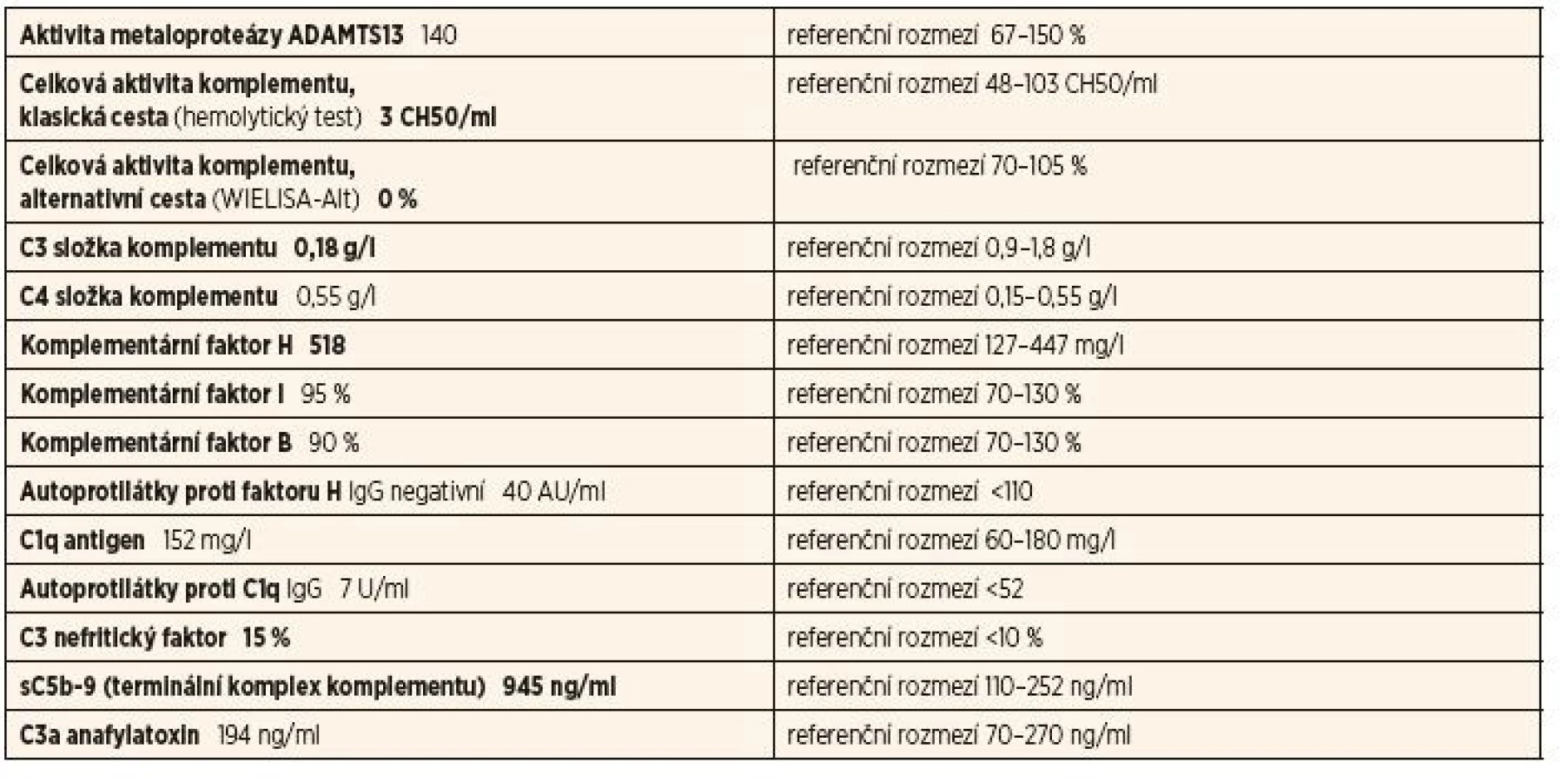
Ve 2. vzorku byl pozitivní C3 nefritický faktor (C3Nef), snížená hladina C3, známky klasické i alternativní cesty aktivace komplementu, nízká hladina faktoru B a elevované aktivační produkty komplementu (C5b-9 – terminální komplex komplementu) a C3a anafylatoxin.
Tyto výsledky svědčily pro C3Nef asociovanou deregulaci alternativní cesty a aktivaci komplementu potvrzující diagnózu C3 glomerulopatie. Genetické vyšetření neprokázalo přítomnost mutace.
Léčebná dávka prednisonu byla postupně snižována a na přání rodiny zcela vysazena. Renální funkce jsou trvale v normě, dochází k vzestupu proteinurie Uprot/Ukreat 60–120 mg/mmol a přetrvává snížená C3 složka komplementu. Doporučené očkování proti opouzdřeným patogenům v době vysazené imunosuprese před zvažovanou biologickou terapií rodina odmítala, nakonec po opakovaném vysvětlení problematiky očkování po konzultaci s očkovacím centrem proběhlo. V současné době je dívka léčená mykofenolát mofetilem a ACE inhibitorem. Je dispenzarizována v naší nefrologické ordinaci, ale rodina přes opakované podrobné poučení se nadále staví k problematice velmi alternativně, spolupráce je obtížná a rodina rezolutně odmítá biologickou terapii.
KAZUISTIKA 2
Pětiletá dívka narozená z fyziologické gravidity, rodinná, osobní i perinatální anamnéza jsou nevýznamné. U pacientky se vstupně objevila makroskopická hematurie a proteinurie při současně snížené C3 složce komplementu (0,64 g/l) a normálních renálních funkcích. Imunologie včetně autoprotilátek byla negativní. Stav byl hodnocen jako poststreptokoková GN. Dívka byla dispenzarizována v nefrologické ordinaci, klinicky bez obtíží, postupně se normalizoval močový nález i hladina C3 (1,06 g/l), renální funkce byly trvale v normě. Po roce se objevila makroskopická hematurie a proteinurie – Uprot/Ukreat 1006 mg/mmol, renální funkce byly normální, opět snížená C3 složka komplementu (0,20 g/l), normální C4 a ASLO, negativní autoprotilátky, extrémně nízké hladiny aktivace klasické (14 %, norma 69–190 %), alternativní (1 %, norma 30–230 %) i lektinové cesty komplementu (4 %). Pro klinický nález akutní faryngitidy a kontakt se spálou byla zahájena terapie penicilinem. Mikrobiologicky byl pozitivní Campylobacter jejuni ve stolici, sérologicky a klinicky aktivní infekce HHV 6. Na základě klinického průběhu a všech provedených vyšetření dívka podstoupila renální biopsii.
Histologické vyšetření:
Mikroskopicky bylo u všech glomerulů prokázáno jasně naznačené lobulární uspořádání, při sporné tloušťce GBM (se segmentálně naznačenými rozštěpy). V lumen kapilárních kliček byly přítomny četné neutrofilní granulocyty a menší množství lymfocytů. Nekrózy kapilárních kliček či celulární srpky nebyly v histologicky vyšetřeném materiálu zastiženy. Tubulární aparát byl bez nápadné patologie, v intersticiu fokálně se smíšenou zánětlivou celulizací. Vaskulární aparát byl bez výraznějších změn. Imunofluorescenční nález vykazoval silnou (3+) granulární pozitivitu mesangiálně a na GBM v C3. Ostatní vyšetřované markery (IgA, IgG, IgM, C1q, lehké řetězce kappa a lambda) reagovaly pouze necharakteristicky mimo glomeruly, či byly kompletně negativní. Ultrastrukturálním vyšetřením elektronovým mikroskopem byla zastižena objemná splývající elektrondenzní depozita lokalizovaná na obou stranách GBM s maximem subendoteliálně (drobná depozita i subepiteliálně a mesangiálně) (obr. 2, 3). Po klinicko-patologické korelaci bylo vysloveno podezření na GN s dominantní C3 depozicí.
Fig. 2. Histological findings in patient 2.

Fig. 3. Histological findings in patient 2. Electron microscopy with the findings of dense deposits.

Před zahájením imunosupresivní terapie dále provedeno vyšetření komplementu v ÚHKT Praha s výsledkem: CFH a CHI v normě, protilátky nejsou přítomny, byl vyloučen deficit ADAMTS13. Zahájena byla pulzní terapie metylprednisolonem, následně podáván prednison, mykofenolát mofetil a ACE inhibitor.
Kompletní vyšetření komplementu:
Vyšetření prokázalo velice nízkou hladinu C3 s chybějící aktivitou alternativní cesty komplementu a pozitivní C3Nef. Zvýšené hladiny produktů terminální cesty aktivace. Negativní protilátky proti faktoru H a C1q.
Tyto výsledky svědčí pro komplementem mediovanou GN. Vzhledem ke stálé aktivaci a konsumpci C3 a přítomnosti C3Nef je doporučena imunosupresivní terapie a pa- cientka je na základě funkčního defektu komplementového systému považována za imunodeficientní. Genetické vyšetření prokázalo, že pacientka je heterozygotním nosičem vzácné (p.A353V) a běžné (varianta MCPggaac haplotyp) mutace v MCP (označovaný jako CD46, membrane cofactor protein NM 002389.4) genu [4].
Onemocnění pacientky může být považováno za komplementem mediované na základě genetických a získaných (C3Nef) predispozičních faktorů. Pokud by u pa- cientky i navzdory imunosupresivní terapii došlo k progresi onemocnění při přetrvávajících známkách aktivace komplementu, je možno zvážit podání eculizumabu.
DISKUSE
C3 glomerulopatie je raritní, poměrně recentně definované onemocnění způsobené dysregulací komplementové kaskády, s popsaným výskytem sporadickým i familiárním. V českém písemnictví nebylo doposud této jednotce věnováno mnoho pozornosti, proto zde prezentujeme dva případy onemocnění u pacientů dětského věku ve snaze oslovit širokou odbornou pediatrickou veřejnost.
C3 glomerulopatie je termín označující dvě jednotky s překrývající se patogenezí, ne vždy zcela jednoznačně rozlišitelné (C3 GN a DDD), klasifikované podle charakteru a lokalizace depozit v elektronové mikroskopii. Popsány byly též i přechodové formy mezi oběma těmito entitami [6] a jednotky familiární DDD s C3 mutací nebo familiární C3 GN s mutacemi CFHR genů [7, 8]. C3 glomerulopatie představuje klinickou jednotku charakterizovanou izolovanou nebo predominantní akumulací fragmentů C3 složky komplementu v glomerulech při absenci či minimální depozici časných komponent klasické cesty komplementu (C1q a C4) a imunoglobulinů, což lze snadno prokázat imunofluorescenčním vyšetřením z renální biopsie.
Samotný morfologický nález v renální biopsii může být značně variabilní od membranoproliferativních a mesangioproliferativních změn, endokapilární proliferace, až po obraz nekrotizující a krescentní GN. Na podkladě dostupných údajů lze předpokládat, že patogeneticky se při rozvoji C3 glomerulopatie uplatňují vrozené predispozice a vlivy faktorů zevního prostředí. Nepřítomnost klinicky manifestního onemocnění u rodinných příslušníků s geneticky detekovanou mutací může být vysvětlena existencí různého spouštěcího mechanismu, kterým pravděpodobně může být např. prodělaná infekce [10, 11]. Stanovení míry uplatnění jednotlivých faktorů při rozvoji onemocnění nejsou známy a bude vyžadovat další rozsáhlé studie.
Naše zkušenosti i dostupné literární údaje ukazují, že pacienti s C3 glomerulopatií jsou v běžné klinické praxi často vedeni a sledováni pod diagnózou postinfekční GN. Teprve přetrvávání patologického močového nálezu, dlouhodobě snížená hladina C3 složky komplementu a někdy i porucha renálních funkcí indukují další diagnostické rozvahy a provedení vyšetření, jež finálně vedou až ke správné diagnóze. Právě nízké hladiny C3 složky komplementu jsou velmi indikativní pro C3 glomerulopatii, avšak normální hladina C3 tuto chorobu nevylučuje [9]. Z praktického hlediska by tedy na C3 glomerulopatii mělo být pomýšleno zejména u dětí s hematurií, proteinurií, nízkou hladinou C3 a normálními hodnotami C4 složky komplementu a při typickém imunofluorescenčním nálezu v renální biopsii. Pro konečné stanovení diagnózy je dále nutno provést i kompletní specializované vyšetření komplementu včetně vyšetření genetických mutací typicky asociovaných s chorobou (mutace v genech pro faktor H, faktor I a faktor B, CFHR1-5, MCP/CD46).
Pro pochopení patogeneze C3 glomerulopatie je důležitá znalost základních údajů o komplementu (obr. 4). Komplement jako první obranná reakce organismu na přítomnost antigenu (kterým může být bakteriální agens, ale i jiný podnět) je přirozená složka imunity rozeznávající vlastní molekuly od cizích [12] a představuje jednu z hlavních efektorových drah zánětu. Aktivace komplementu zahrnuje kaskádový proces a probíhá třemi cestami. Klasická cesta je zahájena na povrchu cílových buněk vazbou protilátek nebo sérových proteinů. U lektinové cesty není aktivátorem protilátka, ale lektin vážící manosu exprimovanou na povrchu mikroorganismů. Alternativní cesta představuje nespecifickou obranu proti infekci a je aktivována cizími složkami povrchu, např. stěnou bakterií. Všechny tři cesty aktivace komplementu vedou ke stejnému cíli – konverzi C3 složky (pomocí enzymu C3 konvertázy) na C3a a C3b. C3a se podílí na chemotaxi, C3b se váže na povrch mikrobů a pomáhá jejich odstranění opsonizací.

Důležitá je lytická funkce komplementu, kdy vzniká z proteinu C5 konvertázou fragment C5b a společně s dalšími složkami C6, C7, C8 a C9 vznikne MAC (membrane attack complex), který vytvoří póry v napadené buňce a dojde k její lýze za současného uvolňování anatoxinů. C3b váže též aktivovaný komplementární faktor B a po různých aktivačních krocích pak působí jako alternativní C3 konvertáza, velmi účinně štěpící C3 na C3a a C3b. Alternativní cesta aktivace komplementu probíhá prakticky trvale, zahrnuje velké množství složitých procesů a může poškodit i vlastní organismus. Je proto nutná ochrana vlastních buněk před takovým poškozením. Touto ochranou jsou H a I faktory cirkulující v plazmě, přičemž komplementový faktor H je hlavním faktorem ochraňujícím povrch buněk před aktivací komplementu. Dalšími ochrannými faktory jsou proteiny na povrchu endotelu jako membránový proteinový kofaktor MCP/CD46 a trombomodulin. C3 glomerulopatie je asociovaná s nekontrolovatelnou aktivací alternativní cesty komplementu. Dysregulace alternativní cesty je buď způsobená genetickými mutacemi, nebo získanými cirkulujícími protilátkami. To vyúsťuje ve ztrátu kontroly nad komplementární alternativní C3 konvertázou a dochází tak k poruše funkce aktivátorů či regulátorů komplementu. U pacientů je nejčastěji detekovanou autoprotilátkou C3Nef (C3 nefritický faktor), prodlužující poločas C3 konvertázy, což má za následek masivní konsumpci C3 složky komplementu (snížení hladiny C3) a zvýšenou tvorbu C3 a C5 konvertázy [13]. K poškození tkání může dojít depozicí cirkulujících aktivních komponent komplementu nebo jejich lokální aktivací. Ledviny jsou orgánem vysoce vulnerabilním pro komplementem mediované poškození, důvodem mohou být morfologické specificity stavby glomerulárních kapilár, především přítomnost fenestrovaného endotelu a expozice GBM substancím cirkulujícím v séru (včetně složek komplementu).
I přes dobře známou patogenezi v současnosti nejsou k dispozici jednoznačná terapeutická doporučení. Literatura uvádí terapeutické postupy opírající se o zkušenosti jednotlivých center s malým počtem pacientů a doporučení expertů [14–17]. Obecně by však terapie měla být založena na podpůrné antiproteinurické, renoprotektivní, imunosupresivní a antikomplementové terapii. V rámci podpůrné terapie se též užívají hypolipidemika, ACE inhibitory a/nebo blokátory angiotenzinového receptoru. Od dříve doporučovaného podávání plazmy nebo plazmaferézy se pro možnou alloimunizaci v současnosti upouští [15]. Eventuální alloimunizace by mohla v dlouhodobém důsledku zkomplikovat případnou transplantaci ledviny, vést k alergickým reakcím, riziková je i kanylace centrálního řečiště. V literatuře byly popsány jednotlivé kazuistiky [18] i soubory pacientů léčených imunosupresivní terapií. Používán je především mykofenolát mofetil, často v kombinaci s prednisonem [19]. Specificky u pacientů s pozitivním C3Nef je doporučována anti-B lymfocytární terapie (jako např. rituximab) snižující jeho produkci. Nadějnou se zdá být i terapie pomocí rekombinantní humanizované monoklonální IgG protilátky (eculizumab), která se specificky váže na C5 složku komplementu a zabraňuje tak štěpení C5 na C5a a C5b složky a tím následně i vznik komplexu C5b-9 (MAC) [20]. U jednotlivých případů bylo při této terapii zaznamenáno i zastavení další progrese onemocnění [13, 14, 21–23]. Zatím však nejde o standardní terapeutické postupy, poněvadž nejsou dostupná prediktivní data úspěšnosti.
I navzdory intenzivní terapii však dochází u velkého procenta pacientů k renálnímu selhání. Transplantace ledviny je provázena velkým rizikem rekurence onemocnění ve štěpu. Přesná data zatím nejsou dostupná (jedná se o relativně novou jednotku).
Při absenci jednoznačných léčebných doporučení tedy nelze rozhodnout, jaká léčba je optimální. U naší první pacientky došlo k významnému zlepšení klinického stavu a laboratorních parametrů při imunosupresivní terapii. Trvale přetrvávající proteinurie a snížení C3 složky komplementu jsou však známkou subklinicky pokračující aktivity procesu. Velkou limitací u této pacientky je konzervativní postoj rodiny k intenzivnější léčbě. U druhé pacientky bylo při imunosupresivní terapii taktéž dosaženo dobrého klinického stavu a stabilních laboratorních výsledků. Imunosuprese je postupně vysazována, v případě opětovného zhoršení stavu je v plánu biologická terapie. Klinický průběh (2 ataky onemocnění s odstupem roku vyvolané infekcí), přítomnost genetické mutace a C3Nef potvrzují literární údaje o genetické predispozici a dalším spouštěcím momentu, kterým je pravděpodobně infekce, což může významně klinický průběh a terapii komplikovat.
ZÁVĚR
C3 glomerulopatie je nově popsaná, vzácná klinická jednotka způsobená dysregulací komplementového systému vyžadující spektrum vyšetření nezbytných ke stanovení konečné diagnózy. Zjistili jsme, že klinický průběh a výsledky imunologického vyšetření neumožní v počátku odlišení této jednotky od postinfekční GN. Pokud tedy u pacienta nedojde k normalizaci C3 složky komplementu během 2 až 3 měsíců po prodělané streptokokové infekci, je indikována renální biopsie. Pokud prokážeme morfologický obraz konzistentní se širokým spektrem popisovaných změn u C3 glomerulopatie a především charakteristický imunofluorescenční obraz a elektronmikroskopický obraz (který může odlišit DDD od C3GN), pak je nezbytné provést kompletní vyšetření komplementu včetně genetických metod.
Za důležité považujeme, aby v rámci časné detekce tohoto často dlouho asymptomatického závažného onemocnění praktiční pediatři důsledně dbali na cytochemické vyšetřování moče po prodělané streptokokové infekci. Na C3 glomerulopatii je třeba myslet a včasně toto onemocnění diagnostikovat, neboť časné zavedení adekvátní léčby významně zlepšuje stav a další osud nemocných dětí. Přesto, že mnohem častějším následkem streptokokové infekce může být posstreptokoková GN s většinou dobrou prognózou, literární i naše zkušenosti potvrzují, že rizikovým faktorem pro rozvoj C3 glomerulopatie může být i neléčená perzistující streptokoková infekce. Cílem našeho sdělení bylo upozornit na tuto neobvyklou závažnou klinickou jednotku a předat naše diagnosticko-léčebné zkušenosti. Obecně přijaté standardy léčby nejsou dosud definovány, ale lepší pochopení role komplementu umožňuje optimálně směrovat výzkum a zavádění nových terapeutických postupů.
Poděkování:
Semmelweis University, 3rd Department of Internal Medicine, Budapest, Hungary, Director: Dr. István Karádi; Research Laboratory, Head: Dr. Zoltán Prohászka; Ústav hematologie a krevní transfuze Praha.
Studie byla podporována výzkumným záměrem MŠMT Progres Q-39.
Došlo: 29. 9. 2017
Přijato: 8. 2. 2018
MUDr. Eva Sládková
Dětská klinika Fakultní nemocnice Plzeň
Univerzita Karlova v Praze
Lékařská fakulta v Plzni
Alej Svobody 80
304 60 Plzeň
e-mail: sladkova@fnplzen.cz
Sources
1. Bomback AS, Appel GB. Pathogenesis of the C3 glomerulopathies and reclassification of MPGN. Nat Rev Nephrol 2012; 8: 634–642.
2. Fakhouri F, Frémeaux-Bacchi V, Noël LH, et al. C3 glomerulopathy: a new classification. Nat Rev Nephrol 2010; 6: 494–499.
3. Seeman T, Podracká L, Štolbová Š, Bláhová K. Trombotické mikroan-giopatie-hemolyticko-uremické syndromy a trombotická trombocytopenická purpura. Čes-slov Pediat 2017; 72: 99–108.
4. Servais A, et al. Primary glomerulonephritis with isolated C3 deposits: a new entity which shares common genetic risk factors with haemolytic uraemic syndrome. J Med Genet 2007; 44: 193–199.
5. Cook HT, Pickering MC. Histopathology of MPGN and C3 glomerulopathies. Nat Rev Nephrol 2014; 11: 14–22.
6. Medjeral-Thomas N, et al. A novel CFHR5 fusion protein causes C3 glomerulopathy in a family without Cypriot ancestry. Kidney Int 2014; 85: 933–937.
7. Barbour TD, Pickering MC, Terence Cook H. Dense deposit disease and C3 glomerulopathy. Semin Nephrol 2013; 33: 493–507.
8. Gale DP, Maxwell PH. C3 glomerulonephritis and CFHR5 nephropathy. Nephrol Dial Transplant 2013; 28: 282–288.
9. Barbour TD, Pickering MC, Cook HT. Recent insights into C3 glomerulopathy. Nephrol Dial Transplant 2013; 28: 1685–1693.
10. Sandhu G, et al. C3 glomerulopathy masquerading as acute postinfectious glomerulonephritis. Am J Kidney Dis 2012; 60: 1039–1043.
11. Sethi S, et al. Atypical postinfectious glomerulonephritis is associated with abnormalities in the alternative pathway of complement. Kidney Int 2013; 83: 293–299.
12. Zipfel PF, et al. The role of complement in C3 glomerulopathy. Mol Immunol 2015; 67: 21–30.
13. Sethi S, et al. C3 glomerulonephritis: clinicopathological findings, complement abnormalities, glomerular proteomic profile, treatment, and follow-up. Kidney Int 2012; 82: 465–473.
14. Barbour TD, Ruseva MM, Pickering MC. Update on C3 glomerulopathy. Nephrol Dial Transplant 2014; 1–9. doi: 10.1093/ndt/gfu317.
15. Pickering MC, et al. C3 glomerulopathy: consensus report. Kidney Int 2013; 84: 1079–1089.
16. Nester CM, Smith RJ. Diagnosis and treatment of C3 glomerulopathy. Clin Nephrol 2013; 80: 395–403.
17. Medjeral-Thomas NR, et al. C3 glomerulopathy: clinicopathologic features and predictors of outcome. Clin J Am Soc Nephrol 2014; 9: 46–53.
18. Pinho A, Ferreira G, Mota C. Successful management of a patient with a C3 glomerulonephritis and crescentic pattern: a case report. BMC Res Notes 2014; 7: 792.
19. Rabasco C, et al. Effectiveness of mycophenolate mofetil in C3 glomerulonephritis. Kidney Int 2015; 88: 1153–1160. doi: 10.1038/ki.2015.227.
20. Nicolas C, et al. C3 nephritic factor associated with C3 glomerulopathy in children. Pediatr Nephrol 2014; 29: 85–94.
21. Gurkan S, et al. Eculizumab and recurrent C3 glomerulonephritis. Pediatr Nephrol 2013; 28: 1975–1981.
22. Le Quintrec M, et al. Eculizumab for treatment of rapidly progressive C3 glomerulopathy. Am J Kidney Dis 2015; 65: 484–489.
23. Vivarelli M, Emma F. Treatment of C3 glomerulopathy with complement blockers. Semin Thromb Hemost 2014; 40: 472–477
Labels
Neonatology Paediatrics General practitioner for children and adolescentsArticle was published in
Czech-Slovak Pediatrics

2018 Issue 3
- What Effect Can Be Expected from Limosilactobacillus reuteri in Mucositis and Peri-Implantitis?
- The Importance of Limosilactobacillus reuteri in Administration to Diabetics with Gingivitis
Most read in this issue
- C3 glomerulopatie – nově definovaná klinická jednotka
- Současný pohled na diagnostiku a léčbu astmatu u dětí
- Novinky v kardiopulmonální resuscitaci – „guidelines 2018“
- Makro AST jako příčina asymptomatické elevace aspartátaminotransferázy