
Diagnostika, léčba a prognóza vrozené hypotyreózy
Diagnostics, treatment and prognosis of congenital hypothyroidism
Congenital hypothyroidism is the most frequent inborn endocrine disorder and the most frequent disease diagnosed by nation-wide newborn screeening. 80–85% of permanent cases is caused by a defective thyroid development – thyroid dysgenesis. Remaining cases are caused by defects of thyroid hormone biosynthesis – dyshormonogenesis. Early and adequate substitution treatment together with a good family compliance are the main factors playing role in psychomotor, mental and somatic development of the majority of children with congenital hypothyroidism.
Key words:
congenital hypothyroidism, neonatal screening, substitution treatment, thyroid dysgenesis, dyshormonogenesis
Authors:
E. Al Taji
Authors‘ workplace:
Klinika dětí a dorostu, 3. lékařská fakulta Univerzity Karlovy a Fakultní nemocnice Královské Vinohrady, Praha
Published in:
Čes-slov Pediat 2018; 73 (3): 158-164.
Category:
Review
Věnováno památce paní profesorky MUDr. Jiřiny Čížkové-Písařovicové, DrSc. (1908–1994) a paní profesorky MUDr. Olgy Hníkové, CSc. (1931–2017)
Overview
Vrozená hypotyreóza je nejčastější vrozené endokrinní onemocnění a vůbec nejčastější choroba diagnostikovaná v rámci celoplošného novorozeneckého screeningu. 80–85 % případů permanentní vrozené hypotyreózy je způsobeno poruchou vývoje, tj. dysgenezí štítné žlázy. V ostatních případech je příčinou porucha tvorby tyreoidálních hormonů, tj. dyshormonogeneze. Časná a adekvátní substituční terapie spolu s dobrou spoluprací rodiny je hlavním limitujícím faktorem psychomotorického, mentálního a somatického vývoje většiny dětí s kongenitální hypotyreózou.
Klíčová slova:
vrozená hypotyreóza, novorozenecký screening, substituční léčba, dysgeneze, dyshormonogeneze
CESTA K ČASNÉ DIAGNOSTICE A LÉČBĚ VROZENÉ HYPOTYREÓZY
Kazuistika
„…Holčička byla poprvé vyšetřená v 10 měsících věku. Byla dlouhodobě spavá a klidná, měla zácpu, neprořezávaly se jí zuby, neseděla. Délkou 63 cm odpovídala 5 měsícům, vážila 7600 g. Měla nápadně hrubou, suchou a ztluštělou kůži, podkožní vazivo řídké a měkké. V obličeji byl patrný edém, oční víčka měla zduřelá, těžko otvírala oči, fontanela široce zela (obr. 1). Měla hrubé, suché, tvrdé vlasy, velký jazyk drsného vzhledu, vyčnívající z úst. Břicho bylo nad úrovní hrudníku, s ochablými svaly a umbilikální hernií. Končetiny, zvláště dolní, byly vzhledem k trupu velmi krátké, s polotuhým nahromaděním pseudolipomatózní tkáně. Štítná žláza byla nehmatná, nad sternem byl slyšitelný hrubý systolický šelest, akce srdeční byla pomalá. Játra přesahovala oblouk žeberní. Na snímku zápěstí nebyla patrná odpovídající osifikace. Po zahájení léčby vyrostla za 9 měsíců o 9 cm, přibrala 3 kg. Váha a výška se normalizovaly po tříleté léčbě ve 4 letech. Koncem 2. roku se začala posazovat, samostatně chodila koncem 3. roku. Řeč se objevila ve 2,5 letech, souvislé věty ale až v 6 letech. Zuby začaly prořezávat po 2. roce, k normalizaci osifikace došlo kolem 7 let. Po psychické stránce jevila stále značnou retardaci, docházela pak do pomocné školy. Dívka se začala léčit relativně pozdě, příslušnou terapií a správnou výživou bylo dosaženo úspěchu po stránce somatického vývoje více než po stránce psychického vývoje… Závěrem je nutné zdůraznit, jak je důležité chorobu včas diagnostikovat a včas ji začít léčit…“
Fig. 1. Untreated case of congenital hypothyroidism at the age of 10 months – adapted from [1].
![Kojenec s neléčenou vrozenou hypotyreózou v 10 měsících věku – převzato z [1].<br> Fig. 1. Untreated case of congenital hypothyroidism at the age of 10 months – adapted from [1].](https://www.prelekara.sk/media/cache/resolve/media_object_image_large/media/image_pdf/9b7c61681192d79ccf00cb0b2fafb01e.jpeg)
Kazuistika byla převzata, s úpravami a zkrácena, z učebnice Klinická endokrinologie dětského věku, kterou v 50. letech 20. století napsala prof. MUDr. Jiřina Čížková-Písařovicová [1]. Diagnóza kongenitální hypotyreózy mohla být u této dívky (narozené v Praze v roce 1941) stanovena až na základě klinicky vyjádřených symptomů hypotyreózy a některých nepřímých laboratorních známek (např. glykemická křivka, krevní obraz). Léčena pak byla sušeným tyreoidálním extraktem až od 10. měsíce věku. I když její další vývoj nebyl zdaleka optimální, neskončil jako u dětí s ještě později léčenou nebo vůbec neléčenou těžší kongenitální hypotyreózou pod obrazem tzv. sporadického kretenismu (těžká mentální retardace, porucha sluchu, řada neurologických a somatických symptomů) (obr. 2). Již v té době, i přes velmi limitované možnosti diagnostiky a léčby, prof. Čížková-Písařovicová velmi prozřetelně kladla důraz na nutnost zahájení léčby co nejčasněji v kojeneckém věku: „Čím dříve se s terapií začne, tím spíše lze očekávat příznivé výsledky, obzvláště začne-li se hned po narození nebo aspoň do 3 měsíců...“ [1].
Fig. 2. Untreated case of severe congenital hypothyroidism at the age of 14 years (i.e. sporadic cretenism) – adapted from [1].
![Dívka s neléčenou těžkou vrozenou hypotyreózou ve věku 14 let (tzv. sporadický kretenismus) – převzato z [1].<br>Fig. 2. Untreated case of severe congenital hypothyroidism at the age of 14 years (i.e. sporadic cretenism) – adapted from [1].](https://www.prelekara.sk/media/cache/resolve/media_object_image_large/media/image_pdf/2ca5b833f575cbc7b6833b35c09197d4.jpeg)
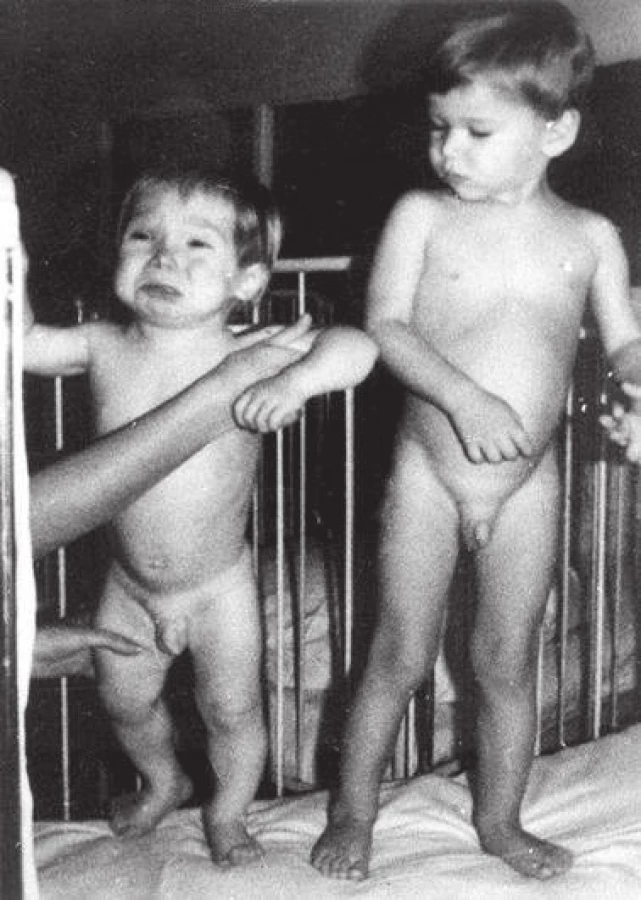
Ještě koncem 60. let 20. století renomované světové učebnice pediatrické endokrinologie uváděly, že určitý stupeň psychomotorické a mentální retardace a ireverzibilita některých neurologických symptomů jsou u kongenitální hypotyreózy i přes léčbu nevyhnutelné [2]. Klíčový význam co nejčasnějšího stanovení diagnózy a léčby pro další prognózu dětí s vrozenou hypotyreózou jednoznačně potvrdily a upřesnily studie v 70. letech 20. století [3]. Ukázaly, že zatímco při zahájení substituční léčby do 3 měsíců věku téměř 80 % dětí dosáhlo hodnoty IQ nad 85, tato hodnota nebyla dosažena u žádného dítěte při zahájení léčby po 6. měsíci věku. Současně potvrdily, že stanovení diagnózy na základě klinických symptomů je možné jen asi u 10 % dětí během prvního měsíce a jen u 1/3 dětí v prvních 3 měsících života. Klinický obraz u novorozence je nenápadný, protože in utero je fetální hypotyreóza do značné míry kompenzovaná transplacentárním přenosem maternálního tyroxinu [4]. U novorozence může být přítomen protrahovaný ikterus nebo perzistuje malá fontanela, další klinické symptomy, zejména váhové neprospívání, nezájem při krmení, obstipace, makroglosie, hypotonie, pupeční kýla, jsou výraznější až kolem 3. měsíce věku (obr. 3). Opoždění psychomotorického vývoje a porucha růstu nastupuje ještě později (obr. 4) [5]. V období před screeningem tak byla diagnóza u některých dětí stanovena až v batolecím nebo dokonce předškolním věku, kdy už byl ireverzibilně poškozen vývoj mozku.
Fig. 3. Untreated case of congenital hypothyroidism at the age of 7 months (provided by prof. MUDr. Olga Hníková, CSc.).
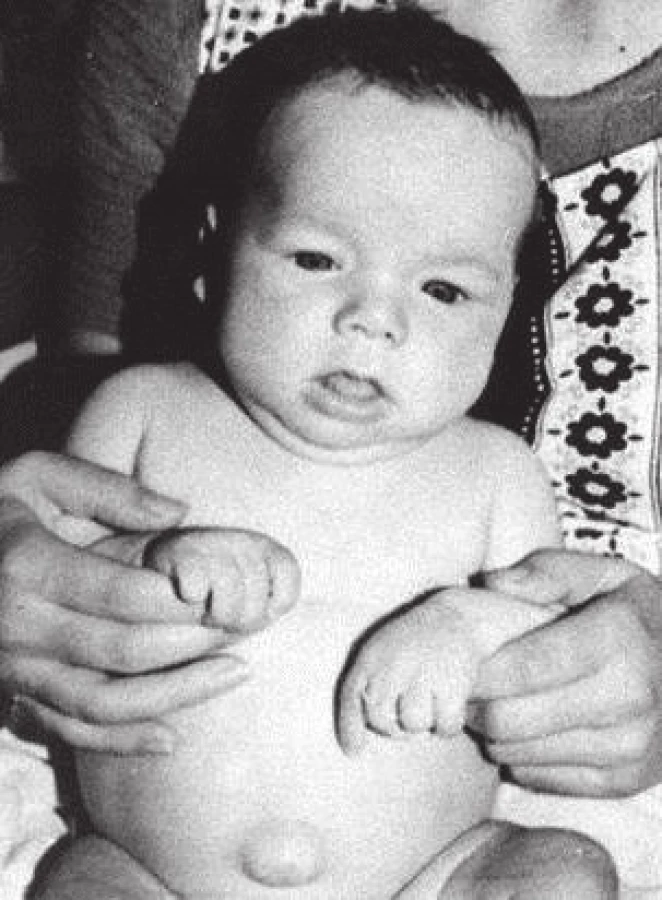
Fig. 4. Untreated case of congenital hypothyroidism at the age of 2 years, compared with a healthy child (provided by prof. MUDr. Olga Hníková, CSc.).
Tyto poznatky spolu s možností stanovení tyroxinu (T4) ze suché krevní kapky, rutinního stanovení sérového tyrotropinu (TSH) a s dostupností moderní léčby syntetickým levotyroxinem od poloviny 70. let 20. století celosvětově vedly k postupnému zavádění celoplošného novorozeneckého screeningu vrozené hypotyreózy (poprvé Kanada, 1974). Prof. MUDr. Olga Hníková, CSc., u nás zahájila pilotní studie už v roce 1975, nejprve stanovením TSH z pupečníkové krve (mezi 156 vyšetřenými novorozenci zachytila jeden případ). Posléze pak zaváděla stanovení T4 ze suché krevní kapky metodou RIA. Vyšetřeno bylo 90 025 novorozenců z 5 pracovišť, diagnostikováno 16 novorozenců a potvrzena incidence vrozené hypotyreózy 1 : 5600 [5]. O průběžných výsledcích publikovala řadu článků i v Česko-slovenské pediatrii. Celoplošný novorozenecký screening se jí značným úsilím podařilo u nás prosadit v roce 1985, v první zemi tehdejšího východního bloku. V letech 1985–1995 se screening prováděl hodnocením hladin T4 ze suché krevní kapky metodou RIA mezi 5.–7. dnem života. Od roku 1996 je založen na hodnocení hladin TSH fluoroimunometricky, nejprve mezi 72.–96., posléze 48.–72. hodinou po narození.
NOVOROZENECKÝ SCREENING VROZENÉ HYPOTYREÓZY
Stanovení TSH je dnes považováno za nejcitlivější metodu k detekci primární hypotyreózy, s optimálním oknem odběru mezi 48.–72. hodinou po narození [6]. Hodnota cut off (koncentrace TSH v suché krevní kapce, která definuje hranici pro negativní nález) není v mezinárodních doporučeních jednoznačně stanovena a liší se v jednotlivých zemích (obvykle se pohybuje kolem TSH 10–15 mIU/l, ale i níže). V České republice jsou v současné době hodnoty TSH vyšší než 15 mIU/l v suché krevní kapce získané z vpichu do patičky mezi 48.–72. hodinou po narození hodnoceny jako suspektní záchyt ve screeningu a vyžadují okamžité vyšetření TSH a fT4 z venózního vzorku krve.
Po zavedení novorozeneckého screeningu se prevalence kongenitální hypotyreózy celosvětově zvýšila z 1 : 6700 na 1 : 4000. Zvyšující se nárůst případů ale nadále pokračuje i v posledních letech (v současné době 1 : 2000–3000, v některých zemích až pod 1 : 2000) [7]. Kumulativní prevalence od roku 2010 v České republice je 1 : 2766 (www.novorozeneckyscreening.cz). Příčiny vzestupu nejsou jednoznačné. Podílí se na něm zejména případy vrozené hypotyreózy se štítnou žlázou in situ, mírnější event. tranzientní formy, pravděpodobně roli hraje i vyšší podíl předčasně narozených dětí. V řadě zemí vzestup úzce souvisí s klesající hodnotou cut off pro TSH [8]. Aktuální velmi diskutovanou otázkou proto je nastavení optimálních hodnot cut off pro TSH tak, aby se nezvyšovala falešná pozitivita a nedocházelo k tomu, že budou léčeni zdraví novorozenci s pouhou variantou normy.
V některých situacích je zvýšené riziko, že vrozená hypotyreóza bude maskována např. farmakologickou supresí TSH (např. po podání dopaminu nebo glukokortikoidů), opožděným vzestupem TSH při nezralosti hypothalamo-hypofyzární osy (typicky u dětí předčasně narozených, s velmi nízkou porodní hmotností, kriticky nemocných), nebo smíšením krve fetálními spojkami u diskordantních monozygotických dvojčat. Ve skupině novorozenců ohrožených falešně negativními výsledky primárního screeningu je proto doporučeno provádět sekundární screening [6]. V České republice se rescreening vrozené hypotyreózy provádí od roku 2002 u následujících vybraných rizikových novorozenců: s porodní hmotností pod 1500 g; jejichž matce byl v posledních 48 hodinách před porodem nebo novorozenci před odběrem screeningu podán celkově přípravek na bázi kortikoidů; jejichž matka byla v posledním trimestru těhotenství léčena tyreostatiky, léky s vysokým obsahem jodu (nikoli však běžnou suplementací jodidu v těhotenství), nebo jí byly podány jodové kontrastní látky; kteří byli léčeni před odběrem screeningu dopaminem, léky s obsahem jodu nebo jim byly podány jodové kontrastní látky; kterým byl podán transfuzní přípravek před odběrem screeningu. Rescreening se provádí mezi 8. a 14. dnem života nebo po dosažení požadované hmotnosti a jeho metodika je přesně, stejně jako metodika novorozeneckého screeningu, specifikována ve Věstníku MZ ČR z roku 2016 [9].
I v zemích, kde je zavedený celoplošný novorozenecký screening i rescreening vrozené hypotyreózy, je třeba na její klinický obraz nezapomínat. Novorozenecký screening založený na detekci zvýšeného TSH nemůže diagnostikovat centrální vrozenou hypotyreózu. Nelze vyloučit ani únik dítěte ze screeningu (porody doma, děti migrantů). Falešně negativní výsledek screeningu byl popsán u diskordantních monozygotických dvojčat (zkreslení screeningových hodnot dvojčete s vrozenou hypotyreózou příměsí krve zdravého dvojčete při fetofetální transfuzi nebo přes placentární vaskulární spojky) [10]. Negativní výsledek screeningu mohou mít děti, u nichž se hypotyreóza, byť na vrozeném podkladě (mírná forma dyshormonogeneze nebo dysgeneze), rozvíjí časně postnatálně [11].
Nelze opomenout, že celoplošný novorozenecký screening vrozené hypotyreózy dlouhodobě slouží také jako nástroj monitorace zásobení populace jodem (tab. 1) [12]. I na základě tohoto kritéria byla Česká republika v oficiální klasifikaci WHO/ICCIDD zařazena v roce 2004 mezi státy s dostatečnou jodovou saturací.
![Hodnoty neonatálního TSH jako ukazatel stupně jodového deficitu v populaci [12].](https://www.prelekara.sk/media/cache/resolve/media_object_image_large/media/image_pdf/37d81223820b20da47600880c0ae8418.jpeg)
KOMPLEXNÍ PÉČE O DĚTI S VROZENOU HYPOTYREÓZOU
Od zavedení novorozeneckého screeningu vrozené hypotyreózy byla mezi endokrinology snaha o jednotný přístup k léčbě a sledování těchto dětí, a proto již v roce 1993 byla publikována doporučení pracovní skupiny pro kongenitální hypotyreózu Evropské společnosti pro dětskou endokrinologii (ESPE) [13]. Dlouholetým členem této pracovní skupiny a spoluautorem doporučení byla také prof. MUDr. Olga Hníková, CSc. V roce 2014 vznikla nová rozsáhlá doporučení ESPE, která odpovídají současným poznatkům medicíny založené na důkazech a zahrnují všechny aspekty týkající se diagnostiky, léčby a komplexní péče o děti, dospívající a mladé dospělé s vrozenou hypotyreózou [6].
Potvrzení diagnózy a léčba vrozené hypotyreózy
Po záchytu zvýšeného TSH ve screeningu (u nás nad 15 mIU/l) je diagnózu nezbytné co nejdříve potvrdit příp. vyloučit stanovením sérové koncentrace fT4 a TSH. Screeningové hodnoty TSH nad 30 mIU/l jsou pro diagnózu vrozené hypotyreózy natolik suspektní, že vyžadují venózní odběr tentýž den.
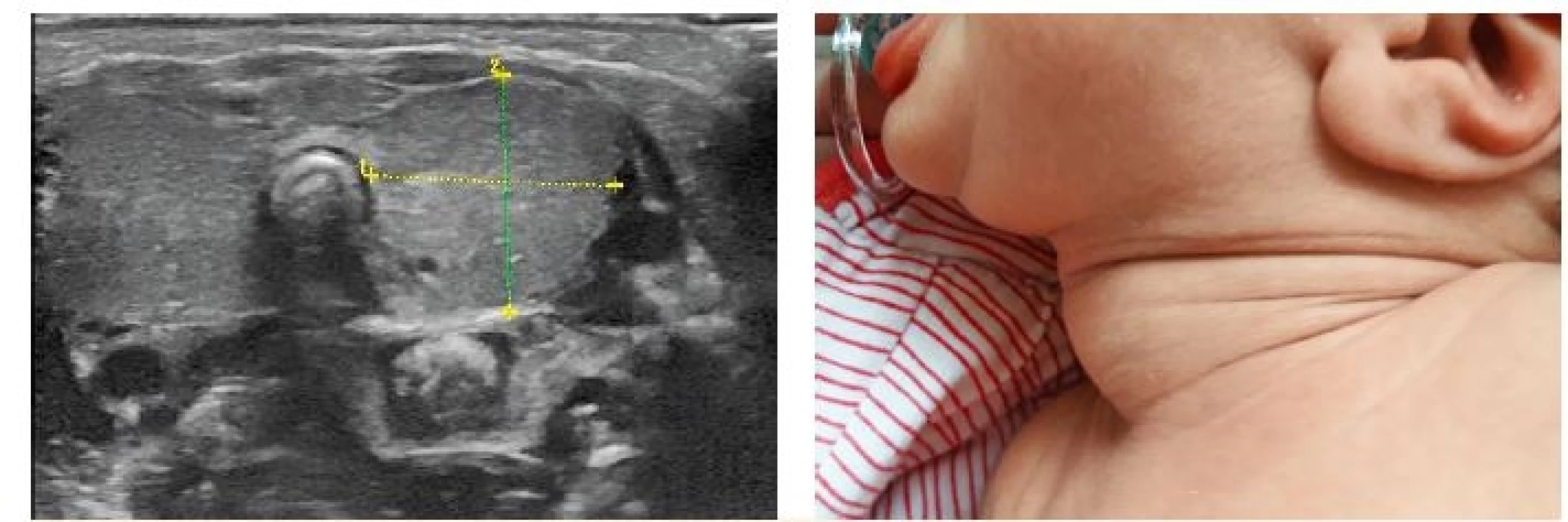
Stupeň závažnosti vrozené hypotyreózy lze posoudit podle klinických symptomů, sérových hormonálních hladin (těžká forma fT4 <5 pmol/l, mírná forma fT4 5–10 pmol/l, lehká forma fT4 >10 pmol/l), výsledku zobrazení štítné žlázy (normální štítná žláza, struma, tyreoidální dysgeneze) a posouzení kostní zralosti. Osifikace (míra opoždění zrání epifýz na RTG kolene podle Sénécala) je ve vztahu k hladině fT4 a slouží jako marker intrauterinní hypotyreózy a prognostický faktor [14]. V době stanovení diagnózy by mělo být provedeno zobrazení štítné žlázy, ale nesmí být důvodem odkladu léčby. Ultrazvukové vyšetření umožňuje stanovení struktury a objemu štítné žlázy (obr. 5) a tím přesnou diferenciální diagnostiku štítné žlázy in situ (struma u dyshormonogeneze, hypoplazie, hemityroidea u dysgeneze, normální štítná žláza). V případě ultrazvukového nálezu atyreózy nelze vyloučit tyreoidální ektopii, nález je třeba posoudit komplexně s ohledem na hladiny fT4, TSH a tyreoglobulinu. Ektopická štítná žláza je nejlépe detekovatelná scintigrafickým vyšetřením.
Fig. 5. Left: ultrasound of a large neonatal goiter – volume 6 ml, normal neonatal thyroid volume about 0.6 ml (provided by MUDr. Jaroslav Zikmund, CSc.). Right: corresponding clinical presentation (with parental consent).
U dětí s kongenitální hypotyreózou je nezbytností podrobné pediatrické vyšetření s cílem časné diagnostiky dalších vrozených vad a případně identifikace dysmorfických syndromů, jejichž součástí kongenitální hypotyreóza může být (pseudohypoparatyreóza, Pendredův syndrom, Downův syndrom, další syndromické formy vrozené hypotyreózy např. na podkladě defektu transkripčních faktorů). Vyšší výskyt a vyšší riziko vrozených vývojových vad byly u dětí s vrozenou hypotyreózou popsány již od první dekády novorozeneckého screeningu, který umožnil shromáždit větší soubory pacientů [15], a potvrdily ho i další rozsáhlé studie [16, 17]. Signifikantně vyšší výskyt strukturálních vrozených vývojových vad (zejména srdečních vad) je asociován s dysgenezí štítné žlázy. Po sdružených vrozených vývojových vadách je třeba aktivně pátrat, je vhodné proto doplnit echokardiografii a ultrazvukové vyšetření uropoetického traktu a centrálního nervového systému. Vůbec nejčastější sdruženou poruchou je senzorineurální porucha sluchu, která postihuje až 10 % dětí s vrozenou hypotyreózou (oproti asi 2,5 % běžné populace). U všech dětí s vrozenou hypotyreózou je proto povinné do 3 měsíců věku provést screening vrozených sluchových vad vyšetřením otoakustických emisí. Nepostačuje pouze vyšetření v novorozeneckém věku, vhodné je další kontrolní vyšetření během dětství [6, 9]. Patogeneze vrozených vad u dětí s kongenitální hypotyreózou je heterogenní, ve většině případů se nejspíše jedná o skutečnou asociaci – ať už v důsledku hypotyreózy per se, nebo v důsledku jednoho geneticky podmíněného molekulárního mechanismu. Je to např. defekt pendrinu vedoucí k poruše sluchu a tyreoidální dyshormonogenezi, defekt transkripčních faktorů NKX2.1 nebo FOXE 1 zodpovědný za poruchu vývoje štítné žlázy a dalších orgánů.
U novorozence a jeho matky je vhodné vyšetřit také tyreoidální autoprotilátky, zejména proti receptoru pro TSH (aTSHR neboli TRAK). Transplacentární přenos aTSHR od matky s atrofickou formou chronické lymfocytární tyreoiditidy může způsobit tranzientní vrozenou hypotyreózu novorozence. Tato situace vyžaduje léčbu obvykle do vymizení protilátek z cirkulace dítěte.
Léčba vrozené hypotyreózy musí být zahájena ihned, pokud je ve venózním vzorku TSH zvýšené a fT4 pod normu k věku, ale také při normálním fT4, pokud je TSH významně zvýšeno nad 20 mIU/l. U hodnot venózního TSH mezi 6–20 mIU/l při bezpečně normálním fT4 je postup individuální, každopádně je nutné sledovat dynamiku TSH a fT4 a přihlédnout k ultrazvukovému nálezu [6]. Léčba vrozené hypotyreózy syntetickým levotyroxinem má být u novorozence zahájena v dávce 10–15 μg/kg/den nejpozději do 14 dnů věku dítěte. Levotyroxin se podává v jedné denní dávce, rozdrcený v malém množství vody nebo mateřského mléka na lžičce nebo stříkačkou [6]. Aktuální doporučení již neuvádí striktní požadavek podávání ráno nalačno, důležité je podávání za stále stejných podmínek. Nicméně u dětí je návyk ranního podávání levotyroxinu nalačno s 30minutovým odstupem od snídaně výhodný. U dětí je doporučené preferovat originální přípravky, je třeba se vyvarovat tekutým formám připravovaným magistraliter a současně nepodávat z důvodů zhoršené absorpce preparáty obsahující železo nebo vápník. Po zahájení léčby kontrolujeme fT4 a TSH po 1–2 týdnech, vždy nejdříve za 4 hodiny po perorálním podání levotyroxinu. Cílem léčby je co nejrychlejší normalizace fT4 a dále udržovat fT4 v horní polovině normy k věku a TSH v mezích normy. Intervaly kontrol se postupně prodlužují (tab. 2) [18]. Elevace TSH jsou jako ukazatel nedostatečné substituce při příliš nízké dávce nebo nepravidelném užívání levotyroxinu nežádoucí. V léčbě je ale nutné se vyvarovat i předávkování a déletrvající suprese TSH s rizikem urychleného kostního zrání, negativního vlivu na kostní denzitu a nejspíše i neurokognitivní vývoj.
![Léčba a sledování dětí s vrozenou hypotyreózou (upraveno dle [18]).](https://www.prelekara.sk/media/cache/resolve/media_object_image_large/media/image_pdf/ee93ca7c75e6a02550eb8adb683d0449.jpeg)
Zajímavý je historický pohled na substituční léčbu u vrozené hypotyreózy. Prof. Čížková-Písařovicová, která k léčbě vrozené hypotyreózy mohla užívat pouze sušenou štítnou žlázu (preparát Thyreoidin), doporučovala: „S dávkami začátečními nutno zacházet opatrně vzhledem k ev. přecitlivělosti dítěte… U starších kreténů pak je lépe dávat menší dávky, neboť většími dávkami měnili bychom individuum, které je netečné, mírné a přístupné v individuum popudlivé, svéhlavé a nepřístupné...“ [1]. Prof. Hníková ve své monografii v roce 1989 [5] sice uvádí tehdy celosvětově doporučené dávkování levotyroxinu u novorozence 5–7 μg/kg/den, ale zdůrazňuje, že často bývá nutné použít dávky vyšší 8–10 μg/kg/den a předjímá tak pozdější moderní přístup k léčbě relativně vysokými iniciálními dávkami levotyroxinu 10–15 μg/kg/den [6, 13].
DLOHOUDOBÉ SLEDOVÁNÍ A PROGNÓZA DĚTÍ S VROZENOU HYPOTYREÓZOU
U některých dětí po 3. roce věku přistupujeme k reevaluaci tyreoidální osy přerušením léčby na 4–6 týdnů (případně redukcí dávky o 20–30 % na 2–3 týdny) s následným stanovením hormonálních hladin a případně zobrazením štítné žlázy [6]. Cílem je odlišit tranzientní a permanentní hypotyreózu (dojde k signifikantnímu vzestupu TSH) a rozhodnout pak o další potřebě substituční léčby. Reevaluace je indikovaná u dětí, u nichž nebyla provedena kompletní diagnostika v novorozeneckém věku, měly pozitivní tyreoidální autoprotilátky (transplacentárně přenesené), mají štítnou žlázu in situ, nebo vyžadují nízkou dávku levotyroxinu. Reevaluace není nutná u dětí s dysgenezí štítné žlázy, případně pokud došlo po 1. roce života během léčby k významnějším sekundárním vzestupům TSH.
Dlouhodobé sledování dětí s vrozenou hypotyreózou je komplexní, zahrnuje monitoraci hormonálních hladin, ultrazvukové kontroly a sledování somatického vývoje (růstová křivka a rychlost, váhová křivka, prořezávání a výměna dentice, později pubertální vývoj). Nezbytné je také pravidelné hodnocení psychomotorického a mentálního vývoje a vývoje řeči, později školních výsledků [6, 19]. Zvláštní pozornost v tomto ohledu vyžadují děti se zvýšeným rizikem ve smyslu těžké vrozené hypotyreózy nebo horší kompenzace. Při obtížích je nutná časná rehabilitace, nácvik pozornosti, paměti, logopedická a specifická školní péče. Obecně ale není doporučeno primárně informovat školská zařízení o diagnóze vrozené hypotyreózy, aby se předešlo případné stigmatizaci dítěte.
Více než 90 % dětí s kongenitální hypotyreózou diagnostikovanou novorozeneckým screeningem se vyvíjí normálně a dosahuje hodnot DQ/IQ srovnatelných se zdravými kontrolami. U méně než 10 % časně a adekvátně léčených dětí je vývoj méně příznivý, hlavními rizikovými faktory jsou nízká spolupráce při léčbě, nižší socioekonomický status, asociované vrozené vady nebo jiné komplikace. Pokud je léčba zahájena v prvních dvou týdnech života dávkou levotyroxinu 10–15 μg/kg/den, neliší se prognóza dětí s těžkou a mírnou vrozenou hypotyreózou [15]. Nejnovější studie potvrdily, že se děti s vrozenou hypotyreózou mohou vyvíjet po somatické a mentální stránce stejně jako jejich zdraví sourozenci, pokud se léčí iniciální vysokou substituční dávkou levotyroxinu nad 10 μg/kg//den, vedoucí k rychlé normalizaci fT4 (medián 16 dní věku), jsou intenzivně sledovány s častými kontrolami a bez výkyvů TSH a nemají genetická (Downův syndrom, defekt transkripčního faktoru) nebo jiná přídatná rizika [20]. Výzvou proto nadále zůstává co nejčasnější rozpoznání právě těchto rizikových jedinců, u nichž i přes časnou a adekvátní substituční léčbu lze očekávat méně příznivý průběh. Typicky jsou to děti se syndromickou formou vrozené hypotyreózy a asociovanými morfologickými a/nebo funkčními defekty CNS (Downův syndrom, pseudohypoparatyreóza, defekty transkripčních faktorů NKX2.1, FOXE 1 a další) [15, 21].
O něco méně optimistické jsou studie u mladých dospělých, ale je třeba vzít v potaz, že se jedná o první generaci dětí z počátku screeningových programů, kdy nastavení a časování léčby ještě nebylo zcela optimální. U mladých dospělých bylo popsáno vyšší riziko mírně nebo středně závažného chronického onemocnění – encefalopatie, epilepsie, psychiatrické onemocnění (u 5,7 % vs. 2,9 % u kontrol), postižení sluchu (9,5 % vs. 2,5 % u kontrol), problémů se zrakem (55,4 % vs. 47,9 % u kontrol) a nadváhy (22,8 % vs. 15,7 % u kontrol) [22]. Snížení plodnosti bylo zaznamenáno jen u žen s těžkou formou kongenitální hypotyreózy [23]. Současně studie ukázaly suboptimální kompenzaci hypotyreózy se zvýšením TSH u 20 % mladých dospělých [22].
I výše zmíněná data o suboptimální kompenzaci mladých dospělých s vrozenou hypotyreózou podtrhují fakt, že naprosto nezbytnou složkou v systému péče o děti s vrozenou hypotyreózou je kvalitní a kontinutální edukace na všech úrovních, tj. od zdravotnického personálu (velmi častá je nízká adherence k aktuálním doporučením), přes rodinu a dítě samotné [6, 15, 19]. Zcela klíčová je pak edukace a psychosociální podpora v období dospívání a při předání do dospělé péče. U dívek a mladých žen je nutné se zaměřit na budoucí graviditu, informovat je o nutnosti co nejčasnější kontroly tyreoidálního profilu a navýšení substituční dávky o 25–30 %.
Rodině by mělo být poskytnuto také genetické poradenství zahrnující informaci o dvou hlavních formách vrozené hypotyreózy (tyreoidální dysgeneze a dyshormonogeneze) a vysvětlení rizika opakování na základě morfologie štítné žlázy a výskytu v rodině. Molekulárně-genetické vyšetření není zejména pro svou nízkou výtěžnost u tyreoidální dysgeneze plošně dostupné a rutinně prováděné. Vždy mu má předcházet podrobná fenotypizace pacienta. Vhodnými kandidáty pro molekulárně-genetické vyšetření jsou zejména jedinci se syndromickou kongenitální hypotyreózou, jednak s cílem identifikace nových genů, jednak s ohledem na význam genetického poradenství v rodině.
Prenatální diagnostika vrozené hypotyreózy je vyhrazena pro případy závažné fetální strumy. Stanovení tyreoidální funkce plodu kordocentézou je indikováno jen v případech zvažované prenatální intervence, tj. intraamniotické aplikace levotyroxinu. Ta je nutná výjimečně, pokud má plod eutyroidní matky objemnou strumu s progredujícím polyhydramnionem, hrozící tracheální kompresí a rizikem předčasného porodu [6].
Do budoucna směřuje léčba vrozené hypotyreózy nejen k další optimalizaci, ale také k individualizaci – ke stanovení optimální individuální dávky levotyroxinu. Zatím k tomu ale nejsou dostupné vhodnější biochemické markery eutyroidního stavu, než je TSH. U jedinců s atyreózou se zvažuje možný efekt chybějící frakce T3 (trijodtyroninu) přímo syntetizované žlázou (až 25 % T3) a možnosti léčebného ovlivnění i preparáty T3 s delším poločasem [21].
Z globálního hlediska v péči o děti s vrozenou hypotyreózou však nadále hlavním cílem zůstává zavedení celoplošného novorozeneckého screenigu ve všech zemích. Screening vrozené hypotyreózy je totiž celosvětově realizován jen u 29,3 % novorozenců: 84,2 % narozených v Evropě, 82,3 % narozených v Americe, 37,8 % narozených v Africe a Oceánii a 24,4 % narozených v Asii. Každoročně tak má šanci na případnou časnou diagnózu a léčbu jen asi 37 ze 127 milionů narozených dětí. Mezi 90 miliony dětí, které tuto šanci nemají, je bohužel i ve 21. století (při incidenci vrozené hypotyreózy 1 : 3000) kolem 30 000 dětí s pozdně léčenou nebo vůbec neléčenou vrozenou hypotyreózou [8].
Došlo: 2. 2. 2018
Přijato: 3. 2. 2018
MUDr. Eva Al Taji, Ph.D.
Klinika dětí a dorostu
3. lékařská fakulta Univerzity Karlovy
Fakultní nemocnice Královské Vinohrady
Šrobárova 50 100 34 Praha 10
e-mail: evataji@email.cz
Sources
1. Čížková-Písařovicová J. Štítná žláza – thyreoidea. In: Čížková--Písařovicová J. Klinická endokrinologie dětského věku. Praha: Státní zdravotnické nakladatelství, 1954 : 37–40.
2. Hubble D. Diseases of the thyroid gland. In: Hubble D. Paediatric Endocrinology. Oxford and Edinburgh: Blackwell Scientific Publication, 1969 : 113–154.
3. Klein AH, Meltzer S, Kenny FM. Improved prognosis in congenital hypothyroidism treated before age three months. J Pediatr 1972; 81 (5): 912–915.
4. Vulsma T, Gons MH, de Vijlder JJ. Maternal-fetal transfer of thyroxine in congenital hypothyroidism due to a total organification defect or thyroid agenesis. N Engl J Med 1989; 321 (1): 13–16.
5. Hníková O, Kračmar P. Kongenitální hypotyreóza. 1. vyd. Praha: Avicenum, 1989 : 1–100.
6. Léger J, Olivieri A, Donaldson M, et al. On behalf of ESPE-PES-SLEP--JSPE-APEG-APPES-ISPAE, and the Congenital hypothyroidism consensus conference group. European society for paediatric endocrinology consensus guidelines on screening, diagnosis, and management of congenital hypothyroidism. Horm Res Paediatr 2014; 81 (2): 80–103.
7. Grosse SD, Van Vliet G. Prevention of intellectual disability through screening for congenital hypothyroidism: how much and at what level? Arch Dis Child 2011; 96 (4): 374–379.
8. Ford G, LaFranchi SH. Screening for congenital hypothyroidism: a worldwide view of strategies. Best Pract Res Clin Endocrinol Metab 2014; 28 (2): 175–178.
9. Metodický návod k zajištění novorozeneckého laboratorního screeningu a následné péče. In: Věstník Ministerstva zdravotnictví České republiky 2016; částka 6 : 2–12.
10. Perry R, Heinrichs C, Bourdoux P, et al. Discordance of monozygotic twins for thyroid dysgenesis: implications for screening and for molecular pathophysiology. J Clin Endocrinol Metab 2002; 87 (9): 4072–4077.
11. Al Taji E, Biebermann H, Límanová Z, et al. Screening for mutations in transcription factors in a Czech cohort of 170 patients with congenital and early-onset hypothyroidism: identification of a novel PAX8 mutation in dominantly inherited early-onset non-autoimmune hypothyroidism. Eur J Endocrinol 2007; 156 (5): 5212–5229.
12. Indicators for asssessing iodine deficiency disorders and their control through salt iodization. Geneva: WHO, ICCIDD, UNICEF, 1994 : 1–53.
13. Grüters A, Delange F, Giovannelli G, et al. Guidelines for neonatal screening programmes for congenital hypothyroidism. Working group on congenital hypothyrodisim of the European society for paediatric endocrinology. Eur J Pediatr 1993; 152 (12): 974–975.
14. Wasniewska M, De Luca F, Cassio A, et al. In congenital hypothyroidism bone maturation at birth may be a predictive factor of psychomotor development during the first year of life irrespective of other variables related to treatment. Eur J Endocrinol 2003; 149 (1): 1–6.
15. Grüters A, Jenner A, Krude H. Long-term consequences of congenital hypothyroidism in the era of screening programmes. Best Pract Res Clin Endocrinol Metab 2002; 16 (2): 369–382.
16. Olivieri A, Stazi MA, Mastroiacovo P, et al. A population-based study of the frequency of additional congenital malformations in infants with congenital hypothyroidism: data from the Italian registry for congenital hypothyroidism (1991-1998). J Clin Endocrinol Metab 2002; 87 (2): 557–562.
17. Azar-Kolakez A, Ecosse E, Dos Santos S, et al. All-cause and disease-specific mortality and morbidity in patients with congenital hypothyroidism treated since the neonatal period: a national population-based study. J Clin Endocrinol Metab 2013; 98 (2): 2012–2731.
18. Lebl J, Al Taji E, Koloušková S, et al. Tyreopatie. In: Lebl J, Al Taji E, Koloušková S, et al. Dětská endokrinologie a diabetologie. Praha: Galén, 2016 : 189–235.
19. Donaldson M, Jones J. Optimising outcome in congenital hypothyroidism, current opinions on best practice in initial assessment and subsequent management. J Clin Res Pediatr Endocrinol 2013; 5 (Suppl 1): 13–22.
20. Albert BB, Heather N, Derraik JSB, et al. Neurodevelopmental and body composition outcomes in children with congenital hypothyroidism treated with high-dose initial replacement and close monitoring. J Clin Endocrinol Metab 2013; 98 (9): 3663–3670.
21. Krude H, Kühnen P, Biebermann H. Treatment of congenital thyroid dysfunction: achievements and challanges. Best Pract Clin Endocrinol Metab 2015; 25 : 399–413.
22. Léger J, Ecosse E, Roussey M. Subtle health impairment and socioeducational attainment in young adult patients with congenital hypothyroidism diagnosed by neonatal screening: a longitudinal population-based cohort study. J Clin Endocrinol Metab 2011; 96 (6): 1771–1782.
23. Hassani Y, Larroque B, Dos Santos S, et al. Fecundity in young adults treated early for congenital hypothyroidism is related to the initial severity of the disease: a longitudinal population-based cohort study. J Clin Endocrinol Metab 2012; 97 (6): 704–710
Labels
Neonatology Paediatrics General practitioner for children and adolescentsArticle was published in
Czech-Slovak Pediatrics

2018 Issue 3
- What Effect Can Be Expected from Limosilactobacillus reuteri in Mucositis and Peri-Implantitis?
- The Importance of Limosilactobacillus reuteri in Administration to Diabetics with Gingivitis
Most read in this issue
- C3 glomerulopatie – nově definovaná klinická jednotka
- Současný pohled na diagnostiku a léčbu astmatu u dětí
- Novinky v kardiopulmonální resuscitaci – „guidelines 2018“
- Makro AST jako příčina asymptomatické elevace aspartátaminotransferázy