
Pheochromocytoma and paraganglioma syndromes: going on 2010
Pheochromocytomas and paragangliomas are chromaffin cell tumors that produce, store (e.g. in vesicles), metabolize (e.g. to yield metabolites such as normetanephrine or metanephrine), and secrete (release) catecholamines (e.g. norepinephrine and epinephrine, or dopamine). The metabolism of catecholamines is a more consistent process than that of catecholamine secretion. Pheochromocytomas are found in the adrenal gland; closely related tumors of extra-adrenal location are classified as paragangliomas. Most pheochromocytoma and paraganglioma tumors (PTTs) represent sporadic tumors but about 20–30% of these tumors are familial. Mutations in six genes to date have been identified to be responsible for familial PTTs:
- the von Hippel-Lindau (VHL) gene leading to VHL syndrome;
- the RET gene leading to multiple endocrine neoplasia type 2;
- the neurofibromatosis type 1 (NF-1) gene associated with von Recklinghausen’s disease; and 4 and 5. mutations of genes encoding the B, C, and D subunits of mitochondrial succinatedehydrogenase (SDHB, SDHC, and SDHD) associated with familial PTTs. The presence of catecholamine excess reflects various clinical signs and symptoms associated with PTTs. Hypertension is the most common sign and may be sustained or paroxysmal. Numerous independent studies have now confirmed that measurements of fractionated metanephrines (i.e. normetanephrine and metanephrine measured separately) in urine or plasma provide superior diagnostic sensitivity over measurement of the parent catecholamines. Current localization of PTTs should be based on the use of anatomical as well as specific functional imaging studies to proof that a tumor is indeed pheochromocytoma or paraganglioma. Laparoscopic surgery is now the technique of first choice for resection adrenal and extraadrenal PTTs. All patients with PTTs should receive appropriate preoperative medical management to block the effects of released catecholamines (blocking the synthesis and the action of catecholamines). Currently, there is no curative treatment for malignant PTTs.
Key words:
pheochromocytoma, paraganglioma, catecholamines, metanephrines, neuroendocrine, familial, metaiodobenzylguanidine scintigraphy, postitron emission tomography.
Authors:
Karel Pacak 1; Tamara Prodanov 1; Graeme Eisenhofer 3; Karen Adams 1; Vitaly Kantorovich 2
Authors‘ workplace:
Reproductive Biology and Medicine
Branch, The Eunice Kennedy Shriver National
Institute of Child Health and Human
Development (NICHD), MD, USA
1; Division of Endocrinology and Metabolism
University of Arkansas for Medical
Sciences, Little Rock, Arkansas, USA
2; Departments of Medicine and Clinical
Chemistry, University of Dresden, Dresden
Germany
3
Published in:
Ces Urol 2010; 14(1): 5-15
Category:
Review article
Overview
Pheochromocytomas and paragangliomas are chromaffin cell tumors that produce, store (e.g. in vesicles), metabolize (e.g. to yield metabolites such as normetanephrine or metanephrine), and secrete (release) catecholamines (e.g. norepinephrine and epinephrine, or dopamine). The metabolism of catecholamines is a more consistent process than that of catecholamine secretion. Pheochromocytomas are found in the adrenal gland; closely related tumors of extra-adrenal location are classified as paragangliomas. Most pheochromocytoma and paraganglioma tumors (PTTs) represent sporadic tumors but about 20–30% of these tumors are familial. Mutations in six genes to date have been identified to be responsible for familial PTTs:
- the von Hippel-Lindau (VHL) gene leading to VHL syndrome;
- the RET gene leading to multiple endocrine neoplasia type 2;
- the neurofibromatosis type 1 (NF-1) gene associated with von Recklinghausen’s disease; and 4 and 5. mutations of genes encoding the B, C, and D subunits of mitochondrial succinatedehydrogenase (SDHB, SDHC, and SDHD) associated with familial PTTs. The presence of catecholamine excess reflects various clinical signs and symptoms associated with PTTs. Hypertension is the most common sign and may be sustained or paroxysmal. Numerous independent studies have now confirmed that measurements of fractionated metanephrines (i.e. normetanephrine and metanephrine measured separately) in urine or plasma provide superior diagnostic sensitivity over measurement of the parent catecholamines. Current localization of PTTs should be based on the use of anatomical as well as specific functional imaging studies to proof that a tumor is indeed pheochromocytoma or paraganglioma. Laparoscopic surgery is now the technique of first choice for resection adrenal and extraadrenal PTTs. All patients with PTTs should receive appropriate preoperative medical management to block the effects of released catecholamines (blocking the synthesis and the action of catecholamines). Currently, there is no curative treatment for malignant PTTs.
Key words:
pheochromocytoma, paraganglioma, catecholamines, metanephrines, neuroendocrine, familial, metaiodobenzylguanidine scintigraphy, postitron emission tomography.
Introduction
While being a part of the diffuse neuroendocrine system, pheochromocytoma and paraganglioma tumors (PPTs) occupy a unique niche in human pathology due to diverse clinical presentations, possible cure, but potentially fatal course if undiagnosed. Prevalence of PPTs in Western countries is estimated around 1 : 6.500 to 1 : 2.500, with an annual incidence in the United States of 500 to 1,600 cases per year (1). About 85% of PPTs are caused by adrenal tumors, while the majority of the rest are extraadrenal that commonly arise from a collection of chromaffin tissue around the origin of the inferior mesenteric artery (the organ of Zuckerkandl) or aortic bifurcation (2, 3). Although 20–30% of PPTs are familial and display multicentric and bilateral features, histopathological characteristics of all PPTs are uniform. Metastases may be rare for adrenal (about 10%) and familial (less than 5%) pheochromocytomas (4), the prevalence increases to 36–50% for extra - adrenal abdominal pheochromocytomas, especially ones related to SDHB mutations (5). Finally, up to 10% of intra-adrenal pheochromocytomas show local recurrence (6, 7).
PPTs concurrently show number of similarities and dissimilarities to other neuroendocrine tumors (NETs), including carcinoids (Table 1). These are rare tumors requiring a high index of suspicion for diagnosis and are known to fiercely masquerade as common benign conditions. Despite being part of diffuse neuroendocrine system, PPTs are excluded from “regular” NET classification and seem to have even inconsistencies with their own. None of the proposed classifications equally captures most significant characteristics of PPTs: localization and neuronal origin. When one concentrates on localization only, than all PPTs are called paragangliomas and further subdivision has them as adrenal and extra-adrenal. This classification misses very significant features of neuronal cell origin, where intra - and extra-adrenal intra-abdominal pheochromocytomas are appreciated as derived from sympathetic nervous systemassociated chromaffin tissue, typically produce catecholamines and are clinically symptomatic; while head and neck paragangliomas derived from parasympathetic tissue are rarely active biochemically and symptoms are mostly related to space-occupying phenomena.
In addition, PPTs express a striking number of dualities within themselves (Table 1) and this update will review current state of the art through these dualities.
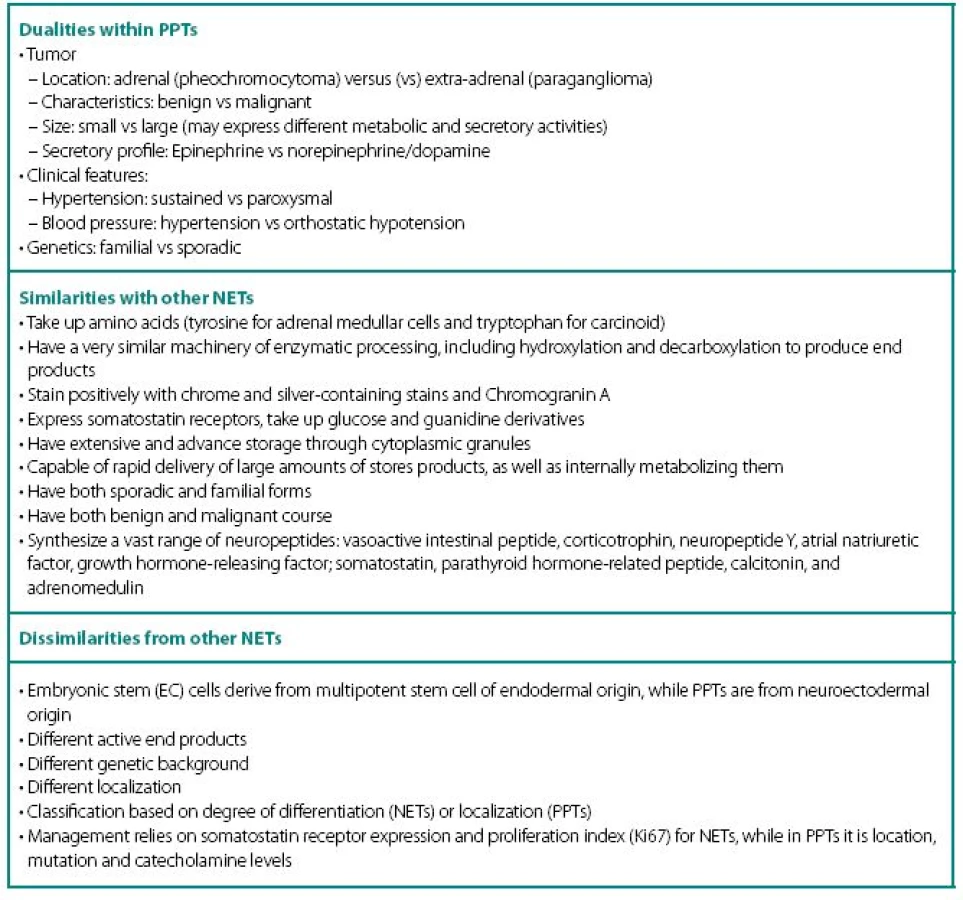
Clinical Features
There is an appearing paradox with clinical assessment of PPTs. Recurrent episodes of anxiety with transient increases in blood pressure are readily suspected as pheochromocytoma, but rarely found to be ones. On the other hand, sustained hypertension with somewhat fluctuating readings is rarely seen as potential PPTs and over the years can progress to cardiac failure, result in hypertensive crisis during surgery or become malignant. One also needs to remember that in familial cases both presentation and the course significantly differ from sporadic PPTs. On the other hand, 4–10% of adrenal insidentalomas show up to be PPTs, while another 5% are diagnosed during unrelated surgeries (8, 9).
Despite a wide variety of clinical features of PPTs, the main remains to be hypertension (Table 2). It can be sustained, paroxysmal or even a combination of both (10, 11). Headache is common feature of PPTs and when combined with palpitations and sweating in patients with hypertension, should readily rise a red flag (10, 12–16). Episodes of sweating and pallor are common, but depend on timing of observation in relation of hypercatecholaminemia and co-secreted neuropeptides (see below). While classic attack will present with “cold sweats” and pallor, post-attack vasodilatation usually shows local hyperemia with flushing, which may raise suspicion of other NETs, like carcinoid. Hypercatecholaminemic spells of PPTs occur with variable frequency and usually last less than an hour. Attacks may be precipitated by palpation of the tumor, postural changes, exertion, anxiety, trauma, pain, ingestion of foods or beverages containing tyramine (certain cheese, beer, and wine), use of certain drugs (histamine, glucagon, tyramine, phenothiazine, metoclopramide, adrenocorticotropic hormone), intubation, induction of anesthesia, chemotherapy, endoscopy, catheterization, and micturition or bladder distention (with bladder tumors) (12).
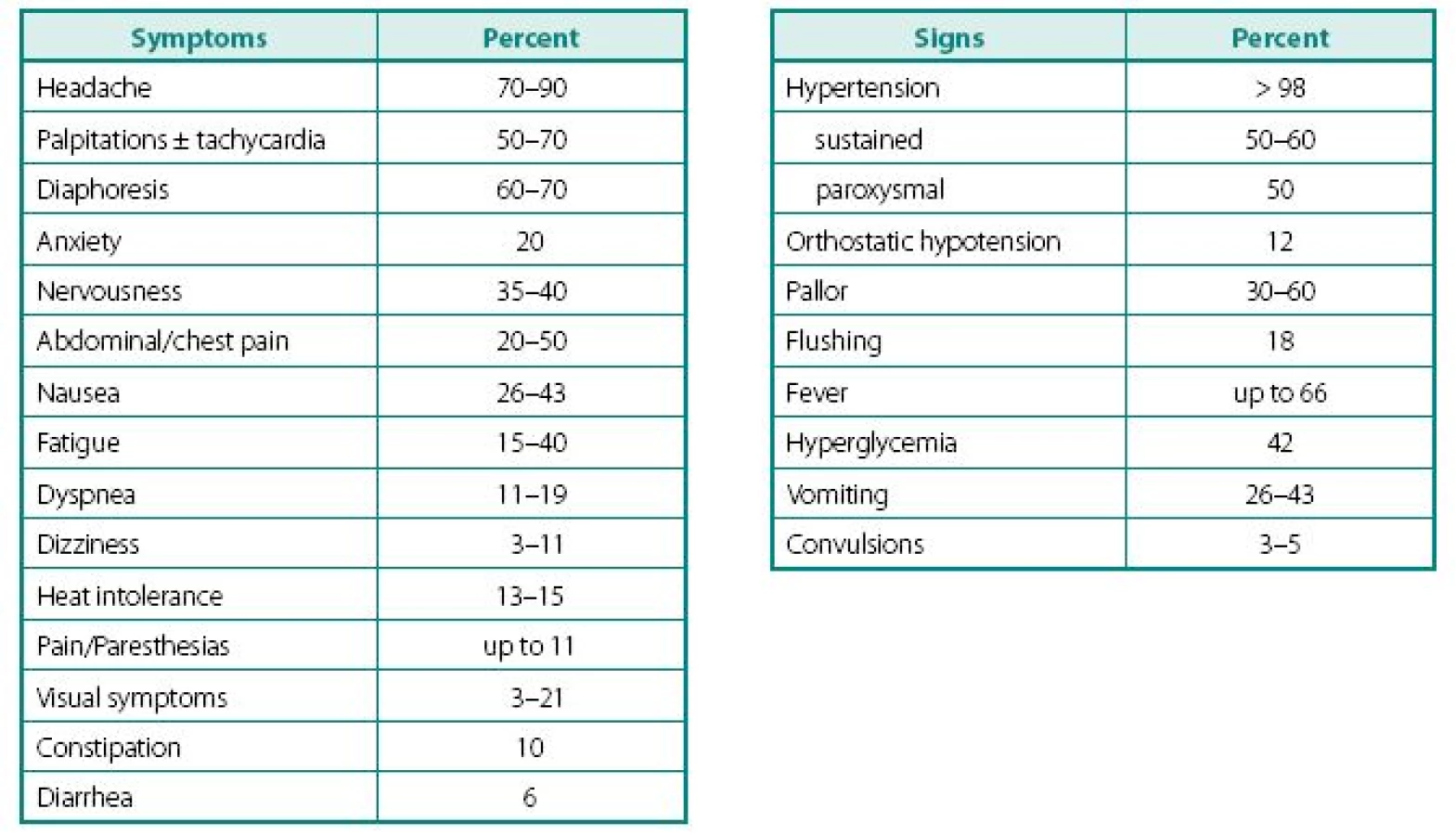
Other common complaints are palpitations and dyspnea, weight loss despite normal appetite (caused by catecholamine-induced glycogenolysis and lipolysis), visual problems during an attack and profound tiredness and polyuria most commonly experienced after an attack. Most patients also present with severe episodes of anxiety, nervousness, or panic attacks. Less frequent clinical manifestations include fever of unknown origin (hypermetabolic state) and constipation (15, 17).
Highly variable symptomatology of PPTs relate to hypercatecholaminemic content, as well as co-secretion of neuropeptides, characteristic to other NETs: vasoactive intestinal peptide (watery diarrhea and hypokalemia), corticotrophin (Cushing’s syndrome), neuropeptide Y, atrial natriuretic factor, growth hormone-releasing factor (acromegaly); somatostatin (diabetes), parathyroid hormonerelated peptide (hypercalcemia), calcitonin (flushing, diarrhea), etc. Structural complications of PPTs include pain from bony and other metastases, as well as signs and symptoms of compression of neighboring structures by mass itself. It is also thought that smaller tumors are metabolically more active and might secrete higher content of catecholamines, while the larger ones overgrow own blood supply and have lower metabolic activity, lesser secretory capacity, but higher probability of malignant transformation. As with other NETs, distant metastases may have unique secretory profile that may differ from the original tumor.
Because most of symptoms and signs of PPTs are nonspecific and relatively common in general population, proper diagnosis relay on high index of suspicion as well as working through extensive differential diagnosis (Schema 1).

Sporadic versus Familial PPT
There is a recent shift in the popular paradigm that familial PPTs are rare. Discovery of genetic basis of paragangliomas and deeper look into frequency of familial pheochromocytoma syndromes has established the prevalence of close to third of all PPTs (2, 18). Most importantly, between 12–24% of tumors with no obvious syndrome or family history appear to be due to otherwise unsuspected germline mutation. The significance of this fact lies not only in overall frequency, but also in difference of the disease phenotype (see below). In addition, discovery of cellular events that lead to familial syndromes significantly advanced our understanding PPTs tumorogenesis.
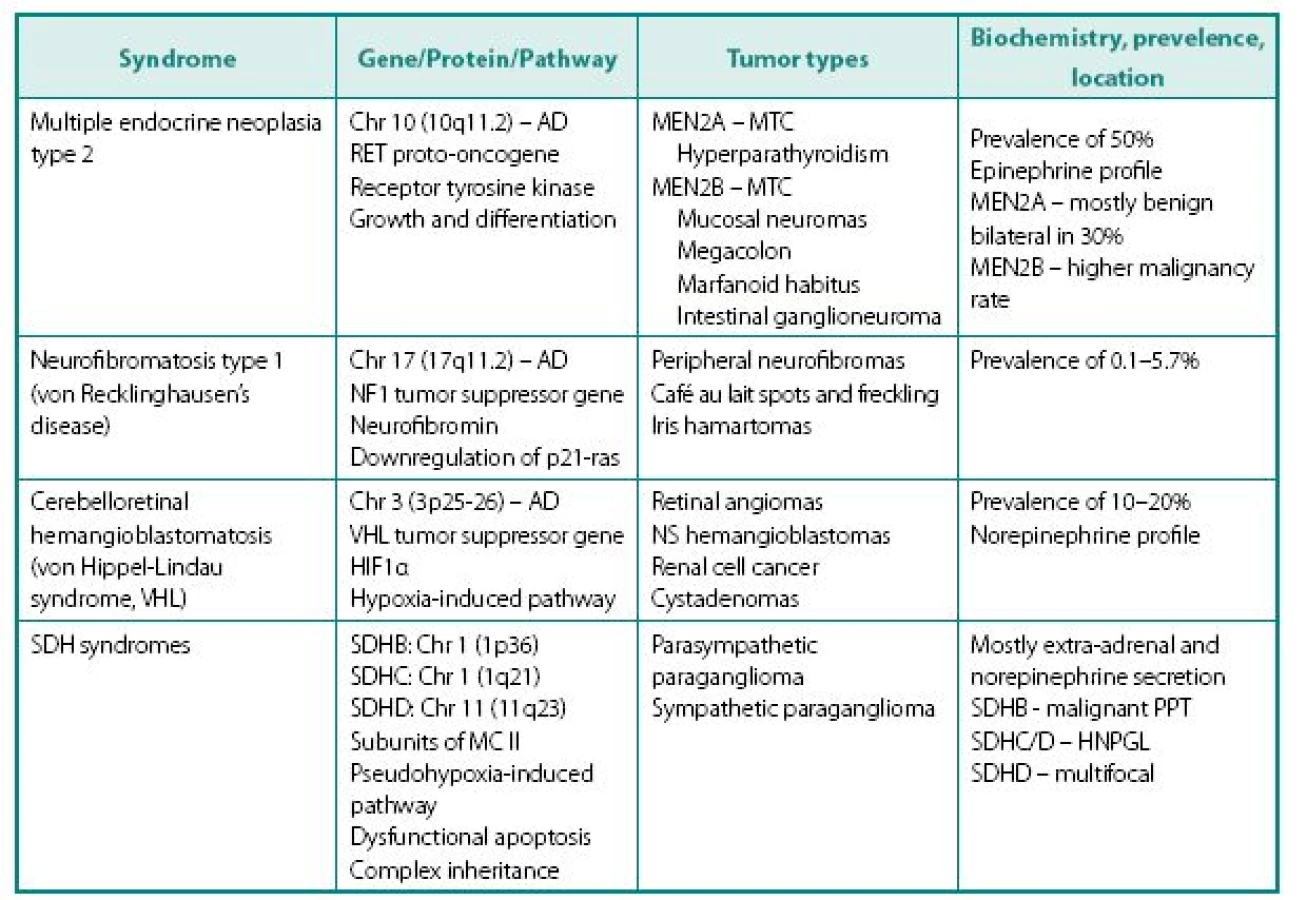
Following distinct familial syndromes are associated with PPTs (see Table 3 for details): von Hippel-Lindau (VHL) syndrome, von Recklinghausen’s disease (NF1), multiple endocrine neoplasia type 2, and PPT associated with mutations in distinct subunits of succinate dehydrogenase (SDHB, SDHC, and SDHD). Mutation testing, now routinely available for four of the above genes (RET, VHL, SDHB, and SDHD). Pheochromocytoma can rarely occur in multiple endocrine neoplasia type 1 associated with mutation of menin gene.
Clinical presentation of familial PPTs is different from sporadic counterparts in several ways:
- Presence of non-PPT-related features of the syndrome, which can include medullary thyroid cancer (MTC) in patients with MEN 2, renal cell cancer in VHL etc.
- Different frequency of the tumor occurrence in specific syndrome – while paragan gliomas are the mainstay of SDH tumors, they are relatively frequent in MEN2 (50%), but rare in NF1 (about 2%).
- Familial PPTs are diagnosed at younger age, compared to their sporadic counterparts, but this may be related not only to the earlier age of development, but also due to biochemical and genetic screening of proband’s relatives.
- In MEN2A MTC always presents before PPT because of much higher penetrance – this alone prompts further screening.
- Presence of genotype-phenotype correlation: in VHL 98% of PPTs associate with missense rather than other type of gene mutation, while mutations at codons 634 (2A) and 918 (2B) are mostly causing pheochromocytoma in MEN2.
- Cellular enzymatic milieu of familial PPTs is considerably different from sporadic cases, because of much lower expression of tyrosine hydroxylase (rate-limiting enzyme in catecholamine synthesis pathway) and phenylethanolamine-N-methyltransferase (norepinephrine to epinephrine converting enzyme) in VHL results in noradrenergic biochemical profile and low overall catecholamine tissue levels. PPTs of MEN2 have usually an adrenergics biochemical profile. Negligible amounts tyrosine hydroxylase also explain the phenomena of non-secreting paragangliomas that are able to uptake MIBG and have high content of chromogranin A.
- Other characteristics of familial PPTs. In MEN2 tumors are invariable adrenal, frequently bilateral and usually benign. PPTs of VHL are very similar to MEN2, although extra-adrenal tumors in VHL patients are more common and biochemical profile is different (see above). Although mutations of SDHB and SDHD genes are occasionally associated with solitary adrenal tumors, patients with these mutations most commonly present with extra-adrenal PPTs localized throughout the body, including head and neck areas. These tumors can be either biochemically active or silent. PPTs expressing SDHB mutations carry a high risk for malignant disease (19).
Diagnosis of PPT
Current diagnostic approach to PPTs is affected by recent discoveries in physiology of these tumors, and includes biochemical diagnosis and tumor localization studies. The last can be further subdivided into functional and anatomical procedures.
The fact that catecholamines are metabolized within chromaffin cells to metanephrines (norepinephrine to normetanephrine and epinephrine to metanephrine) and this intracellular process occurs independently of catecholamine release, suggests that assessment of metanephrines should have a clear diagnostic superiority. In line with these concepts, numerous independent studies have now confirmed that measurements of fractionated metanephrines (i.e. normetanephrine and metanephrine measured separately) in urine or plasma provide superior diagnostic sensitivity over measurement of the parent catecholamines (20). Measurements of fractionated metanephrines in either urine or plasma are available, but plasma values show better diagnostic accuracy (21). An elevation of more than 4-fold above normal range is associated with close to 100% probability of the tumor, while lower values still suggest high probability of tumor. The conditions under which blood or urine samples are collected can be crucial to the reliability and interpretation of test results and extensively discussed elsewhere. When false-positive results, driven by sympathetic over-stimulation, are suspected clonidine suppression test combined with measurements of plasma catecholamines and normetanephrine can assist with differentiation. Measurements of plasma dopamine or free methoxytyramine (dopamine metabolite) can be used to diagnose rare extra-adrenal dopamine-secreting PPTs (22).
Anatomical tumor localization studies include computed tomography (CT) or magnetic resonance imaging (MRI) (23) and are recommended for initial tumor localization. If there is a strong biochemical evidence of PPTs, initial studies should focus on abdomen and pelvis. If extra-abdominal tumor is suspected, appropriate imaging should include head, neck and chest. One should never forget the possibility of either active or silent metastases and always include appropriate imaging into their workup. Although CT and MRI have excellent sensitivity for detecting most catecholamine-producing tumors, these anatomical imaging approaches lack the specificity required to unequivocally identify a mass as a pheochromocytoma.
Functional localization studies are based on the fact that all NETs are capable of preferable uptake of different metabolites by virtue of specific transporters or receptors expression (23). These metabolites include: 1. guanide analogues (123I-labeled meta-iodobenzylguanide (123I-MIBG) scintigraphy), 2. somatostatin analogues (111In-octreotide scanning), 3. glucose analogue (18F-fluorodeoxyglucose positron emission tomography (PET), and 4. catecholamine analogues (18F-fluorodopamine, 18F-fluorodopa or 11C-hydroxyephedrine PET) (24, 25). Of the above studies, only 123I-MIBG scintigraphy and catecholamine analogues PET are specific for PPTs, while only 123I-MIBG scintigraphy is widely available and should be used as an initial functional localization study. Octreoscan can be positive in all NETs, while 18F-fluorodeoxyglucose PET shows generally increased metabolic rate and can be positive in any malignancy. Despite their lower specificity, these imaging techniques have high sensitivity and can be used in cases, where there is a high possibility of PPT (positive biochemical tests) and negative 123I-MIBG scintigraphy. The discussion on need for functional images is somewhat nonsensical, because of relatively high frequency of extra-adrenal, functionally silent, multifocal, malignant and metastatic PPTs and possibility of these being missed on single-site anatomical imaging study. Currently, additional isotopes are available elsewhere (yttrium, lutetium) and near future will show their superiority to current methodology. Another recent advance in functional imaging is use of fusion studies that combine anatomical and functional techniques. These add anatomical relevance of increased functional uptake and are incredibly informative when positive.
In general, diagnostic approach has to be driven by event and tailored to each specific case rather than standardized into a stiff algorithm (Figure 1). While there are numerous algorithms initiated by clinical suspicion of PPT, the case is different for adrenal incidentaloma. An absolutely different scenario would represent incidental surgical finding of tumor that might represent a PPT. In this case, postsurgical catecholamine levels will be normal (if tumor did not metastasize), while pathology report will be of extreme importance to diagnose possible PPT, functional studies to confirm tumor singularity and possible SDHB mutation analysis (in appropriate cases) top assess prognosis.

Management of PPTs
The definitive treatment of PPTs is surgical, preferably laparoscopic, excision of the tumor. As with other potentially complicated tumors, procedure should be performed by specialized experienced surgeon. With this said, it is hard to underestimate the importance of proper pre - and post-surgical medical management. If inappropriate, uncontrolled hypercatecholaminemia can result in cardiac arrhythmias, high output congestive heart failure (CHF) and other well-feared devastating complications of PPTs, as well as associate with poor surgical outcome. Excessive manipulations with the tumor or inability of adequate ligation of draining veins during the surgery can result in rapid burst of large amount of catecholamines into systemic circulation, which can precipitate hypertensive crises and arrhythmias. Over-medication after surgery can associate with profound hypotension, while failure to recognize multifocal disease can associate with rebound hypertension.
Despite continuous debates about preferable medical management, current approach includes initial adequate α-adrenergic blockade with or without subsequent β-blockade (26, 27). Phenoxybenzamine (Dibenzyline), an α-adrenoceptor blocker, is most commonly used agent, while calcium channel blockers and selective α1-adrenoceptor blocking agents, such as Hytrin and Cardura can also be used for preoperative blockade of catecholamine-induced vasoconstriction. A β-adrenoceptor blocker may be used for preoperative control of arrhythmias, tachycardia or angina, but never alone because loss of β-adrenoceptor-mediated vasodilatation in a patient with unopposed catecholamine-induced vasoconstriction can precipitate a hypertensive crisis. In all patients before surgery and in some patients on whom elevated blood pressure and arrhythmia cannot be controlled otherwise, α-methyl-para-tyrosine (metyrosine, Demser™), a competitive inhibitor of tyrosine hydroxylase, can be used to decrease the amount of synthesized catecholamines. It is imperative to timely suspect a true hypertensive crisis, because lack of immediate aggressive treatment can result in permanent organ damage or vascular catastrophes. Intravenous therapy and appropriate cardiovascular monitoring are essential; medications can include phentolamine, labetalol or sodium nitroprusside.
The long-term follow up is essential for patients with PPTs, especially in familial cases, in order to diagnose recurrence or malignant transformation. Normal postoperative biochemical test results do not exclude remaining microscopic disease and about 17% of tumors recur, with about 50% of these showing signs of malignancy. Currently, there is no methodology that would allow prediction of future recurrence of possible malignancy based on pathological examination of a resected tumor.
Malignant PPT
The incidence of metastatic pheochromocytoma ranges from 3% to 36% or even higher, depending on the genetic background and location of the primary tumor (4, 5, 28–30). Location of metastatic lesions appears to affect patient’s survival: it is significantly shortened in those with metastatic lesions in liver and lungs, compared to bone metastases (31, 32). The overall 5-year survival rate varies between 34% and 60%. Presence of the SDHB mutation, large size or an extra-adrenal location of the primary tumor increase the likelihood of future metastatic disease, but sporadic PPTs can recur and transform into malignant disease too and should not be forgotten.
Overall, the management of malignant PPTs is disappointing – as tumor dedifferentiates, it looses ability to take up guanide analogues, while 131I-MIBG therapy is the single most valuable adjunct to surgical treatment of malignant pheochromocytomas. Tumor debulking palliates symptoms and may facilitate subsequent radiotherapy or chemotherapy, although none of those statements has been proven. External-beam irradiations of bone metastases, tumor embolization, or radiofrequency ablation provide some treatment alternatives. Chemotherapy with a combination of cyclophosphamide, vincristine and dacarbazine can provide tumor regression and symptoms relief in up to 50% of patients, but the responses are usually short (33, 34).
Received: 4. 6. 2009.
Accepted: 25. 6. 2009.
Corresponding author
Karel Pacak, MD, PhD., D.Sc.
Section on Medical Neuroendocrinology,
RBMB, NICHD, NIH Building 10, CRC,
1-East,
Room 1-3140,
10 Center Drive,
MSC-1109 Bethesda,
Maryland 20892-1109
e-mail: karel@mail.nih.gov
Sources
1. Pacak K, Chrousos GP, Koch CA, Lenders JW, Eisenhofer G. Pheochromocytoma: progress in diagnosis, therapy, and genetics. In: Margioris A, Chrousos GP eds. Adrenal Disorders. 1 ed. Totowa: Humana Press 2001; 479–523.
2. Elder EE, Elder G, Larsson C. Pheochromocytoma and functional paraganglioma syndrome: no longer the 10% tumor. J Surg Oncol 2005; 89 : 193–201.
3. Manger WM, Gifford RW. Pheochromocytoma. J Clin Hypertens 2002; 4 : 62–72.
4. Whalen RK, Althausen AF, Daniels GH. Extra-adrenal pheochromocytoma. J Urol 1992; 147 : 1–10.
5. O’Riordain DS, Young WF, Jr., Grant CS, Carney JA, van Heerden JA. Clinical spectrum and outcome of functional extraadrenal paraganglioma. World J Surg 1996; 20 : 916–921.
6. Remine W, Chong G, van Heerden J, Sheps S, Harrison EJ. Current management of pheochromocytoma. Ann Surg 1974; 179 : 740–748.
7. Klingler HC, Klingler PJ, Martin JK, Jr., Smallridge RC, Smith SL, Hinder RA. Pheochromocytoma. Urology 2001; 57 : 1025–1032.
8. Bravo E. Evolving concepts in the pathophysiology, diagnosis, and treatment of pheochromocytoma. Endocr Rev 1994; 15 : 356–368.
9. Mantero F, Terzolo M, Arnaldi G, Osella G, Masini AM, Ali A, Giovagnetti M, Opocher G, Angeli A. A survey on adrenal incidentaloma in Italy. Study Group on Adrenal Tumors of the Italian Society of Endocrinology. J Clin Endocrinol Metab 2000; 85 : 637–644.
10. Manger W, Gifford R. Clinical and Experimental Pheochromocytoma. Cambridge, Massachusetts: Blackwell Science 1996.
11. Streeten DH, Anderson GH Jr. Mechanisms of orthostatic hypotension and tachycardia in patients with pheochromocytoma. Am J Hypertens 1996; 9 : 760–769
12. Lenders JW, Eisenhofer G, Mannelli M, Pacak K. Phaeochromocytoma. Lancet 2005; 366 : 665-675.
13. Bravo EL, Gifford RW Jr. Pheochromocytoma. Endocrinol Metab Clin North Am 1993; 22 : 329–341.
14. Bravo EL, Tagle R. Pheochromocytoma: state-of-the-art and future prospects. Endocr Rev 2003; 24 : 539–553.
15. Ram CV, Fierro-Carrion GA. Pheochromocytoma. Semin Nephrol 1995; 15 : 126–137.
16. Steen RE, Kapelrud H, Haug E, Frey H. In vivo and in vitro inhibition by ketoconazole of ACTH secretion from a human thymic carcinoid tumour. Acta Endocrinol (Copenh) 1991; 125 : 331–334.
17. Bouloux PG, Fakeeh M. Investigation of phaeochromocytoma. Clin Endocrinol (Oxf) 1995; 43 : 657–664.
18. Neumann HP, Bausch B, McWhinney SR, Bender BU, Gimm O, Franke G, Schipper J, Klisch J, Altehoefer C, Zerres K, Januszewicz A, Eng C, Smith WM, Munk R, Manz T, Glaesker S, Apel TW, Treier M, Reineke M, Walz MK, Hoang-Vu C, Brauckhoff M, Klein-Franke A, Klose P, Schmidt H, Maier-Woelfle M, Peczkowska M, Szmigielski C. Germ-line mutations in nonsyndromic pheochromocytoma. N Engl J Med 2002; 346 : 1459–1466.
19. Timmers H, Gimenez-Roqueplo AP, Mannelli M, Pacak K. Clinical aspects of SDHx-related pheochromocytoma and paraganglioma. Endocr Relat Cancer 2009; 16 : 391–400.
20. Lenders JW, Pacak K, Walther MM, Linehan WM, Mannelli M, Friberg P, Keiser HR, Goldstein DS, Eisenhofer G. Biochemical diagnosis of pheochromocytoma: which test is best? JAMA 2002; 287 : 1427–1434.
21. Eisenhofer G, Siegert G, Kotzerke J, Bornstein SR, Pacak K. Current progress and future challenges in the biochemical diagnosis and treatment of pheochromocytomas and paragangliomas. Horm Metab Res 2008; 40 : 329–337.
22. Eisenhofer G, Goldstein DS, Sullivan P, Csako G, Brouwers FM, Lai EW, Adams KT, Pacak K. Biochemical and clinical manifestations of dopamine-producing paragangliomas: utility of plasma ethoxytyramine. J Clin Endocrinol Metab 2005; 90 : 2086–2075.
23. Ilias I, Pacak K. Current approaches and recommended algorithm for the diagnostic localization of pheohcromocytoma. J Clin Endocrinol Metab 2004; 89 : 479–491.
24. Mamede M, Carrasquillo JA, Chen CC, Del Corral P, Whatley M, Ilias I, Ayala A, Pacak K. Discordant localization of 2-[18F]-fluoro-2-deoxy-D-glucose in 6-[18F]-fluorodopamineand [123I] - metaiodobenzylguanidine-negative metastatic pheochromocytoma sites. Nucl Med Commun 2006; 27 : 31–36.
25. Timmers HJ, Eisenhofer G, Carrasquillo JA, Chen CC, Whatley M, Ling A, Adams KT, Pacak K. Use of 6-[18F]-fluorodopamine positron emission tomography as first-line investigation for the diagnosis and localization of non-metastatic and metastatic pheochromocytoma. Clin Endocrinol (Oxf) 2009; 71 : 11–17.
26. Pacak K. Preoperative management of the pheochromocytoma patient. J Clin Endocrinol Metab 2007; 92 : 4069–4079.
27. Pacak K, Eisenhofer G, Ahlman H, Bornstein SR, Gimenez-Roqueplo AP, Grossman AB, Kimura N, Mannelli M, McNicol AM, Tischler AS. Pheochromocytoma: recommendations for clinical practice from the First International Symposium. Nat Clin Pract Endocrinol Metab 2007; 3 : 92–102.
28. Goldstein RE, O’Neill JA, Jr., Holcomb GW, 3rd, Morgan WM, 3rd, Neblett WW, 3rd, Oates JA, Brown N, Nadeau J, Smith B, Page DL, Abumrad NN, Scott HW Jr. Clinical experience over 48 years with pheochromocytoma. Ann Surg 1999; 229 : 755–764.
29. John H, Ziegler WH, Hauri D, Jaeger P. Pheochromocytomas: can malignant potential be predicted? Urology 1999; 53 : 679–683.
30. Proye CA, Vix M, Jansson S, Tisell LE, Dralle H, Hiller W. “The” pheochromocytoma: a benign, intra-adrenal, hypertensive, sporadic unilateral tumor. Does it exist? World J Surg 1994; 18 : 467–472.
31. Yu J, Pacak K Metastatic pheochromocytoma. Endocrinologist 2002; 12 : 291–299.
32. Mundschenk J, Lehnert H. Malignant pheochromocytoma. Exp Clin Endocrinol Diabetes 1998; 106 : 373–376.
33. Averbuch SD, Steakley CS, Young RC, Gelmann EP, Goldstein DS, Stull R, Keiser HR. Malignant pheochromocytoma: effective treatment with a combination of cyclophosphamide, vincristine, and dacarbazine. Ann Intern Med 1988; 109 : 267–273.
34. Huang H, Abraham J, Hung E, Averbuch S, Merino M, Steinberg SM, Pacak K, Fojo T. Treatment of malignant pheochromocytoma/paraganglioma with cyclophosphamide, vincristine, and dacarbazine: recommendation from a 22-year follow-up of 18 patients. Cancer 2008; 113 : 2020–2028.
Labels
Paediatric urologist Nephrology UrologyArticle was published in
Czech Urology

2010 Issue 1
Most read in this issue
- Aggressive prostate cancer in patients with low PSA
- Anatomical implications of sex reassignment surgery in male-to-female transsexualism and follow-up study
- Laparoscopic nephropexis – technique with three non-absorbable stitches
- Prostate cancer incidence, diagnostic and treatment in HIV-positive patients