
Kde končí a začíná diagnóza Ewingova sarkomu - popis dvou neobvyklých kostních nádorů s translokací t(20;22)(EWSR1-NFATc2)
Where does Ewing sarcoma end and begin - two cases of unusual bone tumors with t(20;22)(EWSR1-NFATc2) alteration
The authors present two cases of Ewing-like sarcoma of the humerus and femur of a 12-year-old boy and a 28-year-old male, respectively. Identical morphology in both tumors consisted of multiple solid nests with a mosaic collection of small, round, uniform cells with clear cytoplasm and no apparent nuclear atypia. A monotonous structural arrangement, including both rich vascularity of bordering septae and significant admixtures of eosinophil leucocytes, resulted in a final organoid “neuroendocrine-like” pattern. Immunohistochemistry revealed diffuse strong CD10, CD99 and CD138 positivity. Detailed molecular analysis in both tumors confirmed translocation t(20;22) resulting in an EWSR1-NFATc2 fusion gene. Additionally, this translocation was accompanied by amplification of the proximal part of the genes and surrounding areas. Clinically, both neoplasms behaved aggressively and they were primarily chemoresistant. Four years later, the patient with the lesion in the humerus developed a massive local recurrence with a disruption of osteosynthesis. The last follow-up disclosed suspicious metastatic deposits in the lung. The boy with the femoral tumor underwent a total femoral prosthesis and there are no signs of local or systemic recurrence after 11 months of follow-up.
The authors discuss the taxonomic placement of these rare examples of Ewing-like sarcoma family in the light of new molecular discoveries.
Keywords:
bone – humerus – femur – small blue round cell tumor – Ewing sarcoma – Ewing-like sarcoma – t(20;22)(EWSR1-NFATc2) – amplification
:
Zdeněk Kinkor 1; Tomáš Vaneček 1; Marián Švajdler jr. 2; Petr Mukenšnabl 1; Karel Veselý 3; Jan Baxa 4; Milan Kokavec 5
:
Bioptická laboratoř s. r. o., Šiklův ústav patologie, LF UK, Plzeň
1; Oddělení patologie, Univerzitní nemocnice L. Pasteura, Košice
2; I. patologicko-anatomický ústav, LF MU a FN u sv. Anny, Brno
3; Klinika zobrazovacích metod, LF UK, Plzeň
4; Ortopedická klinika, Univerzitní nemocnice Akademika Dérera, Bratislava
5
:
Čes.-slov. Patol., 50, 2014, No. 2, p. 87-91
:
Original Article
Popisovány jsou dva případy primárního „Ewing-like“ sarkomu ve femuru 12letého chlapce a humeru 28letého muže. Morfologie obou nádorů byla shodná a sestávala ze solidních hnízd mozaikovitě uspořádaných, kulatých buněk se světlou cytoplazmou a pravidelnými jádry. Výsledné organoidní uspořádání, včetně bohaté septální vaskularizace a významné příměsi eozinofilních leukocytů, připomínalo neuroendokrinní tumor. Imunohistochemickým vyšetřením byla zjištěna pozitivita antigenů CD10, CD99, CD138. Molekulární analýza prokázala u obou sarkomů přítomnost fúzního transkriptu EWSR1-NFATc2 vycházející z translokace t(20;22), jež byla zároveň doprovázena amplifikací částí těchto genů a jejich přilehlých oblastí. Oba nádory se chovaly agresívně a byly primárně chemorezistentní. Léze v humeru 4 roky po resekci rozsáhle místně recidivovala s rozpadem osteosyntézy a v plicích byly zjištěny suspektní metastázy. Tumor femuru byl definitivně řešen totální femorální endoprotézou a hoch je po 11 měsících od stanovení diagnózy bez recidivy. Diskutováno je taxonomické postavení těchto vzácných lézí v rámci heterogenní skupiny tzv. „Ewing-like“ sarkomu.
Klíčová slova:
pažní kost – stehenní kost – Ewingův sarkom – Ewing-like sarkom – translokace t(20;22)(EWSR1-NFATc2) – amplifikace
Ewingův sarkom/PNET (ES) je po osteosarkomu druhým nejčastějším maligním tumorem kostí v dětském věku (1). K jeho spolehlivé diagnostice v současné době již nestačí pouze klasický histologický obraz hematoxylín-eozínového preparátu, který může být neodlišitelný od biologicky a histogeneticky rozmanité skupiny tzv. nádorů z malých tmavých buněk jako např. lymfoblastického lymfomu, neuroblastomu, rhabdomyosarkomu, mezenchymálního chondrosarkomu, Wilmsova tumoru, malobuněčného osteosarkomu, adamantinomu, desmoplastického kulatobuněčného malobuněčného tumoru, a dalších. Molekulárně genetický průkaz zlomu/translokace EWSR1 genu společně s detekcí konkrétního fúzního partnera je nyní pokládán za standard přesné diagnostiky ES určující léčbu a prognostické výhledy. Výsledky molekulárních analýz dokládají nejen vysokou „zlomovou potenci“ EWSR1 genu, ale především pestrou škálu možných tzv. „driving“ translokací, široce přesahující kategorii malobuněčného, kulatobuněčného sarkomu Ewingova typu (2-9). Nejčastějším typem molekulární alterace ES jsou translokace t(11;22)(EWSR1-FLI1) a t(21;22)(EWSR1-ERG1), reprezentující více jak 95% vyšetřených tumorů (10-14). Tito fúzní partneři genu EWSR1 náleží do skupiny vysoce homologních tzv. ETS („E26 transforming sequence“) transkripčních faktorů. Vzácně se pak v onkogenezi ES uplatňuje fúze EWSR1 i s dalšími ETS geny, jako např. ETV1, ETV4 a FEV (11,12,15). Celá tato skupina tumorů bývá v literatuře často označována jako tzv. „Ewing-family“, kde dosud nebyl hodnověrně prokázán prognostický ani prediktivní význam jednotlivých aberací ani jejich podtypů (10,16). Plošné vyšetřování molekulárního profilu kulatobuněčných sarkomů Ewingova typu odhalilo posléze další ojedinělé fúzní možnosti, kde však vedle EWSR1 alternovali partneři mimo okruh ETS genů např. t(1;22) (ZGS), t(2;22) (SP3), t(4;22) (SMARCA5), t(6;22) (POU5F1) a t(20;22) (NFATc2) (12,15,17). Pro toto malé uskupení řádově jednotek tumorů se pracovně užívá termín „Ewing-like family“. Publikované léze se vyskytovaly ve skeletu i měkkých tkáních a chovaly se vesměs velmi agresivně. I přesto, že společným jmenovatelem byl malobuněčný vzhled a CD99 pozitivita, mnozí autoři vzápětí spekulují o tom, že ve skutečnosti by se mohlo jednat o jiné neoplázie, byť fenotypově příbuzné.
Předkládáme dvě pozorování vyjímečného sarkomu kosti, u nichž neobvyklý mikroskopický vzhled a identické molekulární pozadí (včetně unikátní amplifikace fúzního genu EWSR1-NFATc2) věrně kopírovaly nálezy čtyř izolovaných případů uveřejněných dosud pouze v jediné práci (17).
MATERIÁL A METODIKA
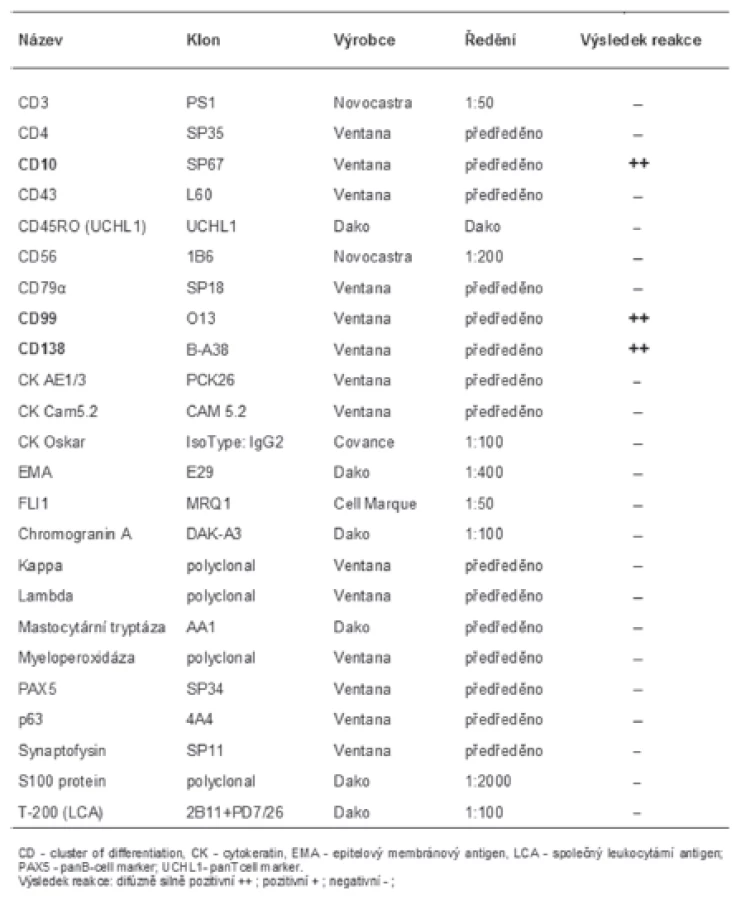
Materiál byl fixován v 10% formolu a zalit do parafínu (FFPE); pro barvení hematoxylinem eozínem a pro fluorescenční „in situ“ hybridizaci (FISH) byly krájeny řezy silné 2 mm, pro imunohistochemické vyšetření řezy silné 4 mm. Imunohistochemické vyšetření bylo prováděno elektronicky nastaveným protokolem v automatu BenchMark ULTRA, VENTANA/Roche. K vizualizaci reakce byl použit diaminobenzidin tetrahydrochlorid, k dobarvení jader metylénová modř. Seznam použitých protilátek, výrobce, klon a ředění jsou uvedeny v tabulce (tab. 1).
FISH analýza byla provedena prostřednictvím EWSR1 „break apart“ sondy (VYSIS/Abbott, Molecular, IL, USA ) a následně též pomocí NFATc2 „break apart“ sondy vyrobené z BAC (bakteriální arteficiální chromosom) klonů RP5-827A12 a RP5-1106N18 (BlueGnome, Cambridge, UK). DNA byla izolována za použití QIAsympohony DNA Mini kitu (Qiagen, Hilden, Germany) na automatizovaném extrakčním systému QIAsymphony SP (Qiagen) dle protokolu výrobce pro FFPE vzorky (Purification of genomic DNA from FFPE tissue using the QIAamp DNA FFPE Tissue Kit and Deparaffinization Solution). Ke komparativní genomové hybridizaci na čipu (aCGH) bylo využito sklo NimbleGen 385K Human CGH WG-T v2.0 (Roche NimbleGen Inc., Madison, WI, USA). RNA byla extrahována pomocí RecoverAll Total Nucleic Acid Isolation kitu (Ambion, Austin, TX, USA). Reverzní transkripce (RT) byla provedena kitem Transcriptor First Strand cDNA Synthesis Kit (Roche Diagnostics, Mannheim, Germany). K detekci fúzního transkriptu EWSR1-NFATc2 byla využita RT-PCR s HotStar Taq PCR Master Mixem (Qiagen) a s primery amplifikujícími oblast předpokládané fúze.
Sekvenační analýza pro ověření specifity fúzního transkriptu byla provedena pomocí Big Dye Terminator Sequencing kitu (Applied Biosystems, Foster City, CA, USA).
POPIS PŘÍPADŮ
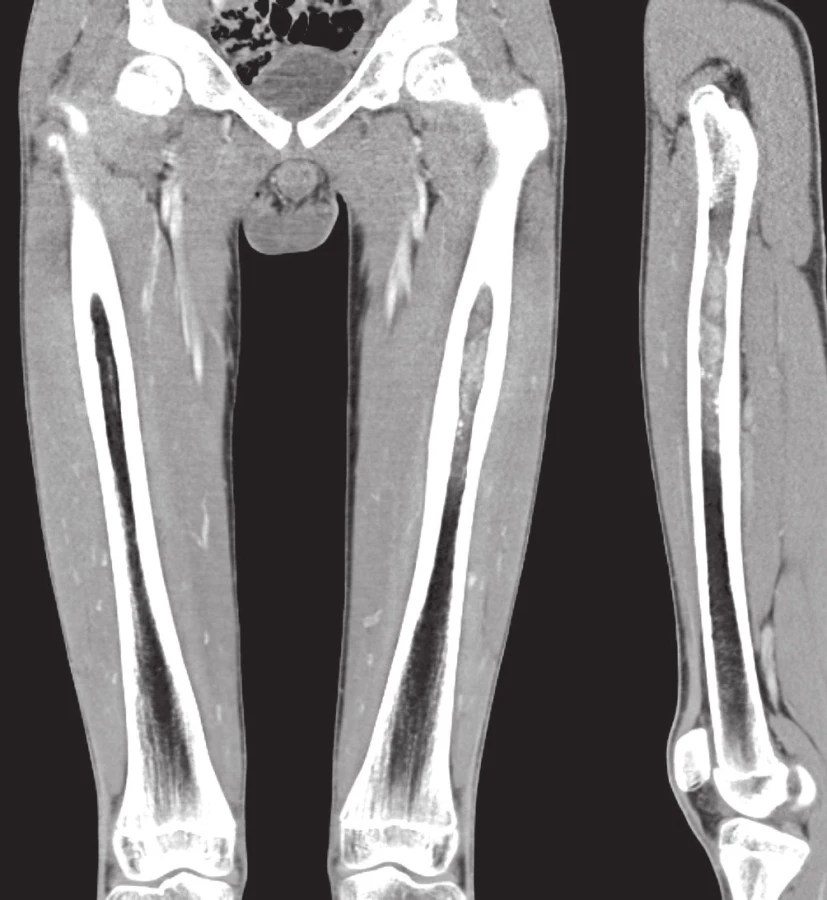
Případ 1. U 28letého muže s neustupujícími bolestmi v levé paži byla prokázána osteolýza střední části diafýzy humeru (obr. 1A). V probatorní biopsii a následné resekci 4 cm dlouhého úseku kosti s náhradou aloštěpem byla s jistými rozpaky stanovena diagnóza difúzního velkobuněčného B maligního lymfomu (DLBCL). „Staging“ potvrdil izolované postižení skeletu a nemocný podstoupil chemoterapii dle standardního protokolu. Po čtyřech letech došlo k rozsáhlé místní recidivě, rozrušení fixační osteosyntézy a masívní měkkotkáňové propagaci tumoru (obr. 1B). Rebiopsie měla obdobný mikroskopický vzhled. Nově přizpůsobené léčebné schéma bylo bez efektu; pacient žije posledních 5 měsíců se suspektním metastatickým rozsevem v plicích.
Případ 2. U 12lého chlapce s trvalou bolestí ve stehně byl zjištěn tumor diafýzy levého femuru v rozsahu 7 cm, bez rozrušení kompakty a známek šíření procesu do okolních měkkých tkání (obr. 1C). Po histologickém závěru v.s. Ewingova sarkomu následovala příslušná neoadjuvantní chemoterapie, která však byla klinicky/radiologicky evidentně neúčinná. Přistoupilo se tedy k totální endoprotéze femuru a hoch je 11 měsíců od stanovení diagnózy bez známek místní či systémové recidivy.


VÝSLEDKY
Morfologický vzhled byl u obou případů totožný. Převažující solidní úpravu nádoru doprovázela bohatá kapilární vaskularizace zvýrazňující dojem organoidního upořádání, které připomínalo neuroendokrinní tumor typu karcinoidu či paragangliomu (obr. 2A). Mozaikovitě seskupené nádorové buňky měly malá kulatá, pravidelná jádra s ojedinělou mitotickou aktivitou bez atypických figur. Objemná, nápadně světlá cytoplazma se zřetelnými konturami výsledných solidních hnízd kontrastovala s výrazně červenou, zrnitě granulovanou cytoplazmou hojné příměsi téměř všudypřítomných eozinofilů (obr. 2B). Až na ojedinělá drobná ložiska nekrózy vypadal tumor blandně, uniformně až monotónně, bez cytologických atypií či jaderného pleomorfizmu. V nádorových elementech se histochemicky nepodařilo prokázat glykogen, hlen ani lipidy.
Imunohistochemickým vyšetřením byla zjištěna difúzní, silná pozitivita CD10, CD99 a CD138 (obr. 2C). Další panel použitých protilátek reagoval kompletně negativně (tab. 1).



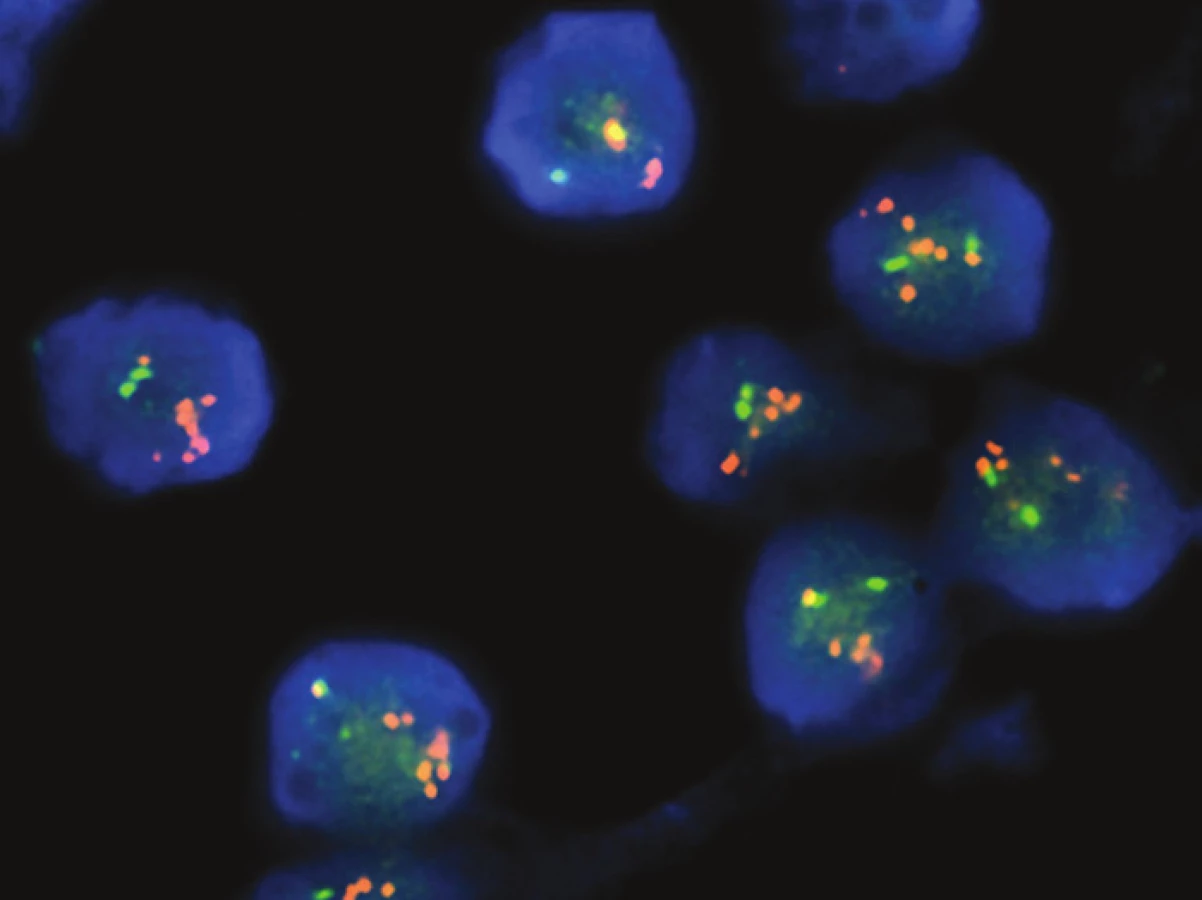
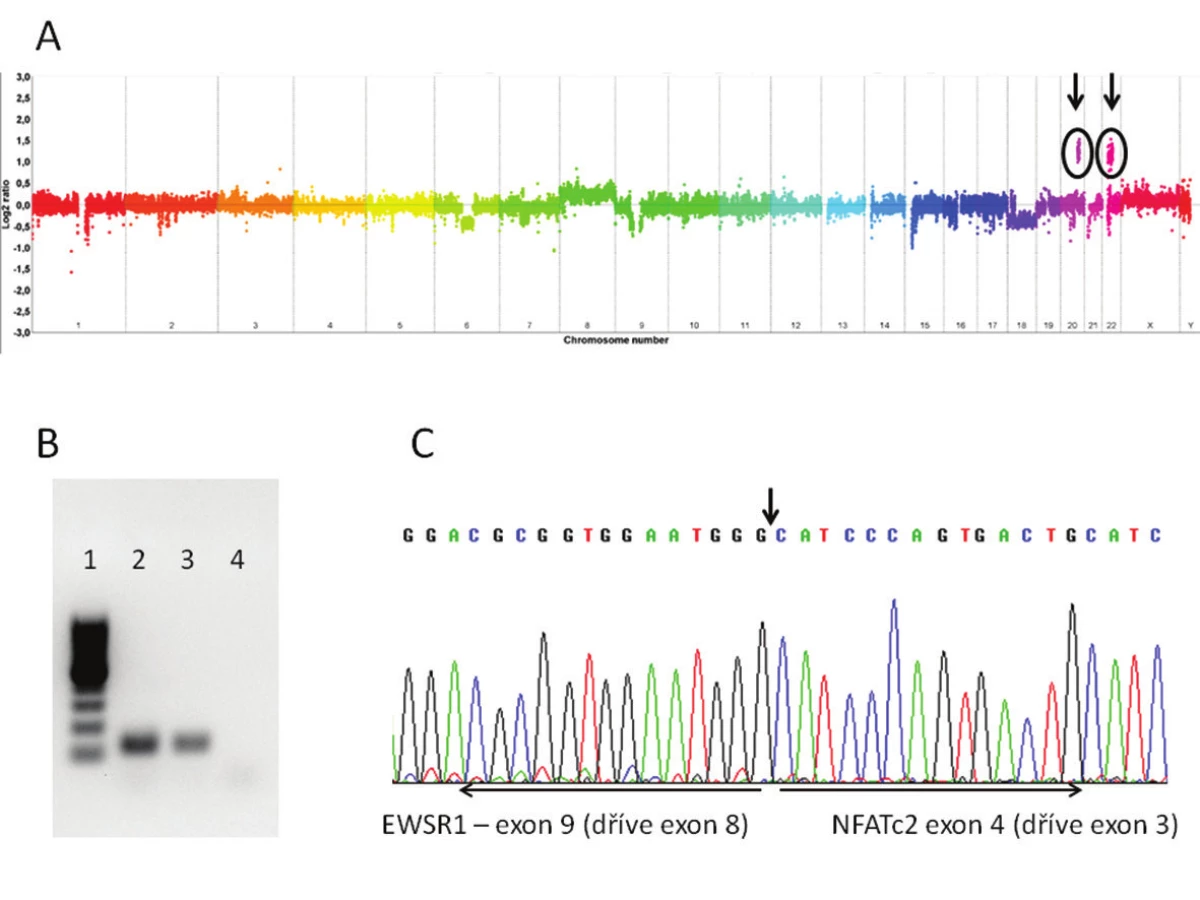
Molekulárně genetickým vyšetření byl nejprve u obou případů pomocí FISH zjištěn zlom genu EWSR1 doprovázený amplifikací proximálně situované části sondy (obr. 2D). Následně byla provedena aCGH, při jejíž analýze byl, mimo dalších změn, nalezen markantní zisk („gain“) části chromosomů 20 a 22 (obr. 3A). Zisk v oblasti chromosomu 20 zasahoval i část genu NFATc2, zisk v oblasti chromosomu 22 zahrnoval též část genu EWSR1. Po provedení této analýzy byla designována „break apart“ sonda k FISH průkazu zlomu genu NFATc2 a současně s tím primery k detekci fúzního transkriptu EWSR1-NFATc2. FISH analýza prokázala zlom genu NFATc2 s amplifikací proximální částí sondy. Současně s tím byl detekován fúzní transkript spojující exon 9 (dříve v literatuře exon 8) genu EWSR1 s exonem 4 (dříve v literatuře exon 3) genu NFATc2 (obr. 3B,C).
DISKUZE
Molekulárně genetická analýza specifických markerů je pro přesnou diagnostiku některých nádorů klíčová a např. diferenciální diagnostika malobuněčných kulatobuněčných tumorů dětského věku jasně ukazuje, že standardní HE preparát se škálou imunohistochemie, byť v rukou zkušeného specialisty, prostě již nestačí. Na druhou stranu poznatky získané rutinním genetickým vyšetřováním naznačují, že molekulární mechanismy jsou velmi složité, a že některé zdánlivě definující genové alterace jsou jen dílčím momentem celého komplexu dějů, dosud neprobádaných, které mohou být patogeneticky někdy významnější než konkrétní známý defekt DNA. Obecná a velmi zjednodušená představa patobiologie Ewingova sarkomu spočívá v tom, že ETS úsek fúzního genu rozhoduje o specifickém vazebném místě na DNA s následnou aktivací/inaktivací příslušných cílových genů. Tvrdí se, že tato specifita je společná/jedinečná pro všechny funkčně homologní geny skupiny ETS jako jsou FLI, ERG, ETV1, ETV4 a FEV (12,17). Proč však výsledný kulatobuněčný, malobuněčný morfo-/fenotyp je pak ale téměř shodný i u spojení EWSR1 s naprosto odlišnými geny mimo ETS spektrum - tedy tzv. „Ewing-like family“, není zcela jasné.
Podobně nesrozumitelnou paralelou též může být histogeneticky různorodá třída úplně jiných „non Ewingovských“ nádorů, kde se EWSR1 patogeneticky uplatňuje - např. světlobuněčný sarkom měkkých tkání a jeho „gastrická analogie“, atypický fibrózní histiocytom, primární plicní myxoidní sarkom, salivární světlobuněčný hyalinizující karcinom, myoepiteliom měkkých tkání atd. (3-9). Zde paradoxně molekulární partnerství totožných genů dá vzniknout histologicky a biologicky naprosto rozdílným nádorům. Jednoznačné vysvětlení tohoto zdánlivě nelogického fenoménu zatím neexistuje; uvažuje se jak o epigenetických, transkripčních či posttranslačních dějích, tak o principielní důležitosti histotypu původního buněčného substrátu, ve kterém příslušná genetická aberace proběhne (12).
Konečně, nedávno se v písemnictví objevilo několik izolovaných sdělení popisujících nově nalezené aberace u nediferencovaného kulatobuněčného „Ewing-like“ sarkomu nezasahující gen EWSR1; jde konkrétně o translokaci t(4;19)(CIC-DUX4) a intrachromozomální fúzi (BCOR-CCNB3) na chromozomu X (18-20). Většina autorů zde již otevřeně obhajuje stanovisko, že v rámci heterogenní tzv. „Ewing-like family“ existují tumory, které díky nespecifické morfologii nelze spolehlivě definovat jinak než geneticky.
Ve světle výše uvedených skutečností se tedy zdá celá skupina molekulárně značně heterogenního tzv. „Ewing-like“ sarkomu poněkud umělá. Méně než dvě desítky dosud publikovaných sporadických případů spojuje nespecifický zlom EWSR1 genu, CD99 pozitivita, měkkotkáňová a skeletální lokalizace, značná agresivita a nediferencovaný malobuněčný, kulatobuněčný vzhled. Je to v současnosti ještě dostatečně definující a vymezující pro toto uskupení?
Čtyři dříve popsané a obě naše vyšetřované léze, charakterizované translokací t(20;22) (EWSR1-NFATc2), se výše zmíněné sestavě přeci jen poněkud vymykají (17). Jeden měkkotkáňový (stehno) a celkem 5 kostních tumorů (4x femur, 1x humerus) vykazovalo shodný vzhled kompaktního, solidně alveolárního, mozaikovitého až organoidního uspořádání buněk s nápadně světlou cytoplazmou a alespoň v našich nádorech s nepřehlédnutelnou, významnou příměsí eozinofilních leukocytů. Originální práce hodnotí morfologii oproti klasickému ES jako atypickou, ale vyjma exprese CD99 detailnějsí imunoprofil neuvádí; my jsme prokázali identickou konfiguraci imunofenotypu s pozitivitou CD10, CD99 a CD138. Nejzajímavější je naprosto souhlasné molekulární pozadí u všech šesti nádorů ve smyslu dispozice místa zlomu EWSR1 resp. NFATc2 genů a detailního uspořádání výsledného fúzního genu, včetně jedinečné amplifikace, která nebyla ještě nikdy v rámci ES popsána.
Pro přesné pochopení biologického významu uváděné raritní genetické poruchy doprovázející tento mimořádný nádor a jeho odpovídající taxonomické začlenění je nutné nejen hlubší molekulárně patologické poznání, ale též širší klinicko-onkologická zkušenost. Oba námi sledované případy se chovaly značně agresívně a běžný léčebný protokol pro ES byl neúčinný.
V této souvislosti si určitě zaslouží pozornost i aktuálně publikovaná práce o benigním hemangiomu kosti s t(18;22)(EWSR1-NFATc1) genotypem (2). Jakkoliv homologní a ze stejné skupiny jako NFATc2 vycházející, NFATc1gen se objevuje na molekulárním pozadí při formování histogeneticky odlišné, benigní „non Ewingovské“ léze. Jedním z možných vysvětlení neočekávané morfologické projekce této genetické aberace by mohl být fakt, že zde, na rozdíl od diskutované EWSR1-NFATc2 fúze, nedochází k amplifikaci nové genové chiméry. Nelze totiž vyloučit, že právě amplifikace EWSR1-NFATc2 fúzního genu společně s koamplifikací sousedních genů může být zodpovědná za výsledné agresívní biologické chování (17).
ZÁVĚR
Prezentovány byly dva neobvyklé kostní nádory se shodným histologickým obrazem a t(20;22) (EWSR1-NFATc2) genotypem. Domníváme se, že jejich začlenění v biologicky neurčité skupině „Ewing-like“ sarkomu je přinejmenším stejně odvážné jako spekulace, že by se ve skutečnosti mohlo jednat o naprosto jiné tumory. Nesporné však je, že existuje celé spektrum neoplázií s neodlišitelnou morfologií, kde zlom EWSR1 genu prokázaný metodou FISH v žádném případě neznamená, že se jedná o proces příbuzný s klasickým ES. Je zřejmé, že bez detailní extenzívní analýzy molekulárního pozadí konkrétní léze není spolehlivé a přesné začlenění v rámci stávající klasifikace možné.
PODĚKOVÁNÍ
Děkujeme prim. MUDr. Igorovi Suškevičovi z Kliniky zobrazovacích metod, LF MU a FN u sv. Anny, Brno za laskavé poskytnutí radiologické dokumentace k prvnímu případu.
Adresa pro korespondenci:
Doc. MUDr. Zdeněk Kinkor, Ph.D.
Bioptická laboratoř s.r.o.
Mikulášské nám. 4, 326 00 Plzeň
tel.: 737 220 449
e-mail: kinkor@medima.cz
Sources
1. Fletcher CDM, Bridge JA, Hogendoorn PCW, Mertens F. Ewing sarcoma. In: WHO classification of tumours of soft tissue and bone (4th ed). Lyon. International agency for research on cancer; 2013: 305-309.
2. Arbajian E, Magnusson L, Brosjö O, et al. A benign vascular tumor with a new fusion gene EWSR1-NFATC1 in hemangioma of bone. Am J Surg Pathol 2013; 37: 613-616.
3. Stockman DL, Miettinen M, Suster S, Spagnolo D. Malignant gastrointestinal neuroectodermal tumor: clinicopathologic, immunohistochemical, ultrastructural and molecular analysis of 16 cases with reappraisal of clear cell sarcoma-like tumors of the gastrointestinal tract. Am J Surg Pathol 2012; 36: 857-868.
4. Thway K, Fisher C. Tumors with EWSR1-CREB1 and EWSR1-ATF1 fusions: the current status. Am J Surg Pathol 2012; 36: e1-11.
5. Thway K, Nicholson AG, Lawson K, et al. Primary pulmonary myxoid sarcoma with EWSR1-CREB1 fusion: a new entity. Am J Surg Pathol 2011; 35: 1722-1732.
6. Cheah AL, Goldblum JR, Billings SD. Molecular diagnostics complementing morphology in superficial mesenchymal tumors. Semin Diagn Pathol 2013; 30: 95-109.
7. Tanguay J, Weinreb I. What EWSR1-ATF1 fusion has taught us about hyalinizing clear cell carcinoma? Head Neck Pathol 2013; 7: 28-34.
8. Boland JM, Folpe AL. Cutaneous neoplasm showing EWSR1 rearrangement. Adv Anat Pathol 2013; 20: 75-85.
9. Tanas MR, Rubin BP, Montgomery EA, et al. Utility of FISH in the diagnosis of angiomatoid fibrous histiocytoma: a series of 18 cases. Mod Pathol 2010; 23: 93-97.
10. Ginsberg JP, de Alava E, Ladanyi M, et al. EWS-FLI1and EWS-ERG gene fusions are associated with similar clinical phenotypes in Ewing´s sarcoma. J Clin Oncol 1999; 17: 1809-1814.
11. Gamberi G, Cocchi S, Benini L, et al. Molecular diagnosis in Ewing family tumors. The Rizzoli experience - 222 consecutive cases in four years. J Mol Diagn 2011; 13: 313-324.
12. Sankar S, Lessnick SL. Promiscuous partnership in Ewing´s sarcoma. Canc Gen 2011; 204: 351-365.
13. Wang W-L, Patel NR, Caragea M. Expression of ERG, an Ets family transcription factor, identifies ERG-rearranged Ewing sarcoma. Mod Pathol 2012; 25: 1378-1383.
14. Berková A, Dundr P, Povýsil C, Melcákova S, et al. A comparison of RT-PCR and FISH techniques in molecular diagnosis of Ewing´s sarcoma in paraffin-embedded tissue. Cesk Patol 2008; 44: 67-70.
15. Wu H-T, Govender D. Ewing sarcoma family of tumours: unusual histological variants and immunohenotypic characteristics. Diagn Histopathol 2012; 18: 348-355.
16. Le Deley MC, Delattre O, Schaefer LK, et al. Impact of EWS-ETS fusion type on disease progression in Ewing´s sarcoma/peripheral primitive neuroectodermal tumor: prospective results from the cooperative Euro-E.W.I.N.G. 99 trial. J Clin Oncol 2010; 28: 19821988.
17. Szuhai K, Ijszenga M, de Jong D, et al. The NFATc2 gene is involved in a novel translocation in a Ewing sarcoma variant that couples its function in immunology to oncology. Clin Canc Res 2009; 15: 2259-2267.
18. Graham C, Chilton-Macneill S, Zielenska M, Somers GR. The CIC-DUX4 fusion transcript is present in a subgroup of primitive pediatric round cell sarcomas. Hum Pathol 2012; 43: 180-189.
19. Italiano A, Sung YS, Zhang L, et al. High prevalence of CIC fusion with double-homeobox (DUX4) transcription in EWSR1-negative undifferentiated small blue round cell sarcomas. Gen Chromosomes Canc 2012; 51: 207-218.
20. Pierron G, Tirode F, Lucchesi C, et al. A new subtype of bone sarcoma defined by BCOR-CCNB3 gene fusion. Nat Genet 2012; 44: 461-466.
Labels
Anatomical pathology Forensic medical examiner ToxicologyArticle was published in
Czecho-Slovak Pathology

2014 Issue 2
Most read in this issue
- WHO classification of tumours of soft tissue and bone 2013: the main changes compared to the 3rd edition
- Where does Ewing sarcoma end and begin - two cases of unusual bone tumors with t(20;22)(EWSR1-NFATc2) alteration
- The current staging for uterine body malignancies and its importance for clinical practice
- Epidermolytic hyperkeratosis of the vulva associated with basal cell carcinoma in a patient with vaginal condyloma acuminatum and vaginal intraepithelial neoplasia harboring HPV, type 42