
Fabryho choroba s kardiovaskulární manifestací u pacienta s terminálním selháním ledvin
Fabry disease with cardiovascular manifestation in a patient with end-stage renal disease
Fabry disease is a rare X-linked hereditary storage disease caused by a mutation of the gene encoding alpha-galactosidase A. The clinical manifestation of the classical disease form is variable depending on the degree of individual organs involvement, including especially kidney, myocardium, central nervous system (CNS) and skin. We report a case of a 51-year-old man whose diagnostic manifestation was cardiac involvement leading to endomyocardial biopsy, which significantly contributed to the diagnosis. Although at that time he was already 9 years dependent on dialysis with terminal renal failure.
Keywords:
Fabry disease – histopathology – kidney failure – hypertrophic cardiomyopathy
Authors:
Hana Skopcová 1; Gabriela Dostálová 2; Tomáš Paleček 2; Aleš Linhart 2; Eva Honsová 3
Authors‘ workplace:
Pracoviště klinické a transplantační patologie IKEM Praha
1; II. interní klinika kardiologie a angiologie 1. LF UK a VFN v Praze
2; AeskuLab Patologie k. s. Praha
3
Published in:
Čes.-slov. Patol., 57, 2021, No. 1, p. 49-52
Category:
Original Article
Overview
Fabryho choroba je vzácné X-vázané hereditární střádavé onemocnění, jehož příčinou je mutace genu kódujícího alfa-galaktosidázu A. Obraz klasické formy je pestrý v závislosti na míře postižení jednotlivých orgánů, mezi které patří především ledviny, myokard, centrální nervový systém (CNS) a kůže. Referujeme případ 51letého muže, jehož diagnostickou manifestací bylo kardiální postižení. Následná endomyokardiální biopsie významně přispěla k diagnóze, ačkoli v té době byl pacient již 9 let v dialyzační léčbě s terminálním selháním ledvin.
Klíčová slova:
Fabryho choroba – histopatologie – selhání ledvin – hypertrofická kardiomyopatie
Fabryho choroba patří do skupiny dědičných střádavých lyzosomálních onemocnění. Jde o progresivní metabolické onemocnění vázané na X chromozom charakterizované intralyzosomální akumulací neutrálních glykosfingolipidů (převážně globotriaosylceramidu, Gb3) v různých buňkách. Akumulace je důsledkem mutace GLA genu kódujícího enzym α-galaktosidázu A (α-Gal A), což vede ke snížené nebo nulové aktivitě enzymu, který za normálního stavu Gb3 degraduje. Akumulace Gb3 vede dosud neznámými mechanismy k rozvoji symptomů spojených s orgánovou dysfunkcí. Klinicky závažné jsou především projevy onemocnění zahrnující životně důležité orgány zvláště myokard, ledviny a CNS. Kvalitu života negativně ovlivňuje postižení periferního nervového systému s bolestivými komplikacemi (pálivé bolesti dlaní a plosek, hypohidróza nebo anhidróza s intolerancí tepla a poruchy GIT) a také postižení smyslových orgánů (oko, vestibulokochleární aparát).
Ačkoli je onemocnění vázané na X chromozom, klinické projevy onemocnění mají často i heterozygotní ženy. Domněnka, že ženy představují zdravé přenašečky onemocnění je mylná. U žen je známá výrazná klinická variabilita, od mírného, téměř asymptomatického průběhu, až po těžké orgánové postižení. Za jeden z možných důvodů rozmanitosti klinických projevů Fabryho nemoci u žen je označován proces náhodné inaktivace chromozomu X v časných fázích embryogeneze (1).
Existují 2 formy Fabryho choroby; tzv. klasická forma (typ 1) charakterizovaná nulovou nebo téměř nulovou aktivitou α-Gal A. Tento typ onemocnění má těžší průběh s klinicky vyjádřenými symptomy již v dětství. Ve 2. a 3. decéniu se přidává postižení myokardu (hypertrofická kardiomyopatie, poruchy srdečního rytmu) a postižení ledvin, které je charakterizované progresivním selháním funkce.
Druhý typ (tzv. late-onset) je mírnější, s reziduální aktivitou enzymu, což vede k tomu, že v dětství a v období dospívání je onemocnění bezpříznakové. Obtíže se objevují až v dospělosti (ve 4. až 6. deceniu), kdy je diferenciální diagnostika komplikovanější vzhledem k dalším běžnějším chorobám.
Z nejasných důvodů mohou být klinické projevy u mužů i u žen velmi variabilní; a to i v rámci jedné rodiny. Studie zabývající se korelacemi genotypu a fenotypu tuto variabilitu nevysvětlují. Navíc se u obou pohlaví mohou vyskytovat formy, které postihují pouze jeden orgán (zvláště kardiální forma choroby). U takto postižených pacientů bývá přítomna reziduální aktivita α-Gal A. Stanovení diagnózy je vzhledem k různorodým projevům systémového onemocnění obtížné a není neobvyklé, že doba od prvních projevů k diagnóze se počítá na roky; v průměru 13,7 roku u mužů a 16,3 roku u žen (2). K potvrzení diagnózy se u mužů používá stanovení aktivity enzymu α-Gal A v plazmě, leukocytech nebo fibroblastech. U žen s reziduální aktivitou enzymu jsou hodnoty aktivity α-Gal A velmi různorodé; a proto je nutné molekulárně genetické vyšetření (3). V posledních desetiletích v zemích s vyspělým zdravotnickým systémem představuje molekulárně genetický průkaz onemocnění u obou pohlaví diagnostický standard. Fabryho choroba (FD) je po Gaucherově nemoci druhým nejčastěji se vyskytujícím lyzosomálním střádavým onemocněním. Prevalence FD je ve světě udávaná v rozmezí 1 : 40 000 – 1 : 60 000 (4). Screeningové studie však ukazují na pravděpodobný vyšší výskyt onemocnění v populaci. V rizikových populacích pro FD, jako jsou dialyzovaní pacienti zvláště bez bioptické verifikace selhání ledvin, mladí pacienti s cévní mozkovou příhodou (CMP), pacienti s hypertrofií levé srdeční komory; se frekvence onemocnění pohybuje od 0,3 % po 1 %. Studie detekující mutaci GLA genu mezi pacienty s ischemickou CMP ve věku od 18 do 55 let odhalila tuto mutaci u 2,4 % žen a 4,9 % mužů (5).
V posledních desetiletích má včasné stanovení diagnózy klíčový význam, protože umožní cílenou léčbu, která dokáže zamezit rozvoji orgánového poškození. FD lze léčit pomocí rekombinantně vyráběného enzymu, který představuje náhradu chybějícího enzymu α-Gal A; a je podáván ve formě infuzního roztoku (6). Od r. 2015 přibyla možnost tzv. chaperonové terapie ve formě tablet. Ta dokáže v případě, že nemocný má mutaci, která umožňuje tvorbu vlastního, ale nedostatečně stabilního enzymu, tento enzym stabilizovat a tím zlepšit jeho funkci (7).
POPIS PŘÍPADU
Pacient, ročník 1967, byl v 37 letech hospitalizován pro psychickou deterioraci a amentní stavy na psychiatrické klinice. V rámci hospitalizace bylo diagnostikováno chronické ledvinné selhání, stadium 5; a výše popsaná neurologická symptomatika byla považovaná za projev uremické encefalopatie. Pacient byl zařazen do pravidelného hemodialyzačního programu a na čekací listinu k transplantaci ledviny. V osobní anamnéze byla uvedena hypertenze, hyperlipidemie, acne rosacea a hypertrofie levé komory srdeční s ejekční frakcí 70 %, bez poruchy diastolické relaxace. Rodinná anamnéza byla bez pozoruhodností.
Po 4 letech na čekací listině podstoupil pacient transplantaci kadaverózní ledviny. Při pitvě dárce byl zjištěn generalizovaný adenokarcinom plic, proto byla indikovaná okamžitá graftektomie a pokračováno v dialyzační léčbě. U pacienta došlo pozvolna k progresi kardiovaskulární symptomatologie, která vedla k hospitalizaci pro stenokardie s kardiálním astmatem a námahovou dušností (NYHA III). Během hospitalizace byla vyloučena koronární příčina potíží. Echokardiografické vyšetření prokázalo těžkou difuzní koncentrickou hypertrofii levé komory bez obstrukce výtokového traktu, s omezenou diastolickou relaxací levé komory a s poklesem ejekční frakce na 50 %. Poprvé bylo vysloveno podezření na střádavé onemocnění. Magnetická rezonance srdce (gadolinium-late enhancement imagining) v diferenciální diagnostice podpořila podezření na střádání a vedla k indikaci endomyokardiální biopsie. Současně byl pacient vyřazen z čekací listiny na transplantaci ledviny.
Mikroskopické vyšetření a stanovení diagnózy
Vyšetřeno bylo 7 vzorků endomyokardu s minimální intersticiální fibrózou do 5 %. Kardiomyocyty byly výrazně hypertrofické a vakuolizované (obr. 1). Ve vzorcích nebyla zánětlivá celulizace ani nekrózy. Průkaz železa a amyloidu (Kongo červeň, Saturnová červeň) byl negativní. Imunofluorescenční detekce amyloidu a ukládání lehkých řetězců (průkaz AA-amyloidu, transthyretinu, lehkých řetězců kappa a lambda) byla negativní. Elektronmikroskopické vyšetření odhalilo v kardiomyocytech četné myelinové figury charakteristického vzhledu s koncentrickou lamelací. Inkluze byly obklopeny jednoduchou membránou identifikující jejich lokalitu v lyzosomech (obr. 3).
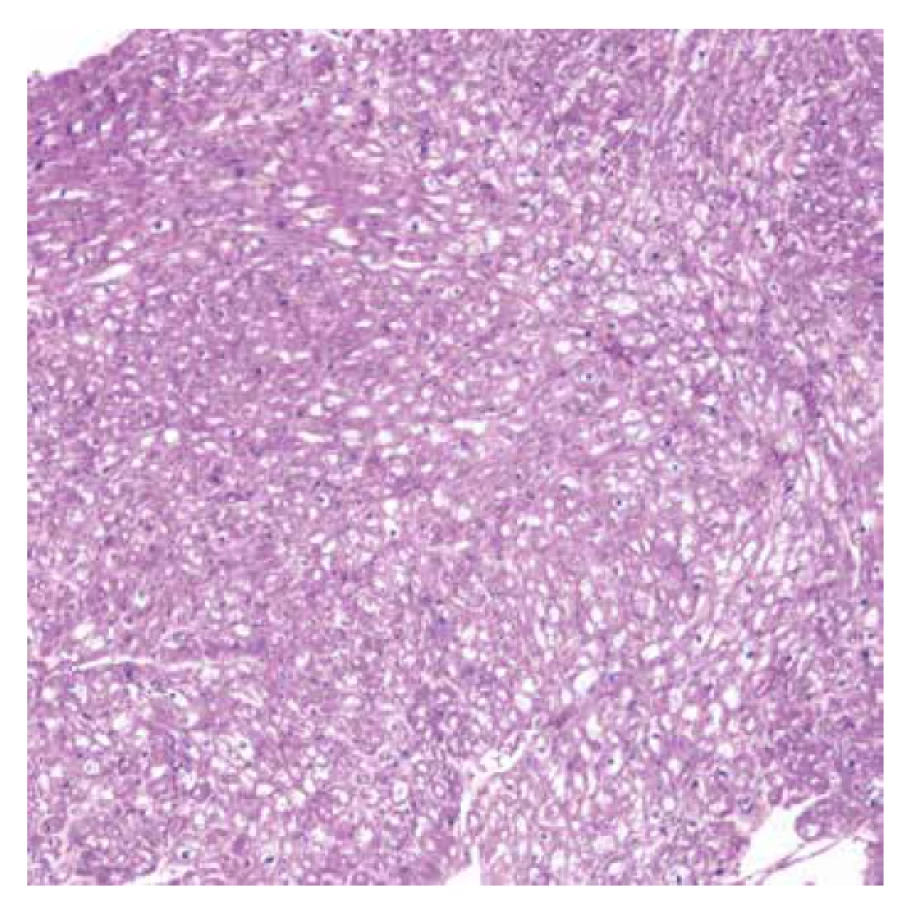
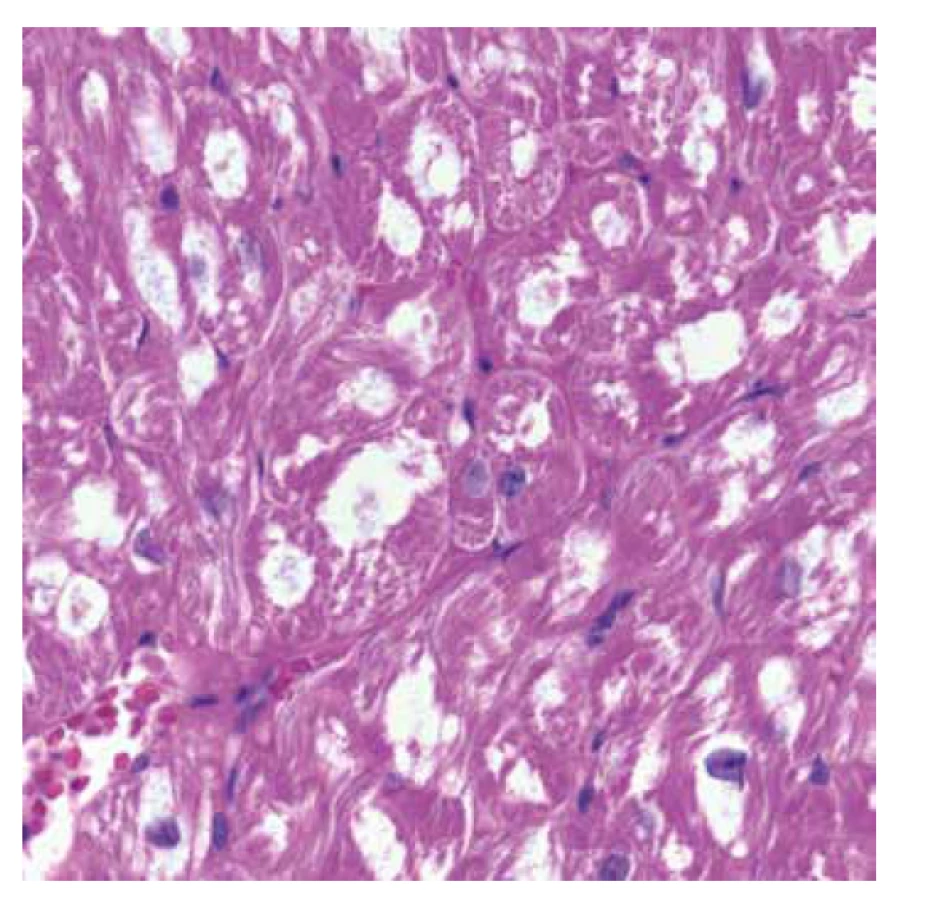
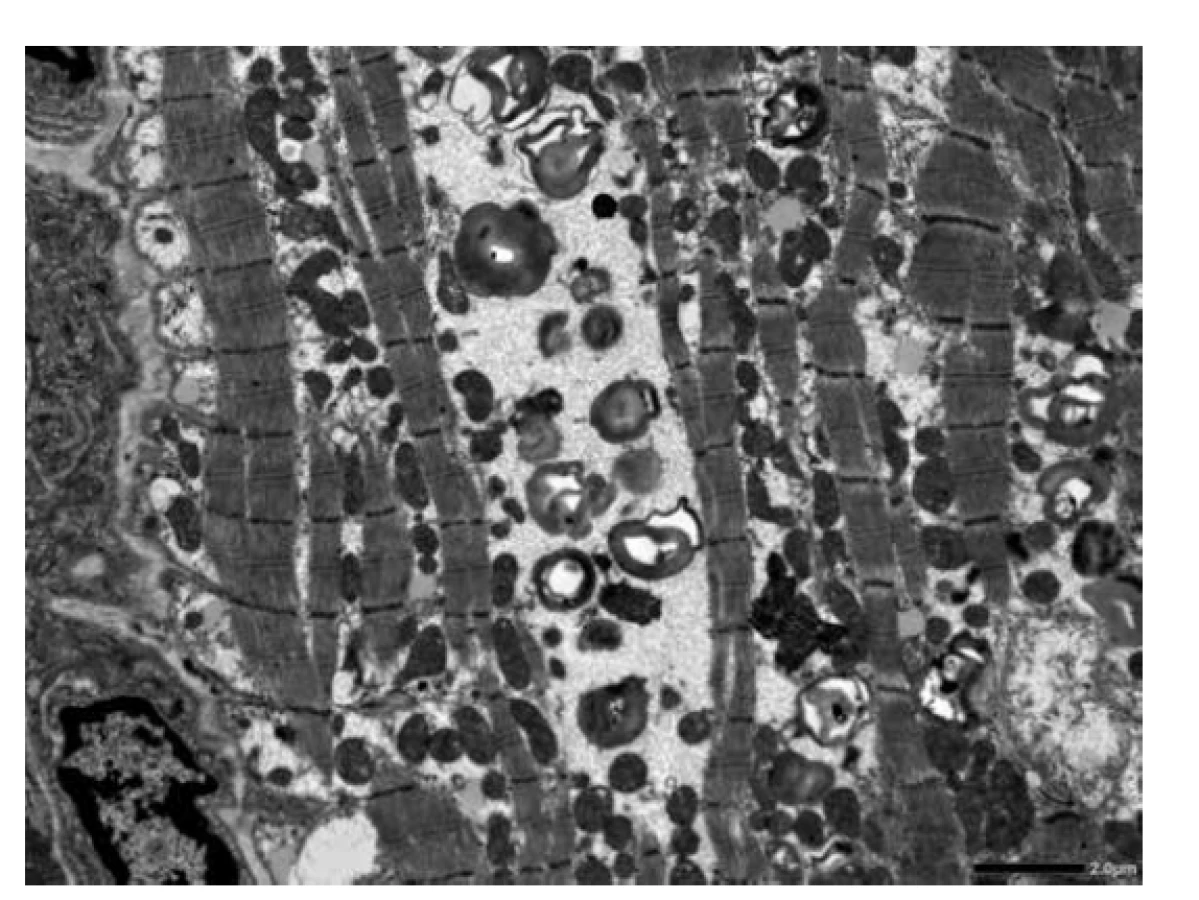
Diagnostický závěr na úrovni morfologie byl vysoce suspektní z Fabryho choroby. Další vyšetření potvrdilo velmi nízkou aktivitu enzymu α-Gal A. Molekulárně genetické vyšetření prokázalo poměrně raritní mutaci c.[488G>T]; [0].
Pacient byl indikován k substituční enzymatické terapii.
Onemocnění bylo v době diagnózy velmi pokročilé s 9 let trvající dialyzační léčbou. Přes enzymatickou léčbu postupně progredovala i kardiální symptomatologie, vyžadující opakované kardiovaskulární intervence, včetně implantace kardiostimulátoru. Přidalo se neurologické postižení v podobě tří tranzitorních ischemických atak, s krvácením do pravé mozečkové hemisféry s reziduální fatickou poruchou a poruchou hybnosti pravé horní končetiny. V 51 letech, po 5 letech od stanovení diagnózy a léčby enzymatickou terapií, spáchal pacient sebevraždu.
DISKUSE
U pacientů s FD představuje postižení myokardu spolu se selháním ledvin a postižením CNS jednu ze 3 hlavních příčin morbidity a mortality.
U referovaného pacienta dominovalo postižení ledvin, které bylo téměř s jistotou součástí FD. Onemocnění nebylo včas rozpoznáno a biopsie ledvin nebyla provedena, přestože v době počátku dialyzační léčby bylo pacientovi pouze 37 let. V tomto věku se klinická diagnóza izolované hypertenzní/vaskulární nefrosklerózy jeví velmi nepravděpodobnou. Stanovení diagnózy z punkční biopsie ledviny není v počátcích onemocnění příliš komplikované. Myelinové figury jsou uloženy převážně v podocytech, což způsobí, že podocyty jsou zvětšené a ve světelné mikroskopii působí jejich cytoplasma vakuolizovaným pěnitým dojmem (obr. 4). V polotenkých řezech a v ultrastruktuře je typická morfologie myelinových figur, tj. koncentricky stočené osmiofilní lamelované struktury nebo paralelně řazená vláknitá depozita tzv. “zebra bodies” (obr. 5, 6). V menším množství lze myelinové figury identifikovat i v dalších buňkách glomerulů, v endotelu kapilár, v mesangiálních buňkách a někdy též v buňkách výstelky Bowmanova pouzdra. Pokud jsou v ultrastruktuře zastižené distální tubuly, lze akumulaci myelinových figur identifikovat i v jejich epitelu. Problém nastává u pokročilých stavů, kdy dochází ke sklerotizaci segmentů a posléze celých glomerulárních trsů. Ve sklerotických úsecích podocyty nejsou (se zhroucením cytoskletu při hyperfiltraci se neudržely na svém místě a odpadly do moči) a proto v pokročilých stádiích občas nelze ani v ultrastruktuře myelinové figury identifikovat. Pak se může stát, že onemocnění je klasifikováno jako fokální segmentální glomeruloskleróza. I když myelinové figury nejsou jen součástí FD a mohou se objevit i u jiných hereditárních chorob, hlavní diferenciální diagnózu představují pacienti léčení lyzosomálními inhibitory (amiodaron, chloroquine). V těchto případech je morfologie prakticky neodlišitelná od FD. Část pacientů s FD může mít současně další imunokomplexové onemocnění ledvin, které klasickou morfologii FD modifikuje a v pozdějších stádiích ji může i překrýt (8). Téměř všichni pacienti, u kterých je podezření na FD vysloveno na základě morfologie v ledvinné biopsii, přichází s jinou klinickou diagnózou.
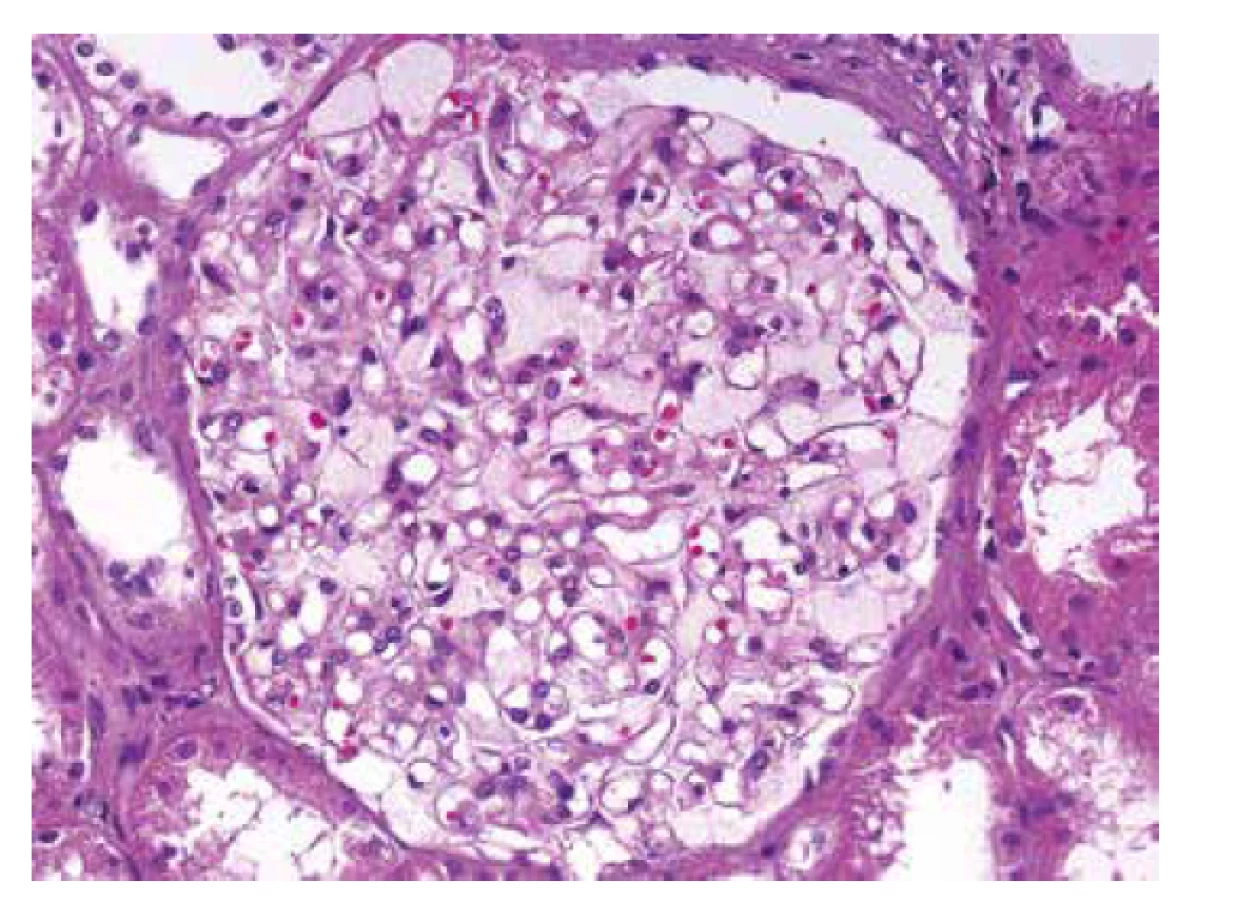
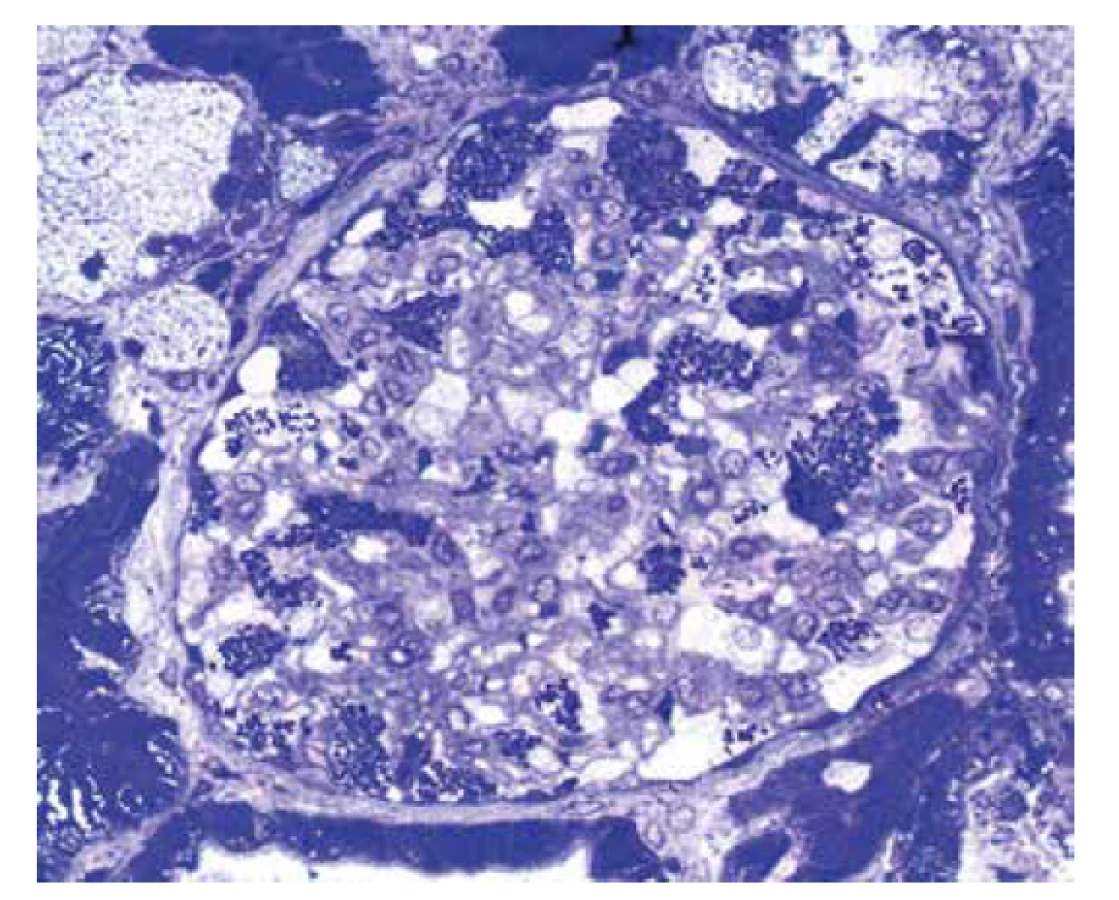
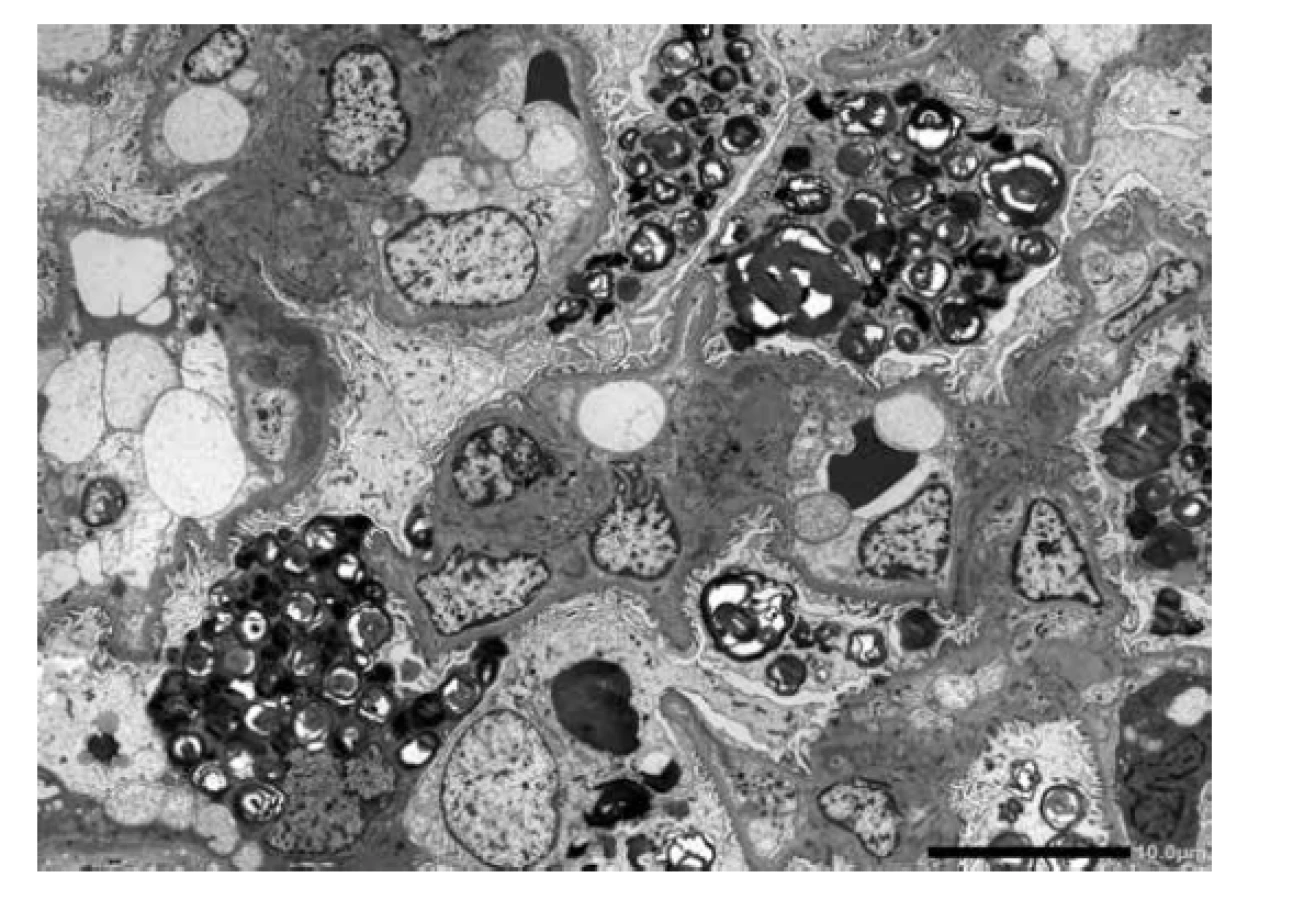
Postižení myokardu je u pacientů s FD běžné a kardiální klinickou manifestaci udává více než 60 % pacientů (9). Většinou jde o progresivní hypertrofii svaloviny s narůstající intersticiální fibrózou. To odpovídá klinickým projevům s mírnou diastolickou dysfunkcí v časných stádiích až po hypertrofickou formu kardiomyopatie včetně obstrukce výtokového traktu u pokročilého onemocnění. Dále jsou běžné převodní poruchy a arytmie, v pokročilých stádiích s bradykardií vyžadující implantaci kardiostimulátoru. Kardiolog má obvykle podezření na chorobu se střádáním, což zahrnuje postižení při různých onemocněních, od amyloidóz po vrozené enzymopatie. Přestože nastřádaného materiálu v lyzosomech je v poměru k celé tkáni myokardu relativně málo (cca 3 %), vede akumulace Gb3 k závažné dysfunkci s různorodou klinickou manifestací. Přesný mechanismus kaskády změn vedoucích k dysfunkci myokardu není znám. Z experimentu je doložena aktivace nejrůznějších metabolických pochodů od tvorby volných radikálů, přes produkci zánětlivých cytokinů, po zvýšenou expresi adhezivních molekul a zvýšenou tvorbu některých proliferačních faktorů, které v experimentu přispívají k hypertrofickým změnám.
Morfologie u FD identifikuje v endomyokardiální biopsii vakuolizaci kardiomyocytů a diferenciální diagnostika je směřována ke kategorii tzv. vakuolárních nebo „vacuolar-like“ kardiomyopatií. Jde o skupinu různorodých onemocnění s obdobnou morfologií na úrovni HE. Bohužel, vakuolizace kardiomyocytů je v endomyokardiálních biopsiích běžná a často se shrne pod termín nespecifické/dystrofické ischemické změny. Tomu odpovídají studie dokládající tento typ morfologie u všech dilatačních kardiomyopatií (10). Další skupiny zahrnují hereditární onemocnění (především Danonovu, Pompeho a Fabryho chorobu) a také toxické poškození myokardu. Mezi případy toxického poškození můžeme zahrnout polékové kardiomyopatie (již zmíněné lyzosomální inhibitory, ale také např. inhibitory tyrosin kinázy nebo chemoterapeutika ze skupiny antracyklinů) a dále kobalt, který se může uvolňovat z kloubních náhrad. V diferenciální diagnostice hereditárních onemocnění zvažujeme také extrakardiální manifestace. V našem případě byla v diferenciální diagnostice na prvním místě jednoznačně Fabryho choroba, protože žádné z dalších onemocnění nemá jako svoji součást progresivní onemocnění ledvin se selháním funkce.
Další závažné komplikace představuje postižení CNS, jde především o ischemické ataky, od tranzitorních po klasické infarkty.
U referovaného pacienta došlo postupně k rozvoji všech závažných projevů FD. Přesto byla diagnóza stanovená velmi pozdě, až po selhání ledvin vyžadujícím mnohaletou dialyzační léčbu; a při rozvinutém kardiálním postižení. Současná léčba umožní lepší kvalitu života pacientům pouze tehdy, pokud je diagnóza stanovena časně. Tam, kde již došlo k nevratným změnám s tvorbou fibrózy, se ani při současných terapeutických možnostech funkce postižených orgánů zlepšit nedá. V takovém případě zůstává metodou léčby orgánová transplantace.
Molekulárně genetická verifikace onemocnění u jednoho pacienta pomůže často odkrýt onemocnění u dalších členů rodiny. Současně se dostáváme mimo čistě racionální medicínskou problematiku, kdy část pacientů z obavy před ostrakizací odmítá širší rodině svoji diagnózu sdělit. V našem případě byl pacient bezdětný a žil s přítelkyní, další členové širší rodiny vyšetřováni nebyli.
ZÁVĚR
Závěrem lze shrnout, že Fabryho choroba je po Gaucherově nemoci druhým nejčastěji se vyskytujícím lyzosomálním střádavým onemocněním. Symptomy onemocnění zahrnují postižení různých orgánů ve velkém věkovém rozmezí. Proto je diagnostika komplikovaná a stojí na diagnostické rozvaze, kterou lze poté verifikovat laboratorními testy; především molekulárně genetickým průkazem. Popisovaný případ dokládá typický průběh onemocnění včetně pokročilého orgánového postižení. Nově dostupná enzymová substituční a chaperonová terapie přinesly do klinické praxe naději pro pacienty, zatím pouze tehdy, pokud je onemocnění diagnostikováno v časných stádiích.
PROHLÁŠENÍ
Autor práce prohlašuje, že v souvislosti s tématem, vznikem a publikací tohoto článku není ve střetu zájmů a vznik ani publikace článku nebyly podpořeny žádnou farmaceutickou firmou. Toto prohlášení se týká i všech spoluautorů.
∗ Adresa pro korespondenci:
doc. MUDr. Eva Honsová, Ph.D.
Aeskulab Patologie, k. s.
Evropská 2589/33b, 160 00 Praha 6
e-mail: honsova.eva @aeskulab.cz
Sources
1. Carrel L, Willard HF. X-inactivation profile reveals extensive variability in X-linked gene expression in females. Nature 2005; 434(7031): 400–404.
2. Mehta A, Ricci R, Widmer U et al. Fabry disease defined: baseline clinical manifestation of 48366 patients in the Fabry Outcome Survey. Eur J Clin Invest 2004; 34(3): 236 – 242.
3. Laney DA., Fernhoff PM. Diagnosis of Fabry disease via analysis of family history. J Genet Couns 2008; 17(1): 79-83.
4. Meikle PJ, Hopwood JJ, Clauge AE et al. Prevalence of lysosomal storage disorders. JAMA 1999; 281(3): 249 – 254.
5. Rolfs A, Bottcher T, Zschiesche M et al. Prevalence of Fabry disease in patients with cryptogenic stroke: a prospective study. Lancet 2005; 366(9499): 1794 – 1796.
6. Beck M, Ricci R, Widmer U et al.: Fabry disease: overall effects of agalsidase alfa treatment. Eur J Clin Invest 2004; 34 : 838-844.
7. Svarstad E and Marti HP. The Changing Landscape of Fabry Disease. Clin J Am Soc Nephrol. 2020 (DOI: https://doi.org/10.2215/CJN.09480819 Online ahead of print)
8. Maixnerová D, Tesař V, Ryšavá R et al. The coincidence of IgA nephropathy and Fabry disease. BMC Nephrol 2013; 14 : 6.
9. Linhart A, Kampmann C, Zamorano JL, et al. Cardiac manifestations of Anderson-Fabry disease: results from the international Fabry outcome survey. Eur Heart J 2007; 28 : 1228-1235.
10. Mitrut R, Stepan AE, Pirici D. Histopathological aspects of the myocardium in dilated cardiomyopathy. Curr Health Sci J 2018; 44 : 243-249.
Labels
Anatomical pathology Forensic medical examiner ToxicologyArticle was published in
Czecho-Slovak Pathology

2021 Issue 1
Most read in this issue
- Základní imunohistochemický panel pro diagnostiku nádorů měkkých tkání
- Ložiskové léze kostí – diagnostické využití imunohistochemie a molekulární patologie
- Sekundárna hypoplázia pľúc asociovaná s kalcifikovaným Meckelovým divertikulom s oseálnou metapláziou
-
Konsensuální doporučení České kooperativní skupiny pro nádory hlavy a krku (2019):
definice resekčních okrajů, reportování krčních disekcí a vyšetřování HPV/p16