
Lidská prionová onemocnění v České republice
Human prion diseases in the Czech Republic
Human prion diseases are a group of very rare diseases with a unique pathogenesis and, due to an inauspicious prognosis and unavailability of therapy, with fatal consequences. The etiopathogenetic background is the presence of pathologically misfolded prion protein, highly resistant to denaturation, the aggregation and presence of which in the brain tissue causes irreversible neuronal damage. The most frequent prion disease in humans is Creutzfeldt-Jakob disease (CJD) which occurs in sporadic, hereditary/familial, or acquired/infectious/iatrogenic forms. A new form of CJD, variant CJD, is considered to be linked to dietary exposure to beef products from cattle infected with bovine spongiform encephalopathy (BSE) and to infection via blood transfusion. The clinical picture of these diseases is characterized by a rapidly progressing dementia, cerebellar and extrapyramidal symptoms, and rather specific MRI, EEG, and CSF findings. Clinically, the diagnosis is described as possible or probable prion disease and needs to be confirmed by neuropathological or immunological investigation of the brain tissue. Epidemiological data from the Czech Republic spanning the last decade are presented.
Key words:
neurodegenerative disease – transmissible spongiform encephalopathy – prion protein – Creutzfeldt-Jakob disease – Gerstmann-Sträussler-Scheinker
:
Z. Rohan 1; R. Rusina 2,3; M. Marešová 4; R. Matěj 1,2
:
Národní referenční laboratoř pro lidská prionová onemocnění, Oddělení patologie a molekulární medicíny, Thomayerova nemocnice, Praha
1; Neurologická klinika a Centrum klinických neurověd, Univerzita Karlova v Praze, 1. lékařská fakulta a Všeobecná fakultní nemocnice v Praze
2; Oddělení neurologie, Thomayerova nemocnice, Praha
3; Protiepidemické oddělení HSHMP, pobočka Praha-jih
4
:
Epidemiol. Mikrobiol. Imunol. 64, 2015, č. 3, s. 115-120
:
Review Article
Lidská prionová onemocnění jsou velmi vzácnou skupinou nemocí s unikátní patogenezí a vzhledem k infaustní prognóze a nedostupnosti terapie i se závažnými důsledky. Etiopatogenetickým podkladem je přítomnost patologicky konformovaného a vůči denaturaci velmi odolného prionového proteinu, jehož agregace a přítomnost v mozkové tkáni vede k nevratnému poškození neuronů. Nejčastějším zástupcem prionových onemocnění v populaci je Creutzfeldtova--Jakobova nemoc, která se vyskytuje ve sporadické, dědičné (familiární) a infekční (náhodně přenesené, iatrogenní) formě. Nová varianta Creutzfeldtovy-Jakobovy nemoci je pak dávána do souvislosti s alimentární expozicí (konzumace hovězího masa z dobytka nakaženého bovinní spongiformní encefalopatií, BSE), s krevní transfuzí z nemocných dárců. Klinický obraz těchto onemocnění je charakterizován rychle progredující demencí, mozečkovou a extrapyramidovou symptomatologií a většinou poměrně specifickým nálezem na magnetické rezonanci, EEG a v mozkomíšním moku. Diagnóza je klinicky na úrovni možného či pravděpodobného prionového onemocnění, definitivní potvrzení přinese až neuropatologické či imunologické vyšetření mozkové tkáně. V článku jsou uvedena i epidemiologická data z ČR za uplynulých 10 let.
Klíčová slova:
neurodegenerativní onemocněn – transmisivní spongiformní encefalopatie – prionový protein – Creutzfeldtova-Jakobova nemoc – Gerstmannův-Sträusslerův-Scheinkerův syndrom
ÚVOD
Lidská prionová onemocnění (transmisivní spongiformní encefalopatie – TSE) jsou vzácnou skupinou neurodegenerativních nemocí, vyznačující se přítomností patologicky konformovaného prionového proteinu. Fyziologicky je prionový protein (PrP) přítomen u mnoha buněčných populací v lidském těle, nejvíce však v neuronech. Za původce prionových onemocnění označil Stanley B. Prusiner v roce 1982 patologicky změněný PrP (tzv. prionová hypotéza, Nobelova cena za rok 1997) [1].
Stěžejním podkladem patologické konformace PrP je změna jeho sekundární struktury (prostorového uspořádání proteinové molekuly) do podoby tzv. beta-skládaného listu. Výsledkem je silně hydrofobní va-rianta PrP s tendencí k agregaci, kdy vznikají oligomery PrPSc (Sc – scrapie), které dále polymerují až do tzv. amyloidových plak. Tyto agregáty PrPSc jsou (na rozdíl od fyziologického, „zdravého“ PrP) velmi odolné vůči fyzikálním a chemickým vlivům a v lidském těle jsou běžnými tkáňovými enzymy v podstatě nerozložitelné. Molekuly PrPSc jsou odolné k proteázám i k působení tepla, na rozdíl od většiny bílkovin nejsou tyto molekuly denaturovány při teplotě 100 °C, jsou také neobyčejně rezistentní vůči dezinfekci a sterilizaci běžnými metodami [2, 3]. PrPSc proto bývá také označován jako PrPres (res – resistant).
Příčina změny konformace PrP v PrPSc je stále neznámá. Proces oligomerizace a následné polymerizace je v současné době považován za příčinu jeho neurotoxicity. Postupně dochází k otoku neuritů (vakuolizace, spongióza); zániku neuronů a reaktivnímu zmnožení astrocytů – glióze. Tyto tři znaky se sdružují pod pojem „spongiformní dystrofie“, která je základním nálezem při standardním neuropatologickém vyšetření mozku [4, 5] – obrázek 1.
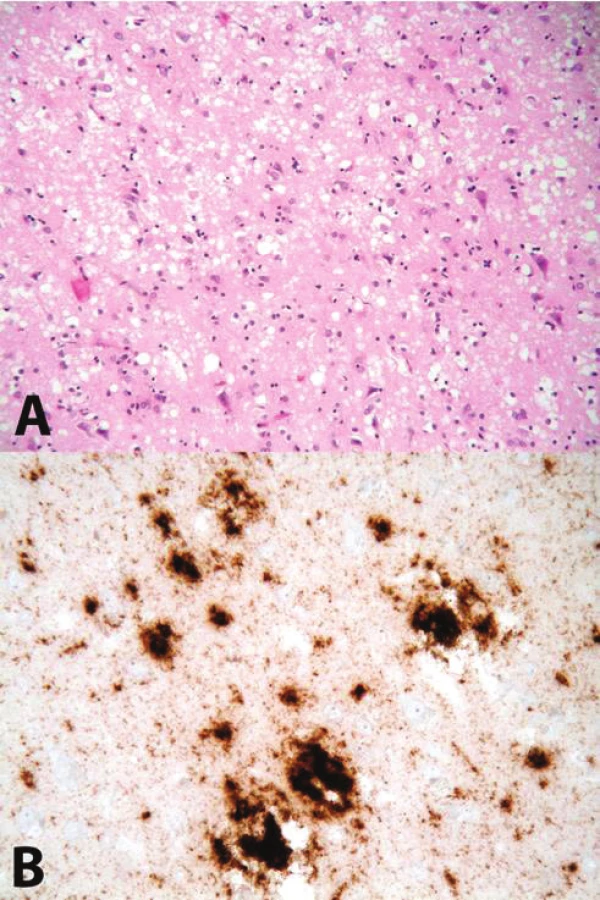
Gen pro prionový protein (PRNP) je kódován na krátkém raménku 20. chromozomu. V jeho sekvenci lze popsat řadu patogenních změn pořadí nukleotidů (polymorfismy) a mutací, které jsou příčinou variability jednotlivých podtypů prionových nemocí, nebo i kauzálními mutacemi u hereditárních forem prionových onemocnění [6].
TYPY PRIONOVÝCH ONEMOCNĚNÍ
Nejčastějším lidským prionovým onemocněním je Creutzfeldtova-Jakobova nemoc (CJN), která se vyskytuje ve třech formách: sporadická, genetická (familiární) a náhodně přenesená v souvislosti s lékařským výkonem (iatrogenní). Nejvíce případů je sporadických.
Mezi hereditární (familiární) prionové choroby vedle genetické formy Creutzfeldtovy-Jakobovy nemoci patří Gerstmannův-Sträusslerův-Scheinkerův syndrom (GSS) a fatální familiární insomnie (FFI).
Nová varianta Creutzfeldtovy-Jakobovy nemoci (vCJN), popsaná ve Velké Británii v roce 1996, která má vztah k bovinní spongiformní encefalopatii (BSE), v ČR dosud nebyla diagnostikována.
Dnes již pouze historický význam má kuru, které se vykytovalo na Nové Guinei v rámci rituálního kanibalismu (od 50. let 20. století se již neobjevily nové případy).
Creutzfeldtova-Jakobova nemoc (CJN)
Nejčastěji se vyskytuje sporadická forma CJN (sCJN) s incidencí přibližně 1–3 případy/1 milion obyvatel/rok.
Klinický obraz se vyznačuje rychle progredující demencí s přidružením nejméně dvou ze čtyř následujících projevů:
- myoklonus,
- mozečkové a zrakově prostorové dysfunkce,
- pyramidové a extrapyramidové (parkinsonské) projevy,
- akinetický mutismus (pacient je nepohyblivý, rigidní, neschopen souvislé řeči).
Onemocnění probíhá nejčastěji několik měsíců, většinou méně než jeden rok (trvání delší než dva roky je vylučujícím kritériem diagnózy sporadické formy CJN).
Diagnózu podporuje elektroencefalografický (EEG) nález generalizovaných trifázických komplexů s periodicitou kolem 1 sec, v mozkomíšním moku pozitivita proteinu 14-3-3 a vysoká hladina h-tau a v magnetické rezonanci (MRI) obraz hyperintenzních signálů v putamen, kaudatu a korových proužcích na sekvencích FLAIR (fluid attenuation and inversion recovery) a/nebo v difuzním vážení [7].
Diagnostická kritéria WHO rozlišují (podobně jako u dalších neurodegenerativních onemocnění) diagnózu možnou (typický klinický obraz), pravděpodobnou (klinický obraz a typický EEG nález, a/nebo MRI obraz a/nebo pozitivita proteinu 14-3-3 v mozkomíšním moku) a jistou (neuropatologicky potvrzená přítomnost prionových depozit v mozkové tkáni).
Termín „pravděpodobná sporadická Creutzfeldtova--Jakobova nemoc“ v podstatě odpovídá klinické jistotě diagnózy (u podezření na prionový původ se rutinně neprovádí mozková biopsie) a je indikací k zahájení komplexní paliativní péče tohoto terapeuticky neovlivnitelného onemocnění.
V rámci sCJN se vyskytují polymorfismy na 129. kodonu PRNP, které se vyznačují specifickým klinicko-patologickými podtypy sCJN s odlišnostmi v neuropatologickém obraze i v klinických projevech (délka trvání nemoci, pozitivita EEG či nálezu v mozkomíšním moku) [8].
Zcela zásadní je, že definitivní diagnózu určuje pouze morfologické – autoptické (vzácně bioptické) vyšetření mozku pacienta s neuropatologickým průkazem PrPres v mozkové tkáni imunohistochemickými metodami. Metoda westernblot využívá obdobných imunochemických postupů a lze ji provést opět pouze na vzorku mozkové tkáně.
Zobrazení mozku MRI je v současné době považováno za zlatý standard a nezbytnou součást diferenciální diagnostiky všech rychle progredujících demencí.
Bioptické vyšetření mozku při klinickém podezření na CJN je vzhledem k riziku iatrogenního přenosu (s důsledkem prakticky nevratné kontaminace operačního instrumentária) nutné považovat za ultimum refugium, s indikací pouze v případě jasné diferenciálně-diagnostické hypotézy léčitelného onemocnění (např. atypická CNS vaskulitida) [9].
Genetické (familiární) formy CJN (fCJN) jsou vel-mi vzácné, typická je u nich přítomnost patogenní mutace PRNP. V různých populacích se na základě historicko-sociálních aspektů udržují typické mutace – např. E200K se vyskytuje častěji na Slovensku a u libyjských Židů [10, 11]. V západní Evropě je pak poměrně častá mutace D178N, nicméně v ČR a v Evropě obecně je nejčastěji zastoupenou mutací E200K [12]. V ČR byla mutace E200K potvrzena v 25 případech, mutace D178N byla potvrzena u 5 případů a velmi vzácná mutace R208H v jednom případě [13]. Pravděpodobnost, že se během života nositele mutace prionové onemocnění projeví (penetrance), roste s věkem, na Slovensku a v Itálii dosahuje přibližně 60 % [11, 14], v jiných populacích pak dosahuje až 100 % [15]. Klinicky se fCJN projevují obdobně jako sCJN, nemusí ale mít pozitivní typický nález na EEG nebo pozitivní protein 14-3-3 v mozkomíšním moku a průběh onemocnění je delší, často přesahuje dva roky (časové omezení sporadických forem).
Iatrogenní (náhodně přenesená) CJN (iCJN) je spojena s invazivními neurochirurgickými výkony, transplantací rohovky a užíváním hormonů (gonadotropin, růstový hormon) izolovaných z lidských hypofýz. Zatím bylo ve světě popsáno přes 400 případů iCJN [16].
Nová varianta CJN (vCJN, nvCJN, Willova nemoc), známá od roku 1996 [17], je spolu s iCJN pravděpodobně epidemiologicky nejzávažnějším prionovým onemocněním. Její výskyt u člověka je vysvětlován alimentárním přenosem při konzumaci hovězího masa z dobytka nakaženého bovinní spongiformní encefalopatií (BSE). Doposud bylo ve světě k červnu 2014 definitivně potvrzeno 229 případů vCJN, z toho většina ve Velké Británii, kde jsou navíc tři případy dávány do souvislosti s transfuzí kontaminované krve [18, 19]. V současné době stále není k dispozici dostatečně senzitivní a specifický test pro přítomnost PrPSc v krvi, který by byl použitelný pro screening krevních derivátů.
Klinicky se vCJN projevuje časnými psychiatrickými projevy, mozečkovou symptomatologií a až pozdějším rozvojem progredující demence [20]. Typický je nález na magnetické rezonanci – hyperintenzita pulvinaru thalamu na FLAIR sekvencích (tzv. pulvinar sign) [21].
U nové varianty CJN (nikoliv u sporadických forem CJN!) je velmi specifická biopsie patrové mandle s imunohistochemickým průkazem PrPres v lymfatické tkáni tonzily, která umožňuje stanovit definitivní diagnózu již za života pacienta bez nutnosti mozkové biopsie [22–24].
Gerstmannův-Sträusslerův-Scheinkerův syndrom (GSS)
GSS je velice vzácné dědičné onemocnění (incidence se odhaduje na 1 případ/100 milionů obyvatel/rok) podmíněné specifickými mutacemi v PRNP. Klinicky u GSS dominuje mozečková symptomatologie a průběh onemocnění je výrazně delší (roky, obvykle kolem pěti let) než u sCJN (měsíce). Nejčastější příčinou GSS je mutace P102L [12]. V ČR bylo doposud autopticky potvrzeno pět případů GSS s její pozitivitou.
Fatální familiární insomnie (FFI)
Toto extrémně vzácné, v ČR se nevyskytující onemocnění, je asociováno s mutací D178N a současnou přítomností methioninu na 129. pozici PrP. Klinicky je charakterizované insomnií, poruchami autonomních funkcí, pyramidovou symptomatologií a demencí [25–27].
Kuru
Kuru bylo endemické onemocnění přenášené kanibalskými rituály kmene Fore na Papui-Nové Guinei, popsané v 50. letech 20. století [28]. Přenos se udával alimentární cestou pojídáním mozků považovaných v rámci rituálů za méně hodnotné – proto je dostávaly děti a ženy, které následně na kuru trpěly nejvíce. S omezením kanibalských praktik se incidence kuru rychle snižovala a v současné době se toto onemocnění považuje za prakticky eradikované.
METODY DEFINITIVNÍ DIAGNOSTIKY PRIONOVÝCH ONEMOCNĚNÍ
Definitivní diagnóza prionového onemocnění se opírá o průkaz PrPres v mozkové tkáni, tedy rutinně z autopsie mozku (mozkové biopsie by měly být indikovány jen zcela výjimečně), v případě vCJN i z patrové mandle [29].
Při vlastním průkazu PrPres je nejprve vyšetřovaná tkáň vystavena účinku nespecifických proteináz (proteináza K), které degradují fyziologický, „zdravý“, PrP. Následně monoklonální protilátky namířené proti PrP rozpoznají zachované molekuly PrPres, které odolaly působení proteinázy K.
Standardně lze užít metod nepřímé imunohistochemie nebo western-blotu. Imunohistochemický přístup umožní určit distribuci PrPres v různých částech mozku a morfologii imunoreaktivit – PrPres se může vyskytovat v podobě difuzních synaptických depozit nebo může vytvářet shluky (agregáty) až vzhledu plaků (tzv. plaque-like struktury).
Doba fixace celého mozku probíhá asi 4 týdny, poté se provádí odběr vzorků, zpracování tkáňových bloků a vlastní neuropatologické vyšetření. Doba od pitvy až po definitivní diagnózu, včetně vyloučení dalších možných neurodegenerativních onemocnění, je proto kolem 6 tý-dnů, může však nezřídka trvat i déle. Western-blot je oproti imunohistochemickým metodám citlivější a také rychlejší. Western blot umožňuje prokázat přítomnost PrPres již do druhého dne po provedení pitvy těla pacienta. Jde však pouze o detekci přítomnosti patologického prionového proteinu bez možnosti kvantifikovat distribuci změn v mozku. Podle kritérií WHO stačí k definitivnímu potvrzení diagnózy prionového onemocnění jedna z výše uvedených metod, ideální je však kombinace obou.
Po potvrzení prionového onemocnění ještě rutinně probíhá molekulárně-genetické vyšetření, cílené na detekci polymorfismů na 129. kodonu a možných mutací PRNP v rámci familiárního výskytu těchto onemocnění. Asi 25 % prionových onemocnění má dědičný podklad, u mnoha rodin i v nepřítomnosti relevantní rodinné anamnézy (!) [12].
Definitivní diagnóza prionového onemocnění má tedy tři části: neuropatologickou, imunologickou (metoda western blot) a molekulárně-genetickou. Pouze kombinací těchto přístupů lze diagnózu uzavřít jako definitivní sporadickou nebo familiární formu daného prionového onemocnění.
Diferenciální diagnostika prionových onemocnění
Klinický obraz je pro typicky probíhající formy poměrně specifický a v kombinaci s doplňkovými vyšetřeními lze dosáhnout vysoké míry diagnostické jistoty (tab. 1). Neuropatologický obraz prionových onemocnění je také velmi specifický (viz výše). Přesto v rámci diagnostiky rychle progredující demence je na místě zvážit i jiná primární neurodegenerativní onemocnění, postižení mozku způsobené v rámci kardiovaskulárních onemocnění, metabolické encefalopatie (jaterní, renální), deficit vitaminu B1 (thiaminu) u chronicky podvyživených a často u alkoholiků, systémové autoimunitní onemocnění (Hashimotova thyreoiditia, systémový lupus erytematodes) či lymfoproliferativní onemocnění. Přítomnost nádorového onemocnění (typicky malobuněčný karcinom plic) může vést k paraneoplastické tvorbě protilátek namířených proti antigenům v mozku, velmi často v mozečku (paraneoplastická cerebelární degenerace) nebo v limbických strukturách (hipokampus, amygdala – tzv. limbická encefalitida). Klinicky se jedná o velmi proměnlivé projevy, které mohou často napodobovat prionová onemocnění či jiné neurodegenerace.
![Modifikovaná WHO diagnostická kritéria pro sporadickou formu CJN (z roku 2009) [7]
Table 1. Modified WHO diagnostic criteria for sporadic CJD (from 2009) [7]](https://www.prelekara.sk/media/cache/resolve/media_object_image_large/media/image/b045374325aeb8c2cd0ce9412a63f519.jpg)
Velmi častý je současný výskyt několika různých primárních neurodegenerativních onemocnění, kdy přítomnost např. Alzheimerovy nemoci nevylučuje možnost prionového onemocnění. K tomuto se pak často přidává různě vyjádřená patologie vaskulární (nejčastěji při postižení penetrujících arterií a arteriol), která může dosahovat až podoby tzv. vaskulární demence.
Z výše uvedeného je patrné, že ač je neuropatologická diagnóza izolovaného prionového onemocnění relativně přímočará, je potřeba dále dovyšetřit další možné patologie v mozku, které mohly modifikovat klinické, biochemické a morfologické projevy pozorované ante mortem, zcela zásadní je pak vliv těchto modifikujících faktorů na profil biomarkerů v mozkomíšním moku [30, 31].
PRIONOVÁ ONEMOCNĚNÍ V ČR
Autoptická verifikace všech podezření na prionová onemocnění je v ČR povinná a podléhá hygienicko-epidemiologickému sledování (surveillance). Vyšetření zajišťuje Národní referenční laboratoř (NRL) pro lidská prionová onemocnění při Oddělení patologie a molekulární medicíny v Thomayerově nemocnici v Praze [31]. Od počátku fungování NRL v roce 2002 bylo do roku 2014 včetně definitivně potvrzeno celkem 181 případů prionových onemocnění, z toho 146 případů sCJN, 31 případů fCJN a 4 případy GSS (obr. 2), což odpovídá úhrnné prevalenci v ČR přibližně 13 případů/rok, nicméně je zde patrný trend ke zvyšování prevalence způsobený postupnou optimalizací diagnostických metod.

V ČR probíhá od 1. 1. 2007 na základě doplňku transplantačního zákona [32] povinné testování mozkové tkáně všech dárců rohovek na přítomnost PrPres metodou westernblot. Vyšetření zajišťuje imunologická laboratoř při NRL a spolupracuje se všemi očními bankami v ČR. Vyšetřeno bylo již přes 3 000 vzorků s negativním výsledkem; u jednoho bylo vysloveno podezření z přítomnost PrPres, nicméně další dovyšetřování tuto možnost zamítlo [33].
EPIDEMIOLOGICKÉ ŠETŘENÍ A PROTIEPIDEMICKÁ OPATŘENÍ
Epidemiologické šetření a protiepidemická opatření se v současnosti stále řídí Metodickým pokynem č. 3 Ministerstva zdravotnictví ČR z roku 2001 [34], nový Metodický návod k zajištění systému epidemiologické bdělosti lidských prionových onemocnění bude součástí novely vyhlášky č. 473/2008, o systému epidemiologické bdělosti (surveillance) pro vybrané infekce.
Je třeba zdůraznit, že klasické epidemiologické šetření se zaměřením na pátrání po zdroji nákazy a cestě přenosu onemocnění je relevantní pouze v případě podezření na iatrogenní formu CJN, kdy v anamnéze pacienta pátráme po rizikových lékařských výkonech, vyšetřeních a léčbě a dále u vCJN, kdy jedině v tomto případě je odůvodněné šetření týkající se stravovacích zvyklostí, popř. údajů o chovu a kontaktu se zvířaty. Šetření stravovacích zvyklostí u sporadické a familiární formy CJN není odborně podloženo. Upravený list epidemiologického šetření bude přílohou nového metodického návodu, v současnosti je k dispozici na stránkách www.hygpraha.cz. Vzhledem k tomu, že iatrogenní přenos CJN byl zaznamenán prostřednictvím infikovaných tkání, orgánů, eventuálně chirurgických nástrojů, je jediným preventivním opatřením tohoto přenosu důsledné dodržování bezpečnostních předpisů souvisejících se zacházením s tkáněmi a lékařskými přístroji či nástroji. Podle platné legislativy jsou z dárcovství krve, lidských tkání, orgánů a buněk vyloučeny osoby s rodinnou anamnézou CJN. Rovněž nelze odebírat orgány od osob, kterým byla transplantována rohovka, nebo štěp tvrdé pleny mozkové nebo byly v minulosti léčeny léčivými přípravky zhotovenými z lidských hypofýz. Jako riziková je rovněž uvedena rychlá progresivní demence nebo degenerativní neurologické onemocnění, z dárcovství krve jsou navíc vyloučeny osoby, které pobývaly ve Velké Británii a ve Francii v letech 1980–1996 po dobu delší než 6 měsíců, popř. jim byla podána transfuze před rokem 1996 v zahraničí. Dohledatelnost dárců prostřednictvím Národního registru dárců a Národního registru transplantací po dobu 30 let je zakotvena v tzv. transplantačním zákoně [32, 35, 36].
Kromě vyloučení rizikových osob z dárcovství krve, tkání a orgánů je pro profylaxi iatrogenního přenosu prionových onemocnění nezbytné důsledné dodržování správných dekontaminačních a sterilizačních postupů.
Pokud jsou u pacienta s předpokládaným nebo potvrzeným lidským prionovým onemocněním prováděny diagnostické a léčebné invazivní výkony, musí být postupováno v souladu s Metodickým listem TSE/CJN a vyhláškou č. 306/2012, kde jsou postupy dezinfekce a sterilizace stanoveny [37–39]. Nástroje, které byly v kontaktu s tkáněmi pacientů s prokázaným onemocněním, již nesmí být znovu použity, běžná sterilizace nepostačuje ke zničení prionů, v případě nástrojů použitých u pacientů s klinicky zjevnou diagnózou (odpovídá „pravděpodobné CJN“ podle WHO kritérií) se nástroje ihned po použití nakládají do dezinfekčního roztoku chlornanu, eventuálně hydroxidu sodného, a následně se sterilizují při teplotě 134 °C po dobu 60 minut. V těchto případech lze také řádně dekontaminované nástroje uložit do tzv. karantény a finální postup určit až po stanovení konečné diagnózy.
ZÁVĚR
V současné době probíhá na území ČR epidemiologická surveillance prionových onemocnění na úrovni srovnatelné s ostatními vyspělými zeměmi. Unikátem je systematický screening mozkové tkáně dárců rohovek na přítomnost PrPres. Rostoucí informovanost diagnostikujících lékařů a širší zavedení magnetické rezonance a analýzy mozkomíšního moku do rutinní klinické praxe vedla během 12 let k nárůstu záchytu těchto onemocnění, z nichž přibližně 17 % má dědičný podklad a jejichž záchyt a následné genetické poradenství je zcela zásadní pro rodiny postižených. Přestože iatrogenní ani nová varianta CJN nebyly v ČR dosud potvrzeny, je jejich riziko stále reálné, zejména v kontextu nárůstu počtu miniinvazivních neurochirurgických výkonů či rizika přenosu krevními deriváty.
Poděkování
Práce byla částečně podpořena granty NT14145-3/2013 a NT12094--5/2011 IGA MZd ČR a P303/12/1791 Grantové agentury ČR.
Do redakce došlo dne 11. 2. 2015.
Adresa pro korespondenci:
doc. MUDr. Radoslav Matěj, Ph.D.
Oddělení patologie a molekulární medicíny
Thomayerova nemocnice
Vídeňská 800
140 59 Praha 4-Krč
e-mail: radoslav.matej@ftn.cz
Sources
1. Prusiner SB. Novel proteinaceous infectious particles cause scrapie. Science, 1982;216(4542):136–144.
2. Pastore A, Zagari A. A structural overview of the vertebrate prion proteins. Prion, 2007;1(3):185–197.
3. Beneš J. Infekční lékařství. Praha: Galén; 2009.
4. Aguzzi A, Sigurdson C, Heikenwaelder M. Molecular mechanisms of prion pathogenesis. Annu Rev Pathol, 2008;3 : 11–40.
5. Aguzzi A, Falsig J. Prion propagation, toxicity and degradation. Nat Neurosci, 2012;15(7):936–939.
6. Capellari S, Strammiello R, Saverioni D, et al. Genetic Creutzfeldt--Jakob disease and fatal familial insomnia: insights into phenotypic variability and disease pathogenesis. Acta Neuropathol, 2011;121(1):21–37.
7. Zerr I, Kallenberg K, Summers DM, et al. Updated clinical diagnostic criteria for sporadic Creutzfeldt-Jakob disease. Brain, 2009;132(Pt 10):2659–2668.
8. Parchi P, Strammiello R, Giese A, et al. Phenotypic variability of sporadic human prion disease and its molecular basis: past, present, and future. Acta Neuropathol, 2011;121(1):91–112.
9. Schuette AJ, Taub JS, Hadjipanayis CG, et al. Open biopsy in patients with acute progressive neurologic decline and absence of mass lesion (Podcast)(CME). Neurology, 2010;75(5):419–424.
10. Hsiao K, Meiner Z, Kahana E, et al. Mutation of the prion protein in Libyan Jews with Creutzfeldt-Jakob disease. N Engl J Med, 1991;324(16):1091–1097.
11. Mitrova E, Belay G. Creutzfeldt-Jakob disease with E200K mutation in Slovakia: characterization and development. Acta Virol, 2002;46(1):31–39.
12. Kovacs GG, Puopolo M, Ladogana A, et al. Genetic prion disease: the EUROCJD experience. Hum Genet, 2005;118(2):166–174.
13. Matej R, Kovacs GG, Johanidesova S, et al. Genetic Creutzfeldt--Jakob disease with R208H mutation presenting as progressive supranuclear palsy. Mov Disord, 2012;27(4):476–479.
14. D’Alessandro M, Petraroli R, Ladogana A, et al. High incidence of Creutzfeldt-Jakob disease in rural Calabria, Italy. Lancet, 1998;352(9145):1989–1990.
15. Chapman J, Ben-Israel J, Goldhammer Y, et al. The risk of developing Creutzfeldt-Jakob disease in subjects with the PRNP gene codon 200 point mutation. Neurology, 1994;44(9):1683–1686.
16. Brown P, Brandel JP, Sato T, et al. Iatrogenic Creutzfeldt-Jakob disease, final assessment. Emerg Infect Dis, 2012;18(6):901–907.
17. Will RG, Ironside JW, Zeidler M, et al. A new variant of Creutzfeldt-Jakob disease in the UK. Lancet, 1996;347(9006):921–925.
18. Hewitt PE, Llewelyn CA, Mackenzie J, et al. Creutzfeldt–Jakob disease and blood transfusion: results of the UK Transfusion Medicine Epidemiological Review study. Vox Sang, 2006;91(3):221–230.
19. Current data on variant CJD cases worldwide. Dostupný na www: http://www.cjd.ed.ac.uk/data.html.
20. Heath CA, Cooper SA, Murray K, et al. Validation of diagnostic criteria for variant Creutzfeldt-Jakob disease. Ann Neurol, 2010;67(6):761–770.
21. Zeidler M, Sellar RJ, Collie DA, et al. The pulvinar sign on magnetic resonance imaging in variant Creutzfeldt-Jakob disease. Lancet, 2000;355(9213):1412–1418.
22. Hill AF, Butterworth RJ, Joiner S, et al. Investigation of variant Creutzfeldt-Jakob disease and other human prion diseases with tonsil biopsy samples. Lancet, 1999;353(9148):183–189.
23. Hill AF, Zeidler M, Ironside J, et al. Diagnosis of new variant Creutzfeldt-Jakob disease by tonsil biopsy. Lancet, 1997;349(9045):99–100.
24. Bruce ME, McConnell I, Will RG, et al. Detection of variant Creutzfeldt-Jakob disease infectivity in extraneural tissues. Lancet, 2001;358(9277):208–209.
25. Lugaresi E, Medori R, Montagna P, et al. Fatal Familial Insomnia and Dysautonomia with Selective Degeneration of Thalamic Nuclei. N Engl J Med, 1986;315(16):997–1003.
26. Medori R, Tritschler H-J, LeBlanc A, et al. Fatal Familial Insomnia, a Prion Disease with a Mutation at Codon 178 of the Prion Protein Gene. N Engl J Med, 1992;326(7):444–449.
27. Montagna P, Gambetti P, Cortelli P, et al. Familial and sporadic fatal insomnia. Lancet Neurol, 2003;2(3):167–176.
28. Gajdusek DC and Zigas V. Degenerative disease of the central nervous system in New Guinea; the endemic occurrence of kuru in the native population. N Engl J Med, 1957;257(20):974–978.
29. WHO. WHO manual for surveillance of human transmissible spongiform encephalopathies, including variant Creutzfeldt-Jakob disease. 2003.
30. Toledo JB, Brettschneider J, Grossman M, et al. CSF biomarkers cutoffs: the importance of coincident neuropathological diseases. Acta Neuropathol, 2012;124(1):23–35.
31. Rohan Z, Parobkova E, Johanidesova S, et al. [Human Prion Diseases in the Czech Republic – 10 Years of Experience with the Diagnosis]. Cesk Slov Neurol N, 2013;76(109)(3):300–306.
32. Vyhláška č. 422/2008 Sb., o stanovení bližších požadavků pro zajištění jakosti a bezpečnosti lidských tkání a buněk určených k použití u člověka.
33. Jirsova K, Krabcova I, Novakova J, et al. The assessment of pathogenic prions in the brains of eye tissue donors: 2-years experience in the Czech Republic. Cornea, 2010;29(9):996–999.
34. Metodický list TSE/CJN, surveillance, diagnóza a terapie transmisivních spongiformních encefalopatií a Creutzfeldt-Jakobovy nemoci. 2000.
35. Vyhláška č. 143/2008 Sb., o stanovení bližších požadavků pro zajištění jakosti a bezpečnosti lidské krve a jejich složek (vyhláška o lidské krvi).
36. Zákon č. 285/2002 Sb., o darování, odběrech a transplantacích tkání a orgánů a o změně některých zákonů (transplantační zákon).
37. Metodický list TSE/CJN, surveillance, diagnóza a terapie transmisivních spongiformních encefalopatií a Creutzfeldt-Jakobovy nemoci.
38. WHO. WHO Guidelines on Tissue Infectivity Distribution in Transmissible spongiform Encephalopathies. WHO Press; 2006.
39. Vyhláška č. 306/2012 Sb., o podmínkách předcházení vzniku a šíření infekčních onemocnění a o hygienických požadavcích na provoz zdravotnických zařízení a ústavů sociální péče.
Labels
Hygiene and epidemiology Medical virology Clinical microbiologyArticle was published in
Epidemiology, Microbiology, Immunology

2015 Issue 3
Most read in this issue
- Human prion diseases in the Czech Republic
- Ganciclovir treatment failure in adult allogeneic hematopoietic stem cell transplant recipients with cytomegalovirus infection – a single centre experience
- Genetic and molecular background in autoimmune diabetes mellitus
- Detection of biofilm formation by selected pathogens relevant to the food industry