
Hypertrofia ľavej komory u pacientov s diabetes mellitus: na čo nemyslíme?
Left ventricular hypertrophy in patients with diabetes mellitus: what we don‘t think about?
Left ventricular hypertrophy (LVH) is a common finding in patients with diabetes mellitus (DM). DM and its comorbidities are common in the population and therefore the presence of another disease leading to LVH in a patient with DM can’t be ruled out. Management and prognosis of LVH vary depending on the underlying cause, so its differential diagnosis is very important. The aim of the article is a brief overview of the etiology and diagnostic procedure in patients with left ventricular hypertrophy with a focus on the most clinically important but less frequently diagnosed diseases.
Keywords:
diabetic cardiomyopathy – cardiac amyloidosis – Fabry disease – left ventricular hypertrophy
Authors:
Miroslav Gbúr
Authors‘ workplace:
I. kardiologická klinika VÚSCH a. s. a LF UPJŠ v Košiciach
Published in:
Forum Diab 2021; 10(2): 114-120
Category:
Overview
Hypertrofia ľavej komory (ĽK) je častým nálezom u pacientov s diabetes mellitus (DM). Prevalencia DM a jeho komorbidít v populácii je vysoká a preto prítomnosť ďalšieho ochorenia vedúceho k hypertrofii ĽK u pacienta s DM nie je vylúčená. Manažment a prognóza hypertrofie ĽK sa líšia v závislosti od vyvolávajúcej príčiny, preto jej diferenciálna diagnostika veľmi dôležitá. Cieľom článku je stručný prehľad etiológie a diagnostického postupu u pacientov s hypertrofiou ľavej komory so zameraním na klinicky najvýznamnejšie, ale menej často diagnostikované ochorenia.
Klíčová slova:
amyloidóza srdca – diabetická kardiomyopatia – hypertrofia ľavej komory – Fabryho choroba
Úvod
Hypertrofia ľavej komory (ĽK) je zvyčajne definovaná zväčšením masy alebo zvýšenou hrúbkou stien ĽK. Zvýšená masa ĽK (LVM) je dôležitý rizikový faktor a silný prediktor kardiovaskulárnych príhod [1,2]. Výskyt hypertrofie ĽK a zastúpenie jednotlivých príčin závisí od charakteristík študovanej populácie (vek, etnikum, socio-ekonomické faktory a s nimi súvisiaca prevalencia hypertenzie, obezity a diabetes mellitus) ako aj použitej diagnostickej metódy. Základnou vyšetrovacou metódou pre identifikáciu a hodnotenie hypertrofie ĽK je vzhľadom k svojej dostupnosti a diagnostickej výťažnosti transtorakálna echokardiografia (TTE). Prevalencia echokardiograficky diagnostikovanej hypertrofie ĽK v bežnej populácii sa udáva v rozmedzí 9–19 % [3,4]. Spektrum možných príčin je široké, od spomínaných najčastejších populačných ochorení cez časté (aortálna stenóza, hypertrofická kardiomyopatia) až po ochorenia vyslovene zriedkavé (vrodené poruchy metabolizmu, vrodené syndrómy). V posledných rokoch bol dosiahnutý veľký pokrok v diagnostike ako aj liečbe niektorých špecifických foriem hypertrofie ĽK. Novšie práce ukazujú, že etiológiu hypertrofie ĽK možno systematickým vyšetrením stanoviť až u 90 % pacientov [5]. Rôzne príčiny hypertrofie si vyžadujú rôzne terapeutické prístupy, či už z pohľadu použitia konvenčnej farmakologickej aj nefarmakologickej liečby ako aj pohľadu indikácie špecifickej liečby zasahujúcej do patogenézy konkrétneho ochorenia. Historicky ide najmä o enzým-substitučnú liečbu napríklad pri Fabryho chorobe (FD) [6], novšie o stabilizátory transtyretínu (TTR) v liečbe transty - retínovej amyloidózy (ATTR) [7] alebo včasnú indikáciu hematologickej liečby pri primárnej amyloidóze srdca (AL) [8]. To pochopiteľne význam presného určenia etiológie hypertrofie u každého pacienta ešte zvýrazňuje.
Z pohľadu diabetickej populácie je dôležité, že diabetes mellitus (DM) nezávisle od iných rizikových faktorov spája so zvýšením LVM. Prítomnosť diabetes DM 2. typu (DM2T) zvyšuje riziko hypertrofie ĽK 1,5-násobne [9]. Diabetická kardiomyopatia (KMP), charakterizovaná ako abnormálna štruktúra a funkcia srdca u diabetika v neprítomnosti iných kardiálnych rizikových faktorov a ochorení bola popísaná pred viac ako 40 rokmi [10]. Napriek tomu sú údaje o jej prevalencii stále obmedzené. Súvisí to so samotnou definíciou a doposiaľ chýbajúcimi špecifickými diagnostickými testami/parametrami na jej identifikáciu ako aj typickým zhlukovaním kardiovaskulárnych rizikových faktorov u diabetikov, čo jej diagnózu per se vylučuje. O jej prítomnosti však nepriamo svedčí známa vysoká incidencia srdcového zlyhávania (SZ) u diabetikov – 2,4-násobne vyššia u mužov a 5,1-násobne u žien v porovnaní s osobami bez DM [11]. Riziko vzniku SZ u diabetikov tiež koreluje s mierou glykemickej kontroly [12]. Zároveň DM svojimi orgánovými komplikáciami (nefropatia, polyneuropatia apod) môže imitovať extrakardiálne prejavy niektorých ochorení, ktoré zvažujeme v diferenciálnej diagnostike hypertrofie ĽK.
Diagnostika hypertrofie ĽK
Zväčšenie LVM môže byť dôsledkom zvýšenej hrúbky stien alebo zväčšenia dutiny ĽK, prípadne oboch. Meranie masy ĽK spočíva v stanovení objemu svaloviny ĽK, ktorý potom násobíme mernou hmotnosťou (hustotou) myokardu (cca 1,05 g/ml). Objem svaloviny možno merať priamo – volumometricky pomocou 3D-echokardiografie, magnetickej rezonancie srdca (CMR) a výpočtovej tomografie (CT – Computed Tomography) alebo v praxi častejšie kalkulovať z TTE meraní hrúbky stien a enddiastolického rozmeru ĽK v M-móde a 2D-zobrazení. Hodnoty LVM závisia okrem použitej metódy aj od pohlavia, veku, telesnej veľkosti a hmotnosti a etnika, preto sa zistené hodnoty rôznym spôsobom indexujú (najčastejšie na povrch tela). Napriek tomu je definícia univerzálnych referenčných hodnôt komplikovaná a je stále predmetom diskusií [13]. Normálne hodnoty akceptované aktuálne platnými odporúčaniami a klasifikáciu geometrických typov hypertrofie ĽK sumarizuje tab. 1. a schéma.
![Normálne hodnoty masy ĽK (LVM), masy ĽK indexovanej na povrch tela (LVMI), relatívnej hrúbky
stien ĽK (RWT), hrúbky interventrikulárneho septa (IVSd) a zadnej steny (ZSd) ĽK v diastole pri TTE.
Upravené podľa [13]](https://www.prelekara.sk/media/cache/resolve/media_object_image_large/media/image_pdf/b9755c91da05c060b3c2f0df91e2d044.png)
![Schéma | Hodnotenie geometrie ĽK na základe
masy (LVM) a relatívnej hrúbky stien ĽK
(RWT). RWT je kalkulovaná ako 2-krát
hrúbka zadnej steny/enddiastolický rozmer
ĽK. Upravené podľa [13]](https://pl-master.mdcdn.cz/media/image_pdf/297b86f21a900d011354fbf6b6ab36fd.png?version=1628847648)
Hodnotenie LVM nie je vhodné na kvantifikáciu hypertrofie ĽK pri jej asymetrickej distribúcii, často napríklad pri hypertrofickej KMP. V týchto prípadoch hodnotíme hypertrofiu ĽK na základe merania hrúbky stien jednotlivých segmentov ĽK. Hypertrofická kardiomyopatia (KMP) je definovaná hrúbkou steny ≥ 15 mm aspoň v jednom segmente zistená akoukoľvek zobrazovacou metódou. Geneticky podmienené aj získané ochorenia sa však môžu prejavovať aj menším zhrubnutím steny (13–14 mm), ktoré si tiež vyžadujú ďalšiu diagnostiku [14].
Elektrokardiografia (EKG) má stále význam ako skríningová metóda v špecifických populáciách (športovci) [15] a do istej miery aj v diferenciálnej diagnostike príčin hypertrofie. Vo všeobecnej populácii je však použitie EKG limitované relatívne nízkou senzitivitou (klasické voltážové kritéria približne 20–40 %, novší index Peguero-Lo Presti približne 62 %) [16].
Naopak CMR je považovaná za zlatý štandard diagnostiky. Umožňuje presné a reprodukovateľné hodnotenie masy, geometrie a funkcie ĽK aj v prípadoch, v ktorých je výťažnosť echokardiografického vyšetrenia limitovaná zlou vizualizáciou. Špeciálnym prínosom CMR je možnosť charakterizácie tkaniva (prítomnosť a lokalizácia fibrózy, hodnotenie objemu extra - a intracelulárneho kompartmentu), čím významne prispieva k určeniu príčiny hypertrofie[17]. Hlavnými limitáciami CMR sú jej kontraindikácie a obmedzená dostupnosť [17,18].
V týchto prípadoch možno na presnejšie zhodnotenie morfológie a funkcie ĽK použiť CT srdca. Výhodou je možnosť súčasne vyšetriť koronárne riečisko, nevýhodou radiačná záťaž a zatiaľ chýbajúca schopnosť dostatočnej charakterizácie tkaniva [18].
Etiológia a diferenciálna diagnostika hypertrofie ĽK
Hypertrofia ĽK môže byť podmienená hypertrofiou kardiomyocytov (zmnoženie a zväčšenie sarkomér, intracelulárne hromadenie rôznych látok), zmnožením extracelulárnej matrix (v dôsledku fibrózy, depozície abnormálnych proteínov) alebo ich kombináciou. U väčšiny pacientov je hypertrofia ĽK zistená pri vyšetrovaní ich klinických ťažkostí. Nezriedka však môže byť aj náhodným nálezom pri TTE alebo inom zobrazovacom vyšetrení vykonanom z rôznych iných dôvodov. Prvým krokom v diferenciálnej diagnostike je vždy vylúčenie najčastejších príčin. Kontrolu hypertenzie a prípadnú dosiaľ nediagnostikovanú hypertenziu je vhodné overiť, resp. potvrdiť ambulantným monitorovaním tlaku krvi. Klinické vyšetrenie a TTE umožní identifikáciu chlopňových a vrodených chýb (hlavne aortálnej stenózy), ktoré vedú k tlakovému alebo objemovému preťaženiu ĽK. Zároveň však prítomnosť takejto bežnej príčiny nevylučuje možnú koincidenciu s ďalším, zriedkavejším ochorením. Fabryho choroba (Fabry Disease – FD) bola zistená takmer u 1 % hypertonikov s hrúbkou steny ĽK ≥ 13 mm [19], ATTR amyloidóza u 16 % pacientov podstupujúcich transkatétrovú implantáciu aortálnej chlopne pre závažnú aortálnu stenózu [20]. Vo všeobecnosti sa systematické došetrenie etiológie hypertrofie ĽK odporúča u pacientov s hrúbkou steny ĽK ≥ 13 mm [21]. Takáto hypertrofia bola pri CMR prítomná u 4,6 % osôb bez zjavného kardiovaskulárneho ochorenia vo veku 45–84 rokov [22].
Ďalším krokom je odobratie kompletnej rodinnej a osobnej anamnézy. Zameriavame sa na extrakardiálne príznaky systémových ochorení, resp. syndrómov, ktoré slúžia ako tzv. red flags a zvyšujú pravdepodobnosť prítomnosti špecifického ochorenia. Ide predovšetkým o neurologické príznaky ako cievna mozgová príhoda v mladom veku, poruchy chôdze a rovnováhy, sluchu, neuropatická bolesť, príznaky autonómnej neuropatie, svalová slabosť, syndróm karpálneho tunela a podobne. Ďalej sú to kožné zmeny, hyper - a hypohidróza, renálne postihnutie, gastrointestinálne príznaky, ale aj DM (pri mitochondriálnych ochoreniach) [14,21]. V rodinnej anamnéze pátrame po kardiálnych manifestáciách u blízkych príbuzných (známa KMP alebo vrodené ochorenie, náhle úmrtie a podobne) s cieľom určiť typ dedičnosti. Autozomálne dominantná dedičnosť je typická pre hypertrofickú KMP (sarkomérové mutácie) a vrodenú ATTR amyloidózu, autozomálne recesívna dedičnosť pre Pompeho chorobu a na X-chromozóm viazaná dedičnosť hlavne pre Fabryho a Danonovu chorobu. Charakteristické nálezy, ktoré usmernia ďalší diagnostický postup, môžeme nájsť aj na EKG. Prítomnosť AV-blokád je častá pri infiltratívnych, metabolických a mitochondriálnych KMP. Extrémne vysokú voltáž QRS, niekedy sprevádzanú hlbokými (gigantickými) negatívnymi T-vlnami, nájdeme pri „strádavých“ ochoreniach (Pompeho a Danonova choroba, FD). Naopak nízka voltáž QRS ukazuje na možnú amyloidózu. Negatívne T-vlny v prekordiálnych zvodoch sú tiež časté pri apikálnej forme hypertrofickej KMP. V laboratórnom náleze sa môžeme stretnúť s eleváciou kreatinínu a proteinúriou pri renálnom postihnutí (FD, amyloidóza). Zvýšené hodnoty transamináz a kreatínkinázy (z postihnutia skeletálneho svalstva) nájdeme pri Danonovej chorobe a mitochondriálnych ochoreniach. Tie sa tiež môžu spájať s laktátovou acidózou a myoglobinúriou [21]. Veľmi dôležitý je opakovaný nález mierneho zvýšenia kardiálnych troponínov, ktorý svedčí pre pretrvávajúce dlhodobé poškodenie myokardu a mal by iniciovať pátranie po jeho možných neischemických príčinách (hlavne amyloidózy, ale aj FD a ďalšie). Srdcové troponíny spolu s nátriuretickými peptidmi tiež predstavujú dôležité prognostické parametre pri srdcovej amyloidóze [23]. Dôležitú úlohu v diferenciálnej diagnostike hrajú zobrazovacie metódy. Ich správny výber, indikácia a interpretácia patrí do rúk kardiológa, ideálne zaoberajúceho sa diagnostikou kardiomyopatií a presahuje zámer tohto článku.
Genetické vyšetrenie by malo byť zamerané na potvrdenie suponovanej diagnózy s cieľom umožniť rodinný skríning a cielenú včasnú diagnostiku ochorenia u blízkych príbuzných. Výsledkom genetického vyšetrenia môže byť nález rôznych genetických variantov, často s nejasnou patogenicitou a klinickým významom, a naopak negatívny výsledok nevylučuje prítomnosť familiárnej hypertrofickej KMP. Genetická analýza sa preto neodporúča ako iniciálny diagnostický alebo skríningový test. Jednou z mála výnimiek je genetické vyšetrenie u žien v rámci skríningu FD (viď nižšie) [14]. Prehľad najdôležitejších príčin hypertrofie ĽK uvádza tab. 2, tri najdôležitejšie špecifické príčiny hypertrofie ĽK aj z pohľadu diabetickej populácie sú bližšie opísané v ďalšom texte.
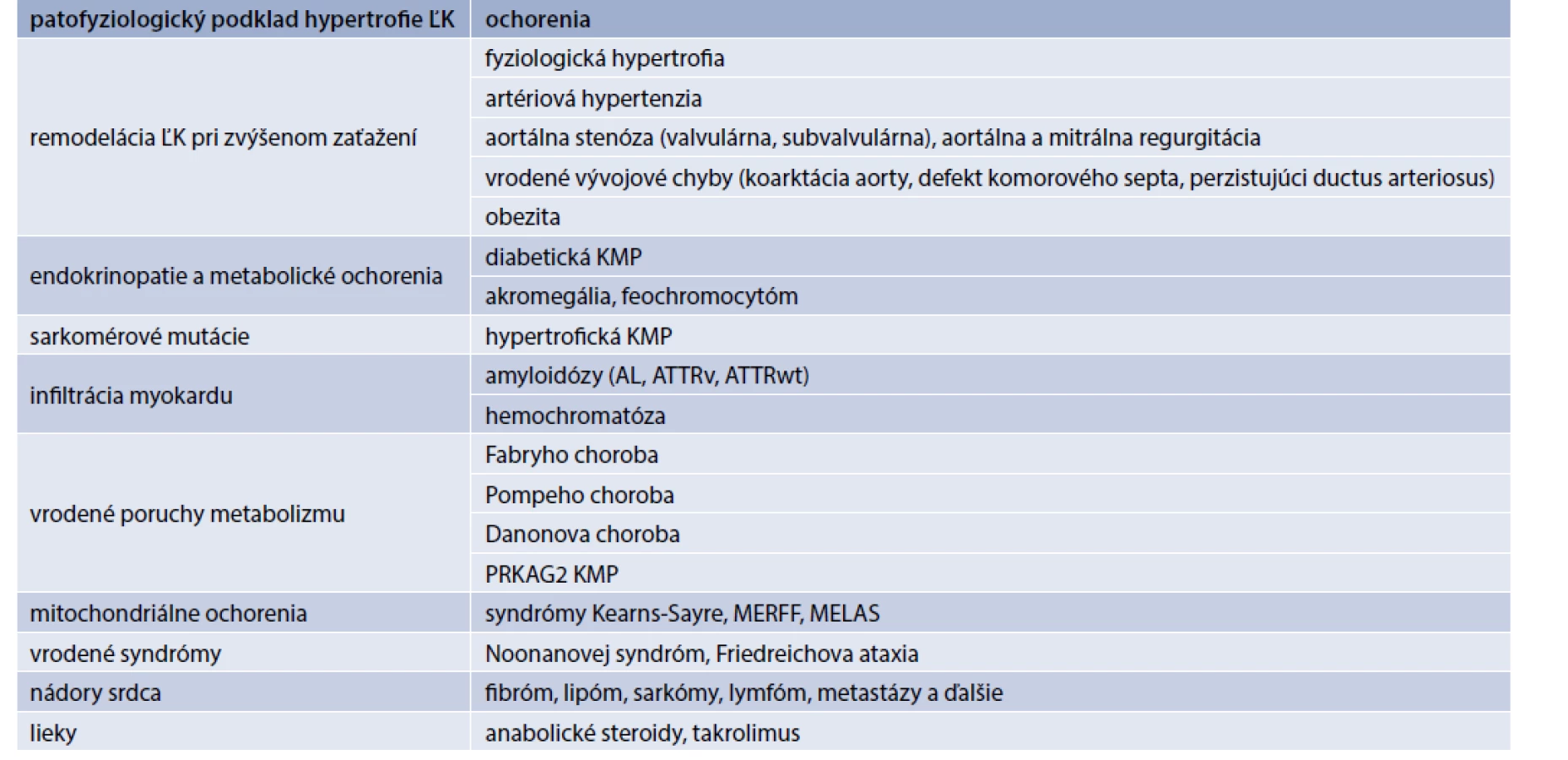
Amyloidóza srdca
Amyloidóza je ochorenie spôsobené extracelulárnym ukladaním nerozpustných fibríl amyloidu v tkanivách. Fibrily vznikajú agregáciou proteinových prekurzorov a obsahujú aj nefibrilárne komponenty (sérový amyloidový proteín a proteoglykán heparánsulfát). Amyloid sa môže ukladať len lokálne alebo častejšie systémovo v rôznych orgánoch. Poznáme viac ako 30 proteínových prekurzorov s rôznou orgánovou distribúciou amyloidu [24]. Z pohľadu kardiálneho postihnutia sú najdôleži tejšie 2 typy amyloidózy. Prvou je primárna systémová amyloidóza (AL), pri ktorej sú proteínom tvoriacim fibrily monoklonálne ľahké reťazce (častejšie lambda ako kappa) produkované patologickým kmeňom plazmatických buniek. Tento klon môže (približne 5–10 % prípadov), ale častejšie nespĺňa diagnostické kritéria pre mnohopočetný myelóm (> 20 % infiltrácia kostnej drene klonálnymi plazmatickými bunkami) [8]. Druhou je transtyretínová amyloidóza (ATTR), pri ktorej sú fibrily tvorené transtyretínom (TTR). TTR je transportný proteín syntetizovaný prakticky výlučne v pečeni. Do obehu sa z pečene dostáva ako tetramér, ktorý viaže a transportuje hormóny štítnej žľazy a retinol. Tetramér destabilizuje buď v dôsledku bodovej mutácie (ATTRv – variant, mutovaný TTR) alebo z nejasných príčin hlavne vo vyššom veku (ATTRwt – wild type TTR, v minulosti senilná amyloidóza) na monomér, ktorý agreguje a vytvára fibrily [24,25]. Pri oboch ochoreniach je postihnutie srdca rozhodujúce pre prognózu pacientov. V obidvoch prípadoch však ide o systémové ochorenia a rozpoznanie extrakardiálnych prejavov môže nasmerovať diagnostiku správnym smerom. Pre AL je typická makroglosia, podkožné krvácania v dôsledku zvýšenej fragility malých ciev a nefrotický syndróm. Podobne ako pri ATTR medzi extrakardiálne príznaky patrí aj polyneuropatia a syndróm karpálneho tunela, ktorý predchádza kardiálnej manifestácii aj o niekoľko rokov [8]. Ešte častejšie sa s ním stretávame pri ATTRwt. V dôsledku ukladania amyloidu do šliach vzniká aj lumbálna spinálna stenóza (prejavujúca sa neurogénnymi klaudikáciami) a spontánna ruptúra šľachy bicepsu s typickou deformitou svalu (príznak Pepka námorníka). Pri oboch formách ATTR sa stretávame aj so senzorickou, motorickou a autonómnou neuropatiou, vrátane postihnutia gastrointestinálneho traktu. Tieto sú typické pre neurologickú formu ATTRv, ktorá sa nazýva familiárna amyloidná polyneuropatia (FAP). Spektrum postihnutia (prevaha kardiálneho alebo neurologického) pri ATTRv závisí od konkrétnej mutácie a jednotlivé mutácie majú väčšinou typický geografický výskyt [25].
Postihnutie srdca sa manifestuje častejšie u mužov, vo veku nad 40 rokov, pri ATTRw zvyčajne viac ako 60 rokov. Pri EKG je typická nízka voltáž QRS komplexu (obr. 1), ktorá je v protiklade s významnou hypertrofiou ĽK pri echokardiografii. Často je prítomný obraz pseudonekrózy s Q-kmitmi, AV-blokády a predovšetkým v neskorších štádiách fibrilácia predsiení. Echokardiograficky nachádzame symetrickú koncentrickú hypertrofiu ĽK (hrúbka stien ≥ 14 mm). Hypertrofia ĽK je sprevádzaná diastolickou dysfunkciou (väčšinou 2.–3. stupňa) s dilatáciou ľavej predsiene, ejekčná frakcia býva spočiatku zachovaná, alebo mierne znížená, častá je malá perikardiálna efúzia. Typická je porucha longitudinálnej funkcie ĽK so znížením globálneho longitudinálneho strainu, ktorá relatívne najmenej postihuje apikálne segmenty ĽK (tzv. obraz apical sparing). Tieto nálezy by u pacienta s hypertrofiou ĽK mali viesť k doplneniu ďalších vyšetrení za účelom potvrdenia a diferenciácie srdcovej amyloidózy. Klinicky sa amyloidóza srdca manifestuje príznakmi SZ, dominuje slabosť, dýchavica a opuchy. Častý je sklon k hypotenzii a intolerancia antihypertenzívnej a štandardnej liečby SZ [23,25]. Podozrenie by preto mala vzbudiť progresia hypertrofie ĽK u hypertonika, ktorému bola redukovaná antihypertenzívna liečba pre „normalizáciu“ tlaku krvi.
Archív autora
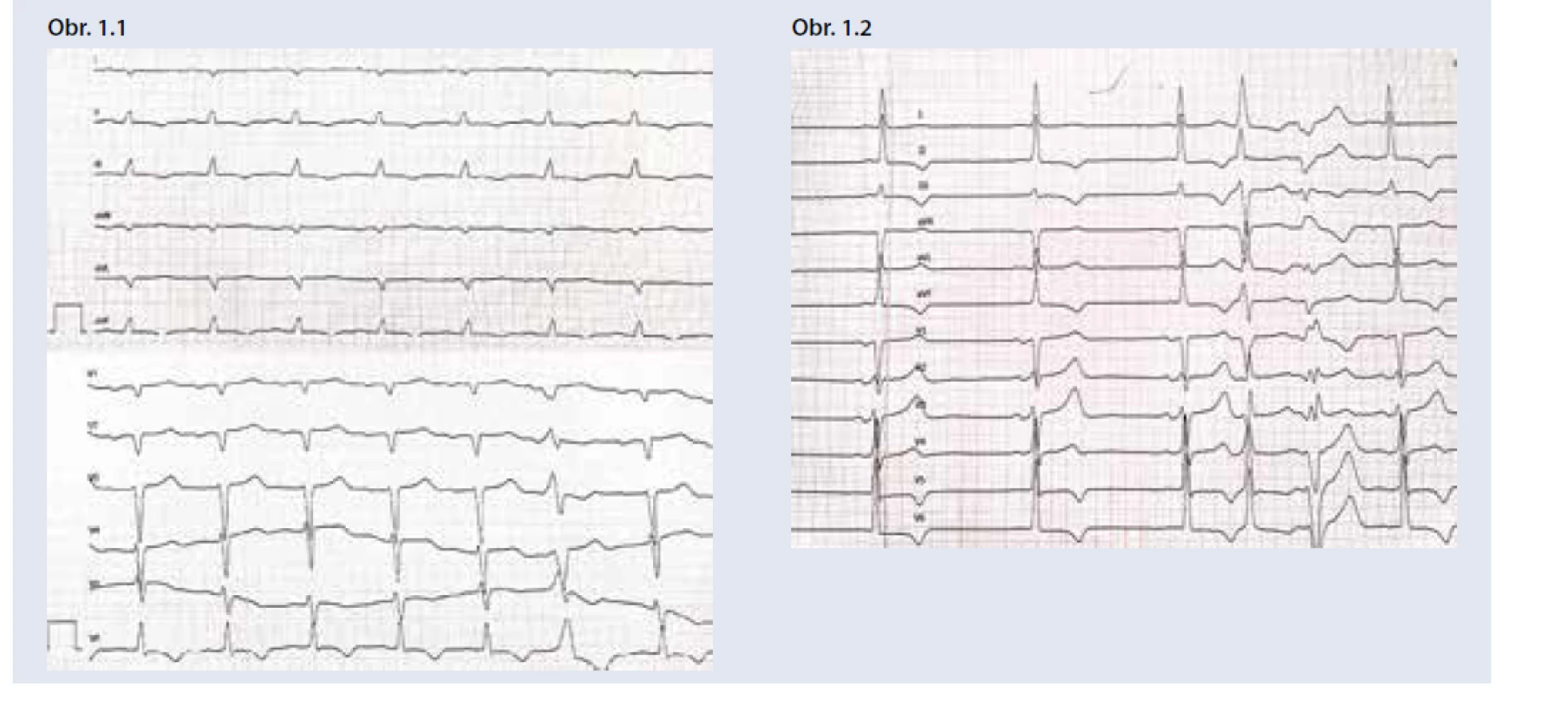
Prvým krokom v diagnostike je skríning monoklonálnej gamapatie (vyšetrenie imunofixácie v sére a moči a voľných ľahkých reťazcov v sére) a scintigrafia skeletu (osteoscan). Fibrily ATTR doposiaľ neznámym mechanizmom viažu osteotropné rádiofarmaká a miera ich vychytávania v myokarde sa hodnotí pomocou vizuálneho 3-stupňového Peruginiho skóre [26]. Nález významnej akumulácie rádiofarmaka (2. a 3. stupeň Peruginiho skóre) u pacienta s vylúčenou monoklonálnou gamapatiou je diagnostický pre srdcovú ATTR [27]. V prípade potvrdenia monoklonálnej gamapatie je indikované hematologické vyšetrenie a biopsia postihnutých orgánov s cieľom potvrdenia a typizácie amyloidu. U pacientov s negatívnym alebo nekonkluzívnym výsledkom prvolíniových vyšetrení je možné podozrenie na amyloidózu podporiť vyšetrením CMR a následne zvážiť histologizáciu, najčastejšie cestou endomyokardiálnej biopsie [8,25].
Fabryho choroba
Fabryho choroba je progresívne, na X-chromozóm viazané genetické ochorenie spôsobené chýbajúcou alebo nedostatočnou aktivitou lyzozomálneho enzýmu alfa-galaktozidázy A (GLA). Tento funkčný defekt má za následok hromadenie glykosfingolipidov, hlavne globotriaozylceramidu (Gb3) a jeho deacylovaného derivátu globotryaosylsfingozínu (lyso-Gb3) v lyzosómoch a telesných tekutinách. Postihnuté sú najmä bunky srdca, obličiek, kože, neuróny a endotel ciev [28]. Ochorenie vo svojej klasickej forme postihuje hlavne mužov (hemizygotov). V súčasnosti ale vieme, že aj heterozygótne ženy (ktoré sú z genetického hľadiska mozaiky) môžu mať závažné klinické prejavy vrátane kardiovaskulárnych [29]. Tie môžu byť sprevádzané, resp. pri nerozpoznanej klasickej forme predchádzané ďalšími príznakmi FD, po ktorých treba anamnesticky pátrať. V detstve ide hlavne o záchvatovité akroparestézie a bolesti končatín, hypohidrózu s intoleranciou tepla a námahy a záchvaty bolestí brucha. Diagnosticky najcennejšími znakmi sú angiokeratómy na trupe a cornea verticillata (šedo-hnedé depozity v rohovke vyskytujúce sa aj pri užívaní niektorých liekov, hlavne amiodarónu). Z orgánových komplikácií je to predovšetkým proteinúria, progredujúca renálna insuficiencia a kryptogénne cievne mozgové príhody v mladej dospelosti (približne v 20.-30. roku veku) [30]. Takzvaná neskorá forma ochorenia sa manifestuje takmer výlučne kardiálnym postihnutím s pomalšou progresiou [28].
Na EKG sú väčšinou prítomné znaky hypertrofie ĽK s repolarizačnými zmenami v zvodoch z ľavého prekordia. V počiatočných štádiách býva skrátený interval PQ bez preexcitácie, neskôr sa prevodové intervaly predlžujú a pacienti často vyžadujú implantáciu kardiostimulátora pre pokročilé atrioventrikulárne (AV) blokády [31].
Hlavnou kardiálnou manifestáciou je symetrická koncentrická hypertrofia ĽK, bez obštrukcie vo výtokovom trakte ĽK. Je prítomná u viac ako polovice mužov s FD (zvyčajne po 30. roku veku) a viac ako tretiny žien (obyčajne po 40. roku života). Diastolická dysfunkcia je prítomná u všetkých pacientov, ejekčná frakcia ĽK býva zachovaná, zhoršuje sa až v štádiu pokročilého ochorenia, kedy sa rozvíja reštriktívny typ plnenia ĽK [32].
Pri CMR je neskoré vychytávanie gadolínia (LGE) z nejasných príčin najčastejšie prítomné v strednej vrstve myokardu bazálne a midpapiplárne inferolaterálne. Ďalším charakteristickým nálezom sú nízke natívne hodnoty v T1-zobrazení, ktoré odlišujú postihnutie pri FD od hypertrofie ĽK pri sarkomérových mutáciách, amyloidóze a hypertenzii [33].
Základom diagnostiky je skríningové vyšetrenie pomocou metódy tzv. suchej kvapky, ktoré je indikované u všetkých pacientov s hypertrofiou ĽK (hrúbka stien ≥ 13 mm u mužov vo veku > 30 rokov a žien > 40 rokov) [34]. Komerčne dostupné testy umožňujú stanovenie aktivity GLA (u mužov), genetickú analýzu (u žien a mužov s potvrdeným deficitom GLA) ako aj vyšetrenie koncentrácie lyzo-Gb3. Podľa štúdií možno pozitívny nález očakávať asi u 0,5–1 % pacientov s hypertrofickou KMP [35].
Diabetická kardiomyopatia
Diabetická KMP je najčastejšie definovaná ako dysfunkcia myokardu u pacienta s DM, ktorá sa vyvinie v neprítomností iných kardiovaskulárnych ochorení ako koronárna choroba, nekontrolovaná hypertenzia, významné valvulopatie a vrodené srdcové chyby. Jej patogenéza nie je plne objasnená [12]. Najväčšiu úlohu (predovšetkým u pacientov s DM2T) pravdepodobne zohráva inzulínová rezistencia, kompenzačná hyperinzulinémia a pretrvávajúca hyperglykémia. Tieto vedú v myokarde k zvýšenému metabolizmu voľných mastných kyselín, lipotoxicite a oxidatívnemu stresu. Ukladanie konečných produktov pokročilej glykácie (AGEs) stimuluje produkciu kolagénu, a tým vznik fibrózy. Na progresii sa tiež podieľa endotelová a mikrovaskulárna dysfunkcia, neurohromonálne zmeny (zvýšená aktivita adrenergného a renín-angiotenzín-aldosterónového systému) a rôzne subcelulárne zmeny (mitochondriálna dysfunkcia, preťaženie – stres endoplazmatického retikula, poruchy homeostázy kalcia a ďalšie), ktoré podporujú rozvoj koncentrickej remodelácie a hypertrofie ĽK a spolu s fibrózou vedú z zvýšenej tuhosti a diastolickej dysfunkcii ĽK [12,36]. Tieto zmeny charakterizujú reštriktívny fenotyp diabetickej KMP, ktorý sa klinicky manifestuje ako SZ zo zachovanou EF ĽK. U pacientov s DM 1. typu dochádza aj k úbytku kardiomyocytov, čo vedie k poruche systolickej funkcie a dilatácii ĽK – tzv. dilatačný fenotyp diabetickej KMP. Či a ako často môže prechádzať reštriktívny fenotyp do dilatačného, nie je jasné [37].
Diagnostika diabetickej KMP je v klinickej praxi ťažká. Klinický ani echokardiografický obraz nie sú špecifické. Úloha CMR a biomarkerov je v štádiu výskumu. V praxi však o nej treba uvažovať u diabetika s echokardiografickým nálezom hypertrofie ĽK a prejavmi SZ po vylúčení koronárnej choroby, nekontrolovanej hypertenzie a iných príčin hypertrofie.
Záver
Hypertrofia ĽK je síce pomerne bežným nálezom, vyžaduje však pozornosť nielen kardiológov, ale aj ďalších odborností zainteresovaných v manažmente pacienta. Aj keď DM a komorbidity, ktoré ho sprevádzajú, môžu samy viesť k rozvoju hypertrofie ĽK, nemali by sme predovšetkým pri náleze významnej hypertrofie (s hrúbkou steny ĽK ≥ 13 mm) zabúdať ani na jej menej časté príčiny. Ich rozpoznanie je založené na poznaní hlavných typických znakov (red flags) a následnej správnej indikácii doplňujúcich zobrazovacích, laboratórnych a genetických vyšetrení. Včasná diagnostika potom v mnohých prípadoch umožní kauzálnu špecifickú liečbu, ktorá môže zásadne ovplyvniť prognózu pacienta.
MUDr. Miroslav Gbúr
www.vusch.sk
Doručené do redakcie 15. 3. 2021
Prijaté po recenzii 13. 4. 2021
Sources
1. Levy D, Garrison RJ, Savage DD et al. Prognostic Implications of Echocardiographically Determined Left Ventricular Mass in the Framingham Heart Study. N Engl J Med 1990; 322(22): 1561–1566. Dostupné z DOI: <http://dx.doi.org/10.1056/nejm199005313222203>.
2. Bluemke DA, Kronmal RA, Lima JA et al. The Relationship of Left Ventricular Mass and Geometry to Incident Cardiovascular Events. The MESA (Multi-Ethnic Study of Atherosclerosis) Study. J Am Coll Cardiol 2008; 52(25): 2148–2155. Dostupné z DOI: <http://dx.doi.org/10.1016/j.jacc.2008.09.014>.
3. Levy D, Anderson KM, Savage DD et al. Echocardiographically detected left ventricular hypertrophy: prevalence and risk factors. The Framingham Heart Study. Ann Intern Med 1988; 108(1): 7–13. Dostupné z DOI: <http://dx.doi.org/10.7326/0003–4819–108–1-7>.
4. Schirmer H, Lunde P, Rasmussen K. Prevalence of left ventricular hypertrophy in a general population; The Tromsø Study. Eur Heart J 1999; 20(6): 429–438. Dostupné z DOI: <http://dx.doi.org/10.1053/euhj.1998.1314>.
5. Beneyto M, Cariou E, Brunel J et al. Tip of the iceberg: a tertiary care centre retrospective study of left ventricular hypertrophy aetiologies. Open Hear 2021; 8(1). Dostupné z DOI: <http://dx.doi.org/10.1136/openhrt-2020–001462>.
6. Baig S, Vijapurapu R, Alharbi F et al. Diagnosis and treatment of the cardiovascular consequences of Fabry disease. QJM 2019; 112(1): 3–9. Dostupné z DOI: <http://dx.doi.org/10.1093/qjmed/hcy120>.
7. Maurer MS, Schwartz JH, Gundapaneni B et al. Tafamidis Treatment for Patients with Transthyretin Amyloid Cardiomyopathy. N Engl J Med 2018; 379(11): 1007–1016. Dostupné z DOI: <http://dx.doi.org/10.1056/NEJMoa1805689>.
8. Falk RH, Alexander KM, Liao R et al. AL (Light-Chain) Cardiac Amyloidosis: A Review of Diagnosis and Therapy. J Am Coll Cardiol 2016; 68(12): 1323–1341. Dostupné z DOI: <http://dx.doi.org/10.1016/j.jacc.2016.06.053>.
9. Eguchi K, Boden-Albala B, Jin Z et al. Association between diabetes mellitus and left ventricular hypertrophy in a multiethnic population. Am J Cardiol 2008; 101(12): 1787–1791. Dostupné z DOI: <http://dx.doi.org/10.1016/j.amjcard.2008.02.082>.
10. Rubler S, Dlugash J, Yuceoglu YZ et al. New type of cardiomyopathy associated with diabetic glomerulosclerosis. Am J Cardiol 1972; 30(6): 595–602. Dostupné z DOI: <http://dx.doi.org/https://doi.org/10.1016/0002–9149(72)90595–4>.
11. Kannel WB, Hjortland M, Castelli WP. Role of diabetes in congestive heart failure: the Framingham study. Am J Cardiol 1974; 34(1): 29–34. Dostupné z DOI: <http://dx.doi.org/10.1016/0002–9149(74)90089–7>.
12. Tan Y, Zhang Z, Zheng C et al. Mechanisms of diabetic cardiomyopathy and potential therapeutic strategies: preclinical and clinical evidence. Nat Rev Cardiol 2020; 17(9): 585–607. Dostupné z DOI: <http://dx.doi.org/10.1038/s41569–020–0339–2>.
13. Lang RM, Badano LP, Mor-Avi V et al. Recommendations for cardiac chamber quantification by echocardiography in adults: an update from the American Society of Echocardiography and the European Association of Cardiovascular Imaging. J Am Soc Echocardiogr 2015; 28(1): 1–39. e14. Dostupné z DOI: <http://dx.doi.org/10.1016/j.echo.2014.10.003>.
14. Dostupné z DOI: . 14. Elliott PM, Anastasakis A, Borger MA et al. 2014 ESC Guidelines on diagnosis and management of hypertrophic cardiomyopathy: the Task Force for the Diagnosis and Management of Hypertrophic Cardiomyopathy of the European Society of Cardiology (ESC). Eur Heart J 2014; 35(39): 2733–2779. Dostupné z DOI: <http://dx.doi.org/10.1093/eurheartj/ehu284>.
15. Pelliccia A, Sharma S, Gati S et al. 2020 ESC Guidelines on sports cardiology and exercise in patients with cardiovascular disease: The Task Force on sports cardiology and exercise in patients with cardio vascular disease of the European Society of Cardiology (ESC). Eur Heart J 2021; 42(1): 17–96. Dostupné z DOI: <http://dx.doi.org/10.1093/eurheartj/ehaa605>.
16. Peguero JG, Lo Presti S, Perez J et al. Electrocardiographic Criteria for the Diagnosis of Left Ventricular Hypertrophy. J Am Coll Cardiol 2017; 69(13): 1694–1703. Dostupné z DOI: <http://dx.doi.org/10.1016/j.jacc.2017.01.037>.
17. Burrage MK, Ferreira VM. Cardiovascular Magnetic Resonance for the Differentiation of Left Ventricular Hypertrophy. Curr Heart Fail Rep 2020; 17(5): 192–204. Dostupné z DOI:<http://dx.doi.org/10.1007/s11897–020–00481-z>.
18. Ommen SR, Mital S, Burke MA et al. 2020 AHA/ACC Guideline for the Diagnosis and Treatment of Patients with Hypertrophic Cardiomyopathy. Circulation 2020; 142(25): e558-e631. Dostupné z DOI: <http://dx.doi.org/10.1161/CIR.0000000000000937>.
19. Terryn W, Deschoenmakere G, De Keyser J et al. Prevalence of Fabry disease in a predominantly hypertensive population with left ventricular hypertrophy. Int J Cardiol 2013; 167(6): 2555–2560. Dostupné z DOI: <http://dx.doi.org/10.1016/j.ijcard.2012.06.069>.
20. Castaño A, Narotsky DL, Hamid N, et al. Unveiling transthyretin cardiac amyloidosis and its predictors among elderly patients with severe aortic stenosis undergoing transcatheter aortic valve replacement. Eur Heart J 2017; 38(38): 2879–2887. Dostupné z DOI: <http://dx.doi.org/10.1093/eurheartj/ehx350>.
21. Linhart A, Cecchi F. Common presentation of rare diseases: Left ventricular hypertrophy and diastolic dysfunction. Int J Cardiol 2018; 257 : 344–350. Dostupné z DOI: <http://dx.doi.org/10.1016/j.ijcard.2018.01.006>.
22. Massera D, McClelland RL, Ambale-Venkatesh B et al. Prevalence of Unexplained Left Ventricular Hypertrophy by Cardiac Magnetic Resonance Imaging in MESA. J Am Heart Assoc 2019; 8(8): e012250. Dostupné z DOI: <http://dx.doi.org/10.1161/JAHA.119.012250>.
23. Kittleson MM, Maurer MS, Ambardekar AV et al. Cardiac Amyloidosis: Evolving Diagnosis and Management: A Scientific Statement From the American Heart Association. Circulation 2020; 142(1): e7-e22. Dostupné z DOI: <http://dx.doi.org/10.1161/CIR.0000000000000792>.
24. Benson MD, Buxbaum JN, Eisenberg DS et al. Amyloid nomenclature 2018: recommendations by the International Society of Amyloidosis (ISA) nomenclature committee. Amyloid 2018; 25(4): 215–219. Dostupné z DOI: <http://dx.doi.org/10.1080/13506129.2018.1549825>.
25. Ruberg FL, Grogan M, Hanna M et al. Transthyretin Amyloid Cardiomyopathy: JACC State-of-the-Art Review. J Am Coll Cardiol 2019; 73(22): 2872–2891. Dostupné z DOI: <http://dx.doi.org/10.1016/j.jacc.2019.04.003.
26. Perugini E, Guidalotti PL, Salvi F et al. Noninvasive etiologic diagnosis of cardiac amyloidosis using 99mTc-3,3-diphosphono-1,2-propanodicarboxylic acid scintigraphy. J Am Coll Cardiol 2005; 46(6): 1076 – 1084. Dostupné z DOI: <http://dx.doi.org/10.1016/j.jacc.2005.05.073>.
27. Gillmore JD, Maurer MS, Falk RH et al. Nonbiopsy Diagnosis of Cardiac Transthyretin Amyloidosis. Circulation 2016; 133(24): 2404 – 2412. Dostupné z DOI: <http://dx.doi.org/10.1161/CIRCULATIONAHA.116.021612>.
28. Linhart A, Germain DP, Olivotto I et al. An expert consensus document on the management of cardiovascular manifestations of Fabry disease. Eur J Heart Fail 2020; 22(7): 1076–1096. Dostupné z DOI: <http://dx.doi.org/https://doi.org/10.1002/ejhf.1960>.
29. Echevarria L, Benistan K, Toussaint A et al. X-chromosome inactivation in female patients with Fabry disease. Clin Genet 2016; 89(1): 44–54. Dostupné z DOI: <http://dx.doi.org/10.1111/cge.12613>.
30. Mehta A, Beck M, Eyskens F et al. Fabry disease: a review of current management strategies. QJM 2010; 103(9): 641–659. Dostupné z DOI: <http://dx.doi.org/10.1093/qjmed/hcq117>.
31. Yousef Z, Elliott PM, Cecchi F et al. Left ventricular hypertrophy in Fabry disease: a practical approach to diagnosis. Eur Heart J 2013; 34(11): 802–808. Dostupné z DOI: <http://dx.doi.org/10.1093/eurheartj/ehs166>.
32. Linhart A, Kampmann C, Zamorano JL et al. Cardiac manifestations of Anderson-Fabry disease: results from the international Fabry outcome survey. Eur Heart J 2007; 28(10): 1228–1235. Dostupné z DOI: <http://dx.doi.org/10.1093/eurheartj/ehm153>.
33. Thompson RB, Chow K, Khan A et al. T1 mapping with cardiovascular MRI is highly sensitive for Fabry disease independent of hypertrophy and sex. Circ Cardiovasc Imaging 2013; 6(5): 637–645. Dostupné z DOI: <http://dx.doi.org/10.1161/CIRCIMAGING.113.000482>.
34. Vardarli I, Rischpler C, Herrmann K et al. Diagnosis and Screening of Patients with Fabry Disease. Ther Clin Risk Manag 2020; 16 : 551–558. Dostupné z DOI: <http://dx.doi.org/10.2147/TCRM.S247814>.
35. Elliott P, Baker R, Pasquale F et al. Prevalence of Anderson-Fabry disease in patients with hypertrophic cardiomyopathy: the European Anderson-Fabry Disease survey. Heart 2011; 97(23): 1957–1960. Dostupné z DOI: <http://dx.doi.org/10.1136/heartjnl-2011–300364>.
36. Paolillo S, Marsico F, Prastaro M et al. Diabetic Cardiomyopathy: Definition, Diagnosis, and Therapeutic Implications. Heart Fail Clin 2019; 15(3): 341–347. Dostupné z DOI: <http://dx.doi.org/10.1016/j.hfc.2019.02.003>.
37. Negishi K. Echocardiographic feature of diabetic cardiomyopathy: where are we now? Cardiovasc Diagn Ther 2018; 8(1): 47–56. Dostupné z DOI: <http://dx.doi.org/10.21037/cdt.2018.01.03>.
Labels
Diabetology Endocrinology Internal medicineArticle was published in
Forum Diabetologicum

Most read in this issue
- Hypertrofia ľavej komory u pacientov s diabetes mellitus: na čo nemyslíme?
- Katétrová ablácia fibrilácie predsiení u pacientov s diabetes mellitus
- Alternatívne spôsoby podávania inzulínu
- Kardioprotektívny efekt liečby GLP1-analógmi vo svetle výsledkov najnovších klinických štúdií