
Transmisivní spongiformní encefalopatie jako příčina demence způsobená priony
Transmissible spongiform encefalopathy as a cause of dementia induced by prion particles
Prion diseases (transmissible spongiform encephalopathies – TSE) are one of the most serious diseases causing a progressive dementia syndrome and life limiting prognosis. These diseases are caused by pathological proteinaceous infectious particles (prions). The most common human prion disease is Creutzfeldt-Jakob disease (CJD). In diagnostic process of prion diseases we can use knowledge of clinical symptoms of diseases and also laboratory examinations (EEG, MRI, cerebrospinal fluid). Final diagnosis of prion diseases is based on neurohistopathology and genetic examination. No causal treatment is currently available and treatment is only symptomatic.
KEYWORDS:
transmissible spongiform encefalopathy – prion – dementia – Creutzfeldt-Jakob disease
:
J. Klán; E. Topinková
:
Geriatrie a Gerontologie 2016, 5, č. 1: 39-43
:
Review Article
Prionová onemocnění (označovaná také jako transmisivní spongiformní encefalopatie – TSE) patří mezi závažná onemocnění s progredující demencí a infaustní prognózou. Původcem onemocnění jsou patologické infekční proteinové částice (priony). Nejčastěji se vyskytujícím lidským prionovým onemocněním je Creutzfeldtova-Jakobova nemoc (CJD). V diagnostice využíváme znalosti klinických příznaků onemocnění a pomocných laboratorních vyšetření (EEG, MRI, mozkomíšní mok). Definitivní diagnóza prionových chorob je stanovena na základě neurohistopatologického a genetického vyšetření. V současnosti neexistuje kauzální léčba prionových onemocnění a využíváme pouze symptomatické postupy.
KLÍČOVÁ SLOVA:
transmisivní spongiformní encefalopatie – prion – demence – Creutzfeldtova-Jakobova nemoc
Definice
Transmisivní spongiformní encefalopatie (TSE) jsou přenosná neurodegenerativní onemocnění, jejichž původcem jsou priony. Postihují zvířata i člověka. Mají obvykle dlouhou inkubační dobu, rychlý průběh a infaustní prognózu(1,2,4).
Prionová hypotéza
Za původce TSE jsou považovány tzv. priony (proteinaceous infectious particle) – infekční bílkoviny, které jsou schopny se samy replikovat i v nepřítomnosti nukleových kyselin(1). Objevitel prionů, americký neuropatolog Stanley B. Prusiner (1982 – objev prionů, 1997 – Nobelova cena za prionovou hypotézu), prokázal, že existují dva druhy prionů(2, 3, 4).
PrPc („normální“ fyziologický prionový protein – celulární), který se nachází na buněčné membráně neuronů, ale i v dalších tkáních (přesná fyziologická funkce v organismu není úplně jasná). PrPSc („infeční“ patologický prionový protein – scrapie) se vyznačuje bodovou mutací genu jedné z aminokyselin. Prionový protein (PrP) je kódován genem pro prionový protein (PRNP) lokalizovaným na krátkém raménku 20. chromozomu(1, 2).
Podstatou onemocnění je konformační přeměna PrPc na PrPSc, která může být spontánní, v důsledku somatické mutace (sporadický typ), patogenní mutace PRNP (genetická forma) nebo expozice normálního proteinu abnormální izoformě (přenosný typ)(1, 4).
PrPSc mají stejné chemické složení, ale jiné prostorové uspořádání. Je známo, že PrPc podléhají proteolýze, naopak PrPSc nikoliv a jsou odolné vůči běžným dezinfekčním a sterilizačním postupům(1, 4).
Patologické priony se dostanou do organismu obvykle alimentární cestou. Není přesně objasněno, jak probíhá cesta PrPSc do nervových buněk (neuronů) centrálního nervového systému, ale zde PrPSc indukují přítomné PrPc na PrPSc, které se množí a destruují neurony. Dochází k hromadění patogenního prionového proteinu, histopatologickým projevům degenerace neuronů a spongiózním (houbovitým) změnám mozku. To je základem patogeneze prionových neurodegenerativních onemocnění(3, 4).
Historie objevů prionových onemocnění
Poznávání prionových chorob má dlouhou a složitou historii, kterou uvádí tabulka 1.

Prionová onemocnění u lidí
V současné době je popsáno několik forem lidských transmisivních spongiformních encefalopatií (tab. 2).

Creutzfeldtova-Jakobova nemoc (CJD)
Nejčastější lidské prionové onemocnění je Creutzfeldtova-Jakobova nemoc (85 % všech případů)(4). V současnosti jsou známy 4 hlavní varianty CJD(1, 2):
- sporadická forma CJD
- genetická (familiární) forma CJD
- iatrogenní – náhodně přenesená forma CJD (accidentally transmitted)
- nová varianta CJD
Sporadická forma Creutzfeldtovy-Jakobovy nemoci
Je nejčastějším variantou TSE u lidí, známá od roku 1920. Příčina onemocnění dosud nebyla objasněna, nebyl prokázán žádný vztah k prionovému onemocnění zvířat. Celosvětová incidence se uvádí 1–2 případy na 1 milion obyvatel za rok(2). Průměrný věk v době vzniku onemocnění je 65 let (s rozpětím 14–92 let). Průměrná doba trvání nemoci se uvádí 4–5 měsíců (s rozptylem 8 měsíců)(1). Do 1 roku po stanovení diagnózy umírá 90 % nemocných a méně než 4% pacientů přežijí déle než 2 roky(2, 4).
V klinickém obrazu dominuje rychle progredující demence kortiko-subkortikální s postižením všech složek kognice v různé kombinaci (porucha orientace v čase, prostoru, porucha paměti, frontální dysfunkce, afázie, agnozie). Asi u 1/3 nemocných mohou předcházet nespecifické prodromální příznaky (únava, nespavost, deprese, hubnutí, cefalea)(1, 2, 4).
K demenci se připojují nejméně 2 ze 4 dalších klinických projevů:
- a) myoklonus – často asymetrický, vyvolatelný zevním podnětem (světlo, dotek, zvuk, spontánně)
- b) mozečkové a zrakově prostorové dysfunkce – ataxie, nystagmus, poruchy pohledu, výpadky zorného pole, zrakové halucinace
- c) pyramidové a extrapyramidové projevy – změny svalového tonu, poruchy chůze, oslabení svalové síly, parézy, plegie
- d) akinetický mutismus – rigidita, neschopnost souvislé řeči
U pacientů v terminálním stadiu onemocnění bývá přítomna imobilita, inkontinence, pseudobulbární syndrom, korová slepota(1, 2, 4, 5).
Genetická (familiární) forma Creutzfeldtovy-Jakobovy nemoci
Je autozomálně dominantně dědičné onemocnění s 50% rizikem postižení v 1. linii.
Příčinou choroby je mutace PRNP genu. Představuje asi 15 %případů CJD. Klinický obraz bývá stejný jako u sporadické formy CJD. Pro stanovení diagnózy musí být splněna diagnostická kritéria CJD u příbuzného v 1. stupni příbuznosti a dále prokázána charakteristická mutace PRNP genu u pacienta genetickou analýzou(1, 2, 4, 5).
Iatrogenní – náhodně přenesená forma Creutzfeldtovy-Jakobovy nemoci (accidentally transmitted)
Nejčastěji k přenosu dochází při transplantaci oční rohovky, při transplantaci tvrdé pleny mozkové, při plastikách ušního bubínku s použitím kadaverózního perikardu, použitím růstového hormonu a gonadotropinu získaného z lidských hypofýz nebo při neurochirurgickém výkonu v důsledku používání nedostatečně sterilizovaných intracerebrálních elektrod. Tvoří přibližně 5 % diagnostikovaných případů CJD(1, 2).
Inkubační doba bývá variabilní (1–30 let), klinický průběh stejný jako u sporadické formy CJD(2). První případ onemocnění byl popsán v roce 1974, v současné době je známo celkem asi 250 případů onemocnění(3).
Nová varianta Creutzfeldtovy-Jakobovy nemoci
Původce této varianty onemocnění je identický s původcem BSE (bovinní spongiformní encefalopatie). Prvně byla diagnostikována v roce 1996 ve Velké Británii. Přenos onemocnění je převážně alimentární cestou (ale byly prokázány i případy přenosu krevní transfuzí)(1, 2).
V současné době je prokázáno celosvětově více než 200 zemřelých na tento typ onemocnění(2). Inkubační doba onemocnění je 10 let a více. Mívá pomalejší průběh než sporadická forma CJD. Průměrná doba trvání nemoci se pohybuje okolo 14 měsíců(1, 2). V České republice doposud nová varianta CJD nebyla prokázána(6). Patologickou formu prionového proteinu lze u této formy prokázat v biopsii orgánů retikuloendoteliálního systému (krční mandle, apendix, slezina)(1).
Klinický obraz bývá odlišný od klasické sporadické formy CJD. Od počátku onemocnění dominují časné psychiatrické projevy – depresivní ladění, výrazná emoční labilita, iritabilita, výbuchy vzteku. Mohou se vyskytovat paranoidně persekuční bludy. Objevují se zrakové poruchy – diplopie. Z neurologických symptomů bývá přítomna ataxie a poruchy čití (parestezie, dysestezie). Postupně se rozvíjí demence s progresí až do formy těžkého kognitivního deficitu a akinetický mutismus(1, 2). Naopak myoklonus nemusí být u této varianty přítomen(1).
Další prionová onemocnění
Gerstmannova-Strausslerova-Scheinkerova nemoc (GSS)
Je vzácné autozomálně dědičné onemocnění, jehož příčinou jsou mnohočetné mutace PRNP genu na 20. chromozomu. Onemocnění mívá delší průběh než CJD (až 5 let). Začíná mezi 30.–40. rokem. V popředí klinického obrazu jsou mozečkové a pyramidové příznaky, v pozdějších fázích kognitivní deteriorace(1, 2).
Fatální familiární insomnie (FFI)
Je vzácné hereditární onemocnění. Příčinou jsou mutace PRNP genu na 20. chromozomu. Variantou onemocnění je sporadická fatální insomnie (FSI) s náhodným výskytem.
V klinickém obrazu dominuje rezistentní nespavost. Později se objevují neurologické příznaky – tremor, rigidita, ataxie, myoklonus. V terminálním stadiu je přítomna prakticky úplná insomnie, těžká demence, mutismus. Doba trvání nemoci se pohybuje v rozmezí 6–36 měsíců(1, 2).
Kuru
V současné době již eradikované prionové onemocnění, popsané v roce 1950 u členů kmene Fore (Papua-Nová Guinea). K přenosu docházelo pojídáním tepelně neupravených mozků zemřelých (rituální kanibalismus). Typickým klinickým příznakem byla progredující mozečková ataxie (kuru = třes), později demence. Inkubační doba byla 12–40 let, trvání nemoci 3–36 měsíců. Eradikace bylo dosaženo epidemiologickými opatřeními (zamezení rituálního kanibalismu)(1, 2).
Diagnostika prionových onemocnění
Dosud není znám žádný diagnostický test, který by jednoznačně potvrdil klinickou diagnózu negenetické formy CJN. Prováděná vyšetření mají za cíl vyloučit jinou příčinu nemoci.
V diagnostice využíváme především typický klinický obraz onemocnění, EEG vyšetření, zobrazovací vyšetření (CT, MRI, SPECT, PET) a vyšetření likvoru. Definitivní diagnózu potvrdí až neurohistopatologické vyšetření či genetické vyšetření(1, 2, 4).
EEG vyšetření (elektroencefalografie)
Téměř u 2/3 pacientů nacházíme generalizované symetrické trifázické nebo polyfázické vlny s hrotnatými komplexy, které se periodicky opakují v intervalech 0,5–2 s, trvají
100–300 s, EEG obraz připomíná EKG křivku. U sporadické formy CJD se uvádí senzitivita 67 % a specificita 86 %. U pacientů s novou variantou CJD se objevuje spíše nespecifický obraz pomalých vln. Při suspekci na CJD je doporučeno EEG vyšetření opakovat (možná změna EEG nálezu u 1 pacienta v průběhu onemocnění)(1, 2, 5).
CT vyšetření mozku (výpočetní tomografie)
Na CT snímku mozku nacházíme obvykle pouze nespecifické známky lokální či difuzní atrofie mozku(2).
MRI vyšetření mozku (nukleární magnetická rezonance)
Při MRI vyšetření mozku se až u 70 % nemocných se sporadickou formou CJD objevují hyperintenzity v T2 váženém obrazu a FLAIR sekvencích v oblasti bazálních ganglií (putamen, ncl. caudatus) a v oblasti korové zóny (frontálně, periinsulárně)(1, 2),které vykazují až 67% senzitivitu a 93% specificitu.
Až u 90 % případů pacientů s novou variantou CJD můžeme nalézt při MRI vyšetření mozku symetrické hyperintenzity v zadní části thalamu ve FLAIR sekvencích(1, 2) – pulvinarový příznak(1).
SPECT (jednofotonová emisní výpočetní tomografie), PET (pozitronová emisní tomografie)
Při vyšetření SPECT či PET nacházíme nepravidelná ložiska hypoperfuze či hypometabolismu difuzně kortikálně a subkortikálně(2).
Mozkomíšní mok
V diagnostice rychle progredujících neurodegenerativních demencí má význam přítomnost 14-3-3 proteinu (beta-podjednotky) v likvoru, který je nespecifickým markerem neuronálního rozpadu(1, 2, 4, 5). Typicky se nachází u sporadické formy CJD. Naopak nebývá pozitivní u nové varianty CJD(1). U pacientů s CJD nacházíme též vysokou hladinu celkového a fosforylovaného tau proteinu (h-tau, p-tau) v likvoru(1, 7).
Neurohistopatologické vyšetření mozkové tkáně, genetické vyšetření (post mortem)
Diagnózu lidských prionových onemocnění potvrdí definitivně neurohistologické vyšetření mozkové tkáně, kde nacházíme 3 typické změny – spongiformní (houbovitá) dystrofie, numerická atrofie (úbytek) neuronů a sekundární izomorfní astroglióza(1).
Imunohistochemická vyšetření využívají protilátek k verifikaci výskytu patogenních prionů ve tkáni (western blot – biochemická typizace subtypu prionového proteinu, protilátky
proti PrP, specifický průkaz PrPSc)(1, 4). U familiárních forem onemocnění se využívá molekulárněgenetické vyšetření (sekvenování genu PRNP, hledání kauzální patogenní variace)(1, 5).
Diagnostická kritéria Creutzfeldtovy-Jakobovy nemoci (CJD)
Pro stanovení diagnózy CJD musí být splněna tzv. diagnostická kritéria (tab. 3).


Diagnostika lidských prionových onemocnění v České republice
Je prováděna od roku 2001 v Národní referenční laboratoři lidských transmisivních spongiformních encefalopatií a Creutzfeldtovy-Jakobovy nemoci České republiky při oddělení patologie a molekulární medicíny Thomayerovy nemocnice v Praze(4, 6).
Od roku 2007 se zde provádí též povinné testování mozkové tkáně všech dárců očních rohovek na přítomnost patologické formy prionového proteinu metodou western blot(4).
Pouhé podezření na prionové onemocnění podléhá povinnému hlášení(2). Pro klinickou diagnostiku jsou vyčleněna lůžka na Neurologické klinice Thomayerovy nemocnice v Praze(4). Pitva zemřelých s podezřením na prionové onemocnění je z hygienicko-epidemiologických důvodů povinná (pitvu provádí výhradně oddělení patologie Thomayerovy nemocnice v Praze)(2, 4).
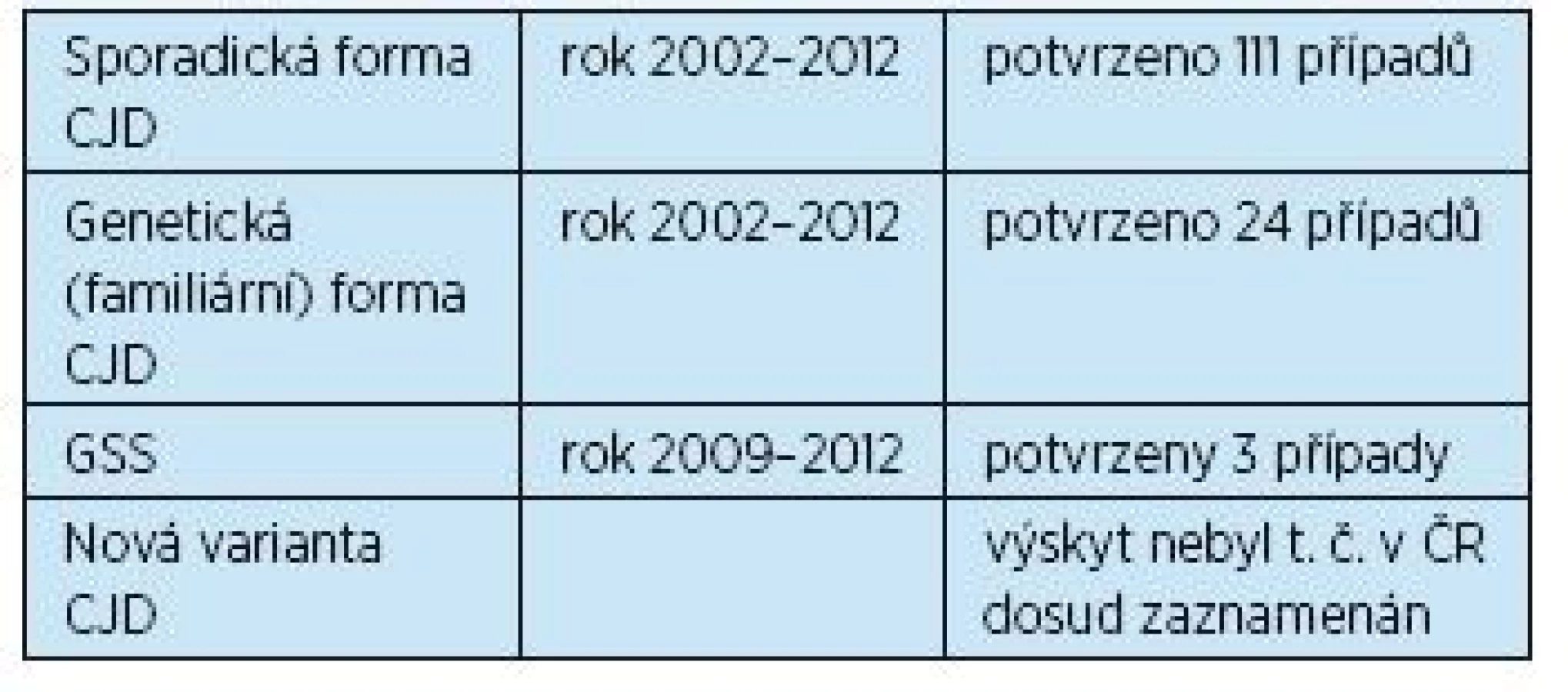
Tabulka 4 uvádí přehled výskytu prionových onemocnění v České republice(6).
Diferenciální diagnostika prionových onemocnění
V diferenciální diagnostice je nutné vyloučit především jiná onemocnění provázená demencí – nejčastěji Alzheimerova nemoc, frontotemporální demence, subkortikální vaskulární demence. Podobný klinický obraz mohou mít encefalitidy, tumory mozku (primární lymfom mozku), dále je třeba myslet též na Huntingtonovu choreu, spinocerebelární ataxii, amyotrofickou laterální sklerózu, roztroušenou sklerózu mozkomíšní, vaskulitidu postihující centrální nervový systém nebo otravu těžkými kovy(2, 5, 7, 8).
Prevence šíření transmisivních spongiformních encefalopatií
Běžný kontakt mezi lidmi (ani sexuální) není spojen se zvýšeným rizikem nákazy.
Při péči o nemocného s podezřením na prionové onemocnění se doporučuje používat jednorázový materiál a nástroje. Použité prostředky je třeba likvidovat jako nemocniční odpad spálením. Při nutnosti opakovaného použití nástrojů a přístrojů se musí dodržovat doporučené předpisy – dezinfekce a sterilizace působením hydroxidu sodného nebo chlornanu sodného s následnou sterilizací v parním sterilizátoru(4). Přenosu TSE ze zvířat na člověka alimentární cestou se snažíme zabránit důsledným testováním poražených zvířat a prevencí šíření onemocnění mezi zvířaty navzájem(2, 4).
Terapeutické aspekty prionových onemocnění
Transmisivní spongiformní encefalopatie mají infaustní prognózu, jejich průběh je terapeuticky neovlivnitelný. Kauzální léčba není známa. Terapie je tedy pouze symptomatická(1, 2, 4, 5).K ovlivnění myoklonu lze užít clonazepam či valproát sodný, při přítomnosti epileptických paroxysmů valproát sodný, u pacientů s psychotickou symptomatikou a neklidem sedativa či neuroleptika, při bolestech analgetika (opioidy), která nemocným přináší úlevu(1, 2, 4). Kognitiva (inhibitory acetylcholinesterázy, memantin) v terapii TSE obecně nejsou doporučována(2). Důležitou součástí terapie je ošetřovatelská péče (dostatečná hydratace, výživa, prevence dekubitů, rehabilitace)(1, 4). V terminální fázi prionových onemocnění je důležité zahájit včasnou a cílenou paliativní péči, zajistit komfort pacienta a podporu jeho rodiny(1).
Závěr
Transmisivní spongiformní encefalopatie (TSE) patří mezi přenosná neurodegenerativní onemocnění způsobená priony (proteinovými infekčními částicemi). Přes relativně vzácný výskyt mají prionové nemoci své významné místo v diferenciální diagnostice demencí. Nejčastěji se vyskytujícím lidským prionovým onemocněním je Creutzfeldtova-Jakobova nemoc, která představuje 85 % případů ze všech TSE u lidí.
Diagnostika prionových chorob je založena na typických klinických projevech a pomocných laboratorních vyšetřeních (EEG, likvor, MRI mozku). Definitivní potvrzení diagnózy TSE umožní až post mortem provedené neurohistopatologické vyšetření (provádí výhradně patologické oddělení Thomayerovy nemocnice v Praze)(2, 4), s výjimkou případů určení diagnózy z biopsie mozkové tkáně(1, 7).
Je nutné vědět, že suspekce na prionové onemocnění podléhá povinnému hlášení do Národní referenční laboratoře trans-misivních spongiformních encefalopatií a Creutzfeldtovy-Jakobovy nemoci České republiky v Thomayerově nemocnici v Praze, kde se též provádí diagnostika prionových onemocnění(2, 4). Významnou úlohu u familiárních forem TSE má molekulárněgenetické vyšetření. Při průkazu dědičného onemocnění má zásadní význam genetické poradenství a podpora rodinných příslušníků(1).
Kauzální terapie prionových chorob v současné době není známa. Hlavní součástí terapeutických postupů u pacientů s TSE je tedy léčba symptomatická a ošetřovatelská péče, v pokročilých fázích onemocnění je indikována paliativní terapie.
Autoři prohlašují, že v souvislosti s publikací článku nejsou ve střetu zájmů a vznik ani publikace článku nebyly podpořeny farmaceutickou firmou.
MUDr. Jan Klán
prof. MUDr. Eva Topinková, CSc.
Geriatrická klinika 1. LF UK a VFN Praha
MUDr. Jan Klán
e-mail: jan.klan@vfn.cz
Od roku 2001 pracuje na Geriatrické klinice 1. LF UK a VFN Praha. V rámci výzkumných aktivit této kliniky spolupracoval pod vedením prof. Evy Topinkové na evropské multicentrické studii AD HOC (Aged in Home Care) zaměřené na zdravotně-sociální problematiku a rizika seniorů v domácí péči. Atestaci z interního lékařství složil v roce 2005, specializovanou způsobilost k výkonu povolání lékaře v oboru geriatrie získal v roce 2008. V rámci pedagogické činnosti se podílí na výukových programech pregraduálního i postgraduálního vzdělávání v oboru geriatrie a gerontologie.
Sources
1. Rusina R, Matěj R. Prionová onemocnění. Neurologie pro praxi 2012; 13(2): 78–82.
2. Krombholz R. Prionové demence. Psychiatrie pro praxi 2007; 15(3): 125–128.
3. Matěj R, Rusina R, Koukolík F, a kol. 5 let činnosti Národní referenční laboratoře lidských prionových onemocnění při Oddělení patologie a molekulární medicíny FTNsP: naše zkušenosti a přehled literatury. Česká a Slovenská neurologie a neurochirurgie 2007; 70/103(6): 637–642.
4. Franková V, Krausová M. Lidské prionové nemoci. Psychiatrie pro praxi 2008; 9(3): 116–118.
5. Franková V, Serbinová I, Matěj R, a kol. Creutzfeldtova-Jakobova nemoc, kazuistika familiární formy onemocnění. Interní medicína pro praxi 2004; 12 : 606–609.
6. Rohan Z, Parobková E, Johanidesová S, a kol. Lidské prionové nemoci v České republice – 10 let zkušeností s diagnostikou. Česká a Slovenská neurologie a neurochirurgie 2013; 76/109(3): 300–306.
7. Růžička E a kol. Diferenciální diagnostika a léčba demencí (příručka pro praxi). Galén 2003; 21–43.
8. Topinková E. Geriatrie pro praxi. Galén 2005; 137–145.
Labels
Geriatrics General practitioner for adults Orthopaedic prostheticsArticle was published in
Geriatrics and Gerontology

2016 Issue 1
- Hope Awakens with Early Diagnosis of Parkinson's Disease Based on Skin Odor
- Metamizole at a Glance and in Practice – Effective Non-Opioid Analgesic for All Ages
- Memantine in Dementia Therapy – Current Findings and Possible Future Applications
- What Effect Can Be Expected from Limosilactobacillus reuteri in Mucositis and Peri-Implantitis?
Most read in this issue
- Deep vein thrombosis and its treatment in questions and answers.
- Diseases of the thyroid gland with a focus on senior age.
- Transmissible spongiform encefalopathy as a cause of dementia induced by prion particles
- Specializace v geriatrii a kompetence geriatra