
Myelodysplastický syndrom v novém tisíciletí. Jak klasifikovat a jak léčit nemocné?
Myelodysplastic syndrome in the new millennium. How to classify and cure patients?
Aim of the study and methods:
A retrospective analysis of the relationship between the initial classification according to either FAB or WHO classification and survival was performed in a group of 197 patients with primary myelodysplastic syndrome (MDS), who were also subdivided into risk groups according to the International Prognostic Scoring System (IPSS). The influence of stem cell transplantation (SCT) or supportive therapy on survival of different subgroups of patients was also studied.
Results:
A classification of 107 patients on the base of WHO criteria revealed a significant difference in median survival between patients with „pure“ RA and RCMD (78.2 months vs. 42.3 months, P = 0.001). Molecular cytogenetics methods (fluorescent in situ hybridization – FISH) were crucial for classification and prognosis of patients with 5q deletion and enabled us to discriminate patients with additional karyotype abnormalities and adverse prognosis from those with isolated deletion of 5q. In patients with advanced MDS (RAEB, RAEB-T), SCT substantially prolonged median survival in both patients with RAEB and RAEB-T (38.4 months vs. 11.5 months, p < 0.001) and patients with intermediate II. and high risk according to IPSS (36.8 months vs. 12.0 months, p < 0.001) in comparison to supportive treatment or/and chemotherapy only. On the other hand, SCT did not significantly prolong median survival either in RA patients (62.3 months vs. 62.4 months, P > 0.8) or in those with low or intermediate I. IPSS risk (57.2 months vs. 60.8 months, P = 0.8), mainly because of a high rate of SCT related mortality. Classification of RA patients according to the WHO criteria showed no benefit from SCT for survival in both RA and RCMD subgroups. Only more detailed subclassification on the base of combined WHO morphologic and IPSS cytogenetic criteria revealed a subgroup of non-transplanted RCMD patients and „poor“ karyotype, who survived only 9.2 months in contrast to transplanted with median survival of 81.8 months. Presence of adverse karyotype abnormalities was the most important independent variable determining survival and leukemic transformation in multivariate regression analysis.
Conclusion:
Combination of classical morphology with molecular cytogenetics methods may improve identification of risk patients within RA subgroup, who may benefit from early SCT despite a high risk of SCT related complications.
Key words:
myelodysplastic syndrome – classification – prognosis – treatment – transplantation – karyotype
:
J. Čermák 1; A. Vítek 1; K. Michalová 1; J. Březinová 1; Z. Zemanová 2
:
Ústav hematologie a krevní transfuze, Praha, ředitel prof. MUDr. Pavel Klener, DrSc.
1; Centrum nádorové cytogenetiky Ústavu klinické biochemie a laboratorní diagnostiky 1. LF UK a VFN, Praha, ředitel prof. MUDr. Tomáš Zima, DrSc.
2
:
Vnitř Lék 2005; 51(1): 20-30
:
Original Contributions
Cíl práce a metodika:
V retrospektivní analýze 197 nemocných s primárním myelodysplastickým syndromem (MDS) byl zkoumán vztah mezi zařazením nemocných dle FAB či WHO klasifikace a do rizikových podskupin dle Mezinárodního prognostického skórovacího systému (IPSS) a délkou přežití. Krom toho byl srovnáván vliv transplantace krvetvorných buněk (SCT) a podpůrné léčby na délku přežití v jednotlivých podskupinách.
Výsledky:
Rozdělení 107 nemocných s refrakterní anémií podle WHO klasifikace prokázalo statisticky významný rozdíl v délce přežití mezi nemocnými s „čistou“ RA a RCMD (78,2 měsíce vs 42,3 měsíce, P = 0,001), pro klasifikaci a prognózu nemocných s delecí dlouhého raménka 5. chromozomu (5q-) měly zásadní význam metody molekulární cytogenetiky (fluorescenční in situ hybridizace – FISH), jež umožnily odlišit nemocné s přídatnými změnami karyotypu a nepříznivou prognózou na rozdíl od nemocných s izolovanou delecí 5q. U nemocných s pokročilými stadii choroby (RAEB, RAEB-T) nepředstavovala samotná chemoterapie statisticky významný přínos pro délku přežití oproti podpůrné léčbě, na druhé straně SCT významně zvýšila délku přežití oproti netransplantovaným nemocným jak u nemocných s RAEB a RAEB-T (38,4 měsíce vs 11,5 měsíce, P < 0,001), tak u nemocných se středním II. a vysokým rizikem dle IPSS (36,8 měsíce vs 12,0 měsíce, P < 0,001). Na druhé straně, SCT významně neovlivnila délku přežití ani u nemocných s RA (62,3 měsíce vs 62,4 měsíce, P > 0,8) ani u nemocných s nízkým a středním I. rizikem (57,2 měsíce vs 60,8 měsíce, P = 0,8), nejspíše díky vysoké incidenci mortality spojené s transplantací. Statisticky významný rozdíl v délce přežití mezi transplantovanými a netransplantovanými nemocnými nebyl přítomen ani při rozdělení nemocných s RA na čistou RA a RCMD dle WHO klasifikace. Teprve podrobnější klasifikace na bázi kombinace WHO klasifikace a cytogenetických rizikových skupin dle IPSS odhalila nemocné s RCMD a „nepříznivým“ karyotypem, kteří bez SCT přežívali pouze 9,2 měsíce na rozdíl od transplantovaných nemocných, u nichž činila průměrná délka přežití 81,8 měsíce. Přítomnost nepříznivých změn karyotypu byla při regresní analýze nejdůležitějším nezávislým parametrem ovlivňujícím délku přežití a riziko leukemické transformace u nemocných s RA.
Závěr:
Užití metod molekulární cytogenetiky v kombinaci s klasickou morfologií může dále zpřesnit identifikaci rizikových nemocných s RA, indikovaných k časné SCT i přes relativně vysoké riziko komplikací spojených s transplantací.
Klíčová slova:
myelodysplastický syndrom – klasifikace – prognóza – léčba – transplantace – karyotyp
Úvod
Myelodysplastický syndrom (MDS) je definován jako klonální porucha krvetvorby. Mutace kmenové krvetvorné buňky vede ke vzniku patologického klonu s růstovou výhodou, jenž může postupem doby zcela nahradit normální krvetvorbu. V časné fázi onemocnění bývá většinou přítomen různý stupeň cytopenie v periferní krvi při současném nálezu buněčně bohaté kostní dřeně s morfologickými známkami dysplazie [5,13]. Tento obraz vzniká v důsledku vystupňovaní procesu apoptózy - buněčné smrti krvetvorných buněk následkem zvýšené sekrece některých cytokinů [13]. Pro pokročilou fázi MDS je naopak charakteristický progresivní nárůst nezralých buněk s velmi nízkým stupněm apoptózy, vedoucí k obrazu nadbytku blastů v kostní dřeni a v periferní krvi. Tato fáze onemocnění je již spojena s vysokou pravděpodobností přechodu choroby do akutní leukémie (AL) [5].
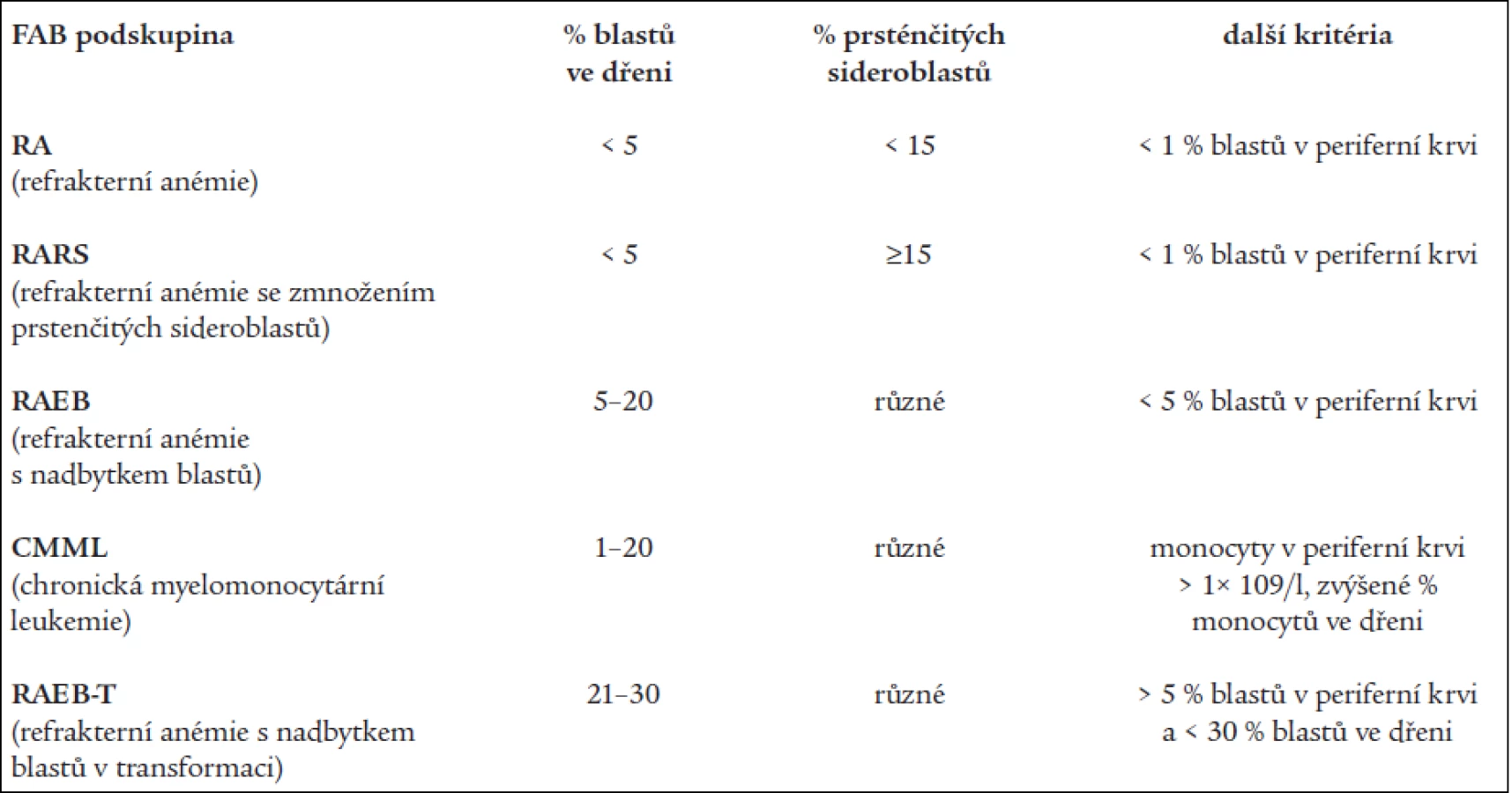
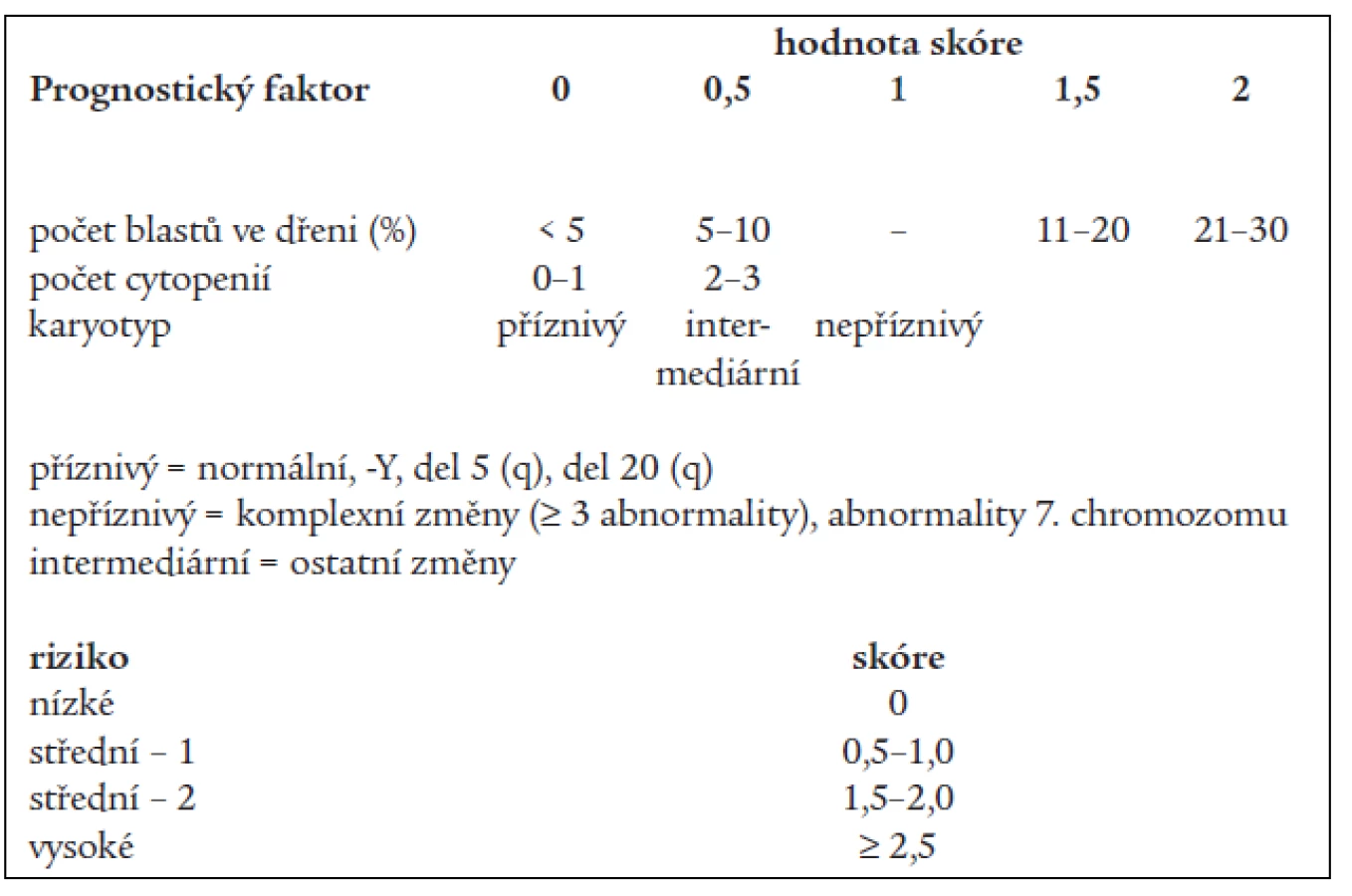
Základní klasifikační schéma nemocných s MDS, tzv. FAB (francouzsko-americko-britská) klasifikace z roku 1982 [2] rozděluje nemocné dle nálezu v kostní dřeni a periferní krvi do 5 podskupin (tab. 1). Toto dělení umožňuje základní odlišení nemocných s obecně lepší prognózou a nižším rizikem leukemické transformace v podskupinách refrakterní anémie (RA) a refrakterní anémie se zmnožením prsténčitých sideroblastů (RARS) od nemocných s nepříznivou prognózou a vysokým rizikem přechodu do AL (refrakterní anémie s nadbytkem blastů - RAEB a refrakterní anémie s nadbytkem blastů v transformaci - RAEB-T). Nicméně jednotlivé podtypy FAB klasifikace tvoří poměrně heterogenní skupiny nemocných, u nichž nejsou zohledněny další významné prognostické faktory. V roce 1997 byl na základě podrobné analýzy řady rizikových faktorů u nemocných s MDS publikován tzv. Mezinárodní prognostický skórovací systém (IPSS) [9], jenž dělí nemocné s MDS na základě procenta blastů v kostní dřeni, přítomnosti a charakteru změn karyotypu a počtu cytopenií v periferní krvi do 4 podskupin s odlišnou pravděpodobností délky přežití a přechodu do AL (tab. 2). Nová klasifikace nádorových onemocnění krvetvorby vypracovaná Světovou zdravotnickou organizací (WHO) a zveřejněná v roce 1999 [10] vyděluje z klasifikace MDS jednak nemocné s chronickou myelomonocytární leukémií (CMML), jež jsou řazeni do skupiny smíšených myelodysplasticko-myeloproliferativních onemocnění, jednak nemocné s více než 20 % blastů v kostní dřeni, kteří jsou zařazeni do skupiny akutních myeloidních leukémií (AML) s dysplazií ve více řadách. Skupinu refrakterních anémií tato tzv. WHO klasifikace dělí s přihlédnutím k některým prognostickým faktorům na tzv. „čistou“ RA a RARS s výhradním postižením erytropoézy, refrakterní cytopenii s dysplazií ve více řadách (RCMD), ze které je někdy ještě vyčleňován RCMD s přítomností prsténčitých sideroblastů (RCMD-RS) a tzv. 5q- syndrom s charakteristickou chromozomální přestavbou a morfologickým nálezem [17], jako nová podskupina byl zařazen tzv. neklasifikovatelný MDS (tab. 3).

Léčba nemocných s MDS se řídí stádiem choroby, prognostickými faktory a věkem nemocných. Konzervativní přístup, zahrnující podpůrnou léčbu (transfuze erytrocytů a krevních destiček, vitaminy, chelatační přípravky) a podávání léků ovlivňujících zejména stupeň buněčné apoptózy (kortikoidy, růstové faktory, imunosupresiva), převládá obecně u nemocných s časnými stadii a nízkým či středním I. rizikem a u starších nemocných [5,15]. Nemocní s nadbytkem blastů a středním II. či vysokým rizikem jsou pak indikování k podávání chemoterapie a k transplantaci krvetvorných buněk, jež dnes představuje jediný kurativní přístup k nemocným s MDS. U mladších nemocných s časnějšími a prognosticky relativně méně závažnými stadii onemocnění může vést transplantace krvetvorných buněk k jejich vyléčení, nicméně představuje stále riskantní výkon spojený s poměrné vysokým stupněm peritransplantační mortality [1,6,16]. Na druhé straně se ukazuje, že při konzervativní léčbě tito nemocní přežívají i bez transplantace v průměru 5-12 let [9]. Je proto třeba na základě analýzy přežití a prognostických faktorů vyčlenit z této skupiny ty nemocné, u kterých představuje transplantace krvetvorných buněk jednoznačný přínos i přes výše zmíněná rizika, a určit též optimální dobu transplantace u těchto nemocných.
V naší studii jsme provedli retrospektivní analýzu nemocných s primárním MDS sledovaných v Ústavu hematologie a krevní transfuze (ÚHKT) v Praze v letech 1980-2000. Byl zkoumán vztah mezi zařazením nemocných dle FAB či WHO klasifikace, délkou přežití a pravděpodobností leukemické transformace, dále byl hodnocen přínos chemoterapie v porovnání s pouhou podpůrnou léčbou u nemocných s pokročilými stadii choroby a srovnáván vliv transplantace krvetvorných buněk a konzervativní léčby na délku přežití nemocných v jednotlivých podskupinách dle FAB či WHO klasifikace a v rizikových skupinách dle IPSS.
Soubor nemocných a metodika
Ve studii bylo hodnoceno 197 nemocných s primárním MDS. Na základě vyšetření periferního krevního obrazu a nálezu v aspirátu a trepanobioptickém vzorku kostní dřeně byli nemocní zařazeni do jednotlivých podskupin dle FAB klasifikace. Při klasifikaci nemocných byla hodnocena vždy nejméně 3 vyšetření krevního obrazu a 2 aspiráty kostní dřeně provedené během 2 měsíců v době diagnózy onemocnění. Jako pomocná metoda pro klasifikaci sloužilo cytochemické vyšetření nátěrů z aspirátu kostní dřeně barvením berlínskou modří na přítomnost železa a vyšetření vzorku kostní dřeně pomocí průtokové cytometrie. Pro klasifikaci nemocných dle WHO klasifikace a IPSS bylo užito výsledků vyšetření karyotypu pomocí standardních metodik čítajících krátkodobou kultivaci krvetvorných buněk z aspirátu kostní dřeně a následné barvení technikou G-pruhování dle Wrighta. K podrobnější analýze karyotypu u nemocných s delecí krátkého raménka chromozomu 5 (5q-) byly užity metody molekulární cytogenetiky - fluorescenční in situ hybridizace (FISH) se specifickou sondou LSI EGR1 (5q31) SpectrumOrange/D5S721, D5S23 (5p15.2) SpectrumGreen (VYSISTM, USA) a celochromozómovými malovacími sondami (CambioTM, Cambridge, Velká Británie) a mnohobarevná FISH (mFISH) s mnohobarevným pruhováním s vysokou rozlišovací schopností (mBAND) se sondami „24 X Cyte“ a „X Cyte 5“ (MetasystemsTM, Altlussheim, SRN).
Ke statistickému zhodnocení byl použit nepárový t-test s Bonferroniho korekcí pro malé soubory a Fisherův test, analýza parametrů ovlivňujících délku přežití byla provedena dle Kaplan-Meiera a pomocí log-rank2 testu a zahrnovala pohlaví, věk, počet cytopenií, hodnotu hemoglobinu (Hb), počet neutrofilů, počet trombocytů, karyotyp (dle IPSS) a přítomnost/nepřítomnost leukemické transformace. Nezávislé proměnné ovlivňující délku přežití a leukemickou transformaci byly studovány regresní analýzou o více proměnných pomocí modelu dle Coxe [3] s hodnocením stejných parametrů jako analýza dle Kaplan-Meiera [11].
Výsledky
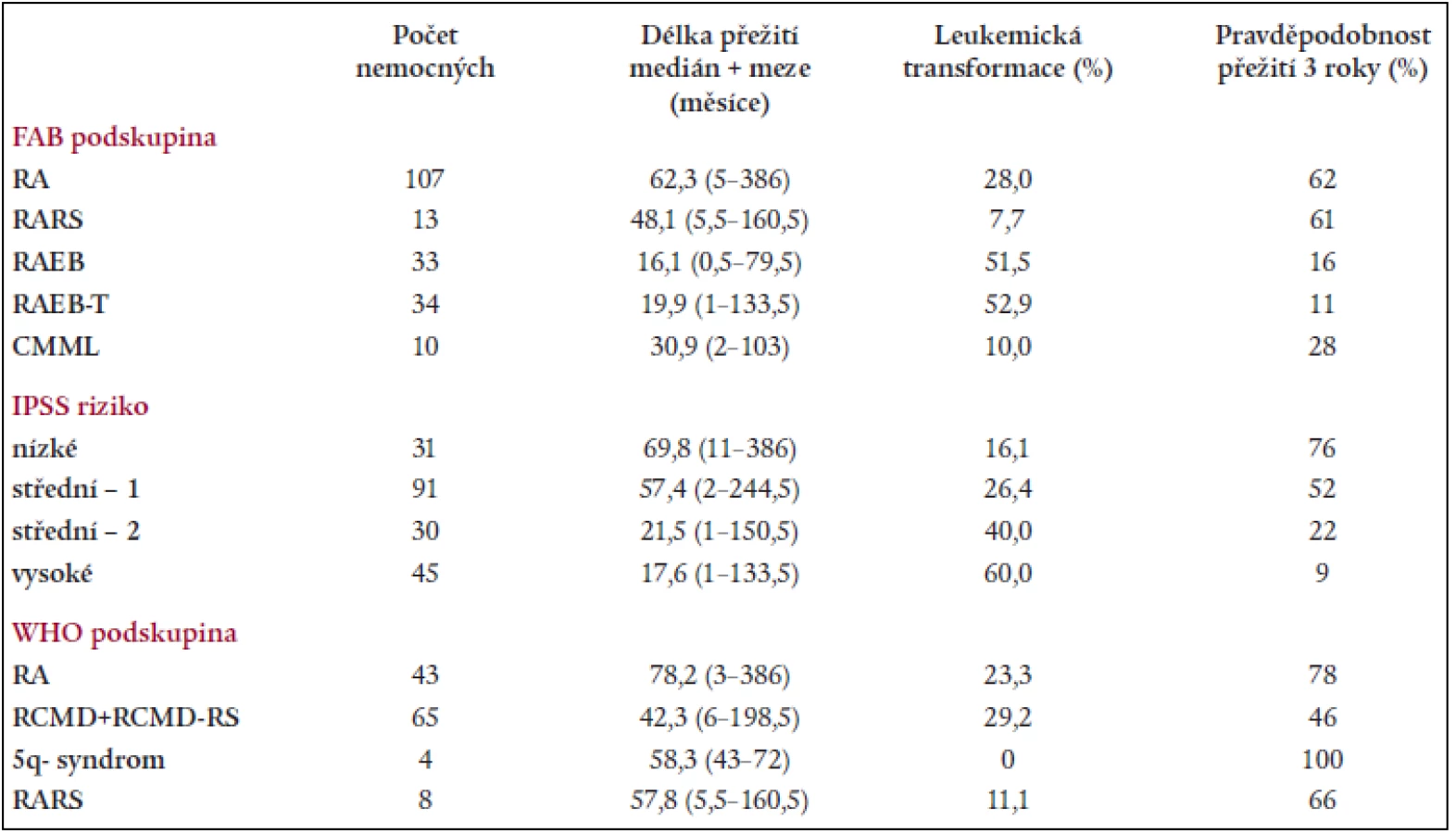
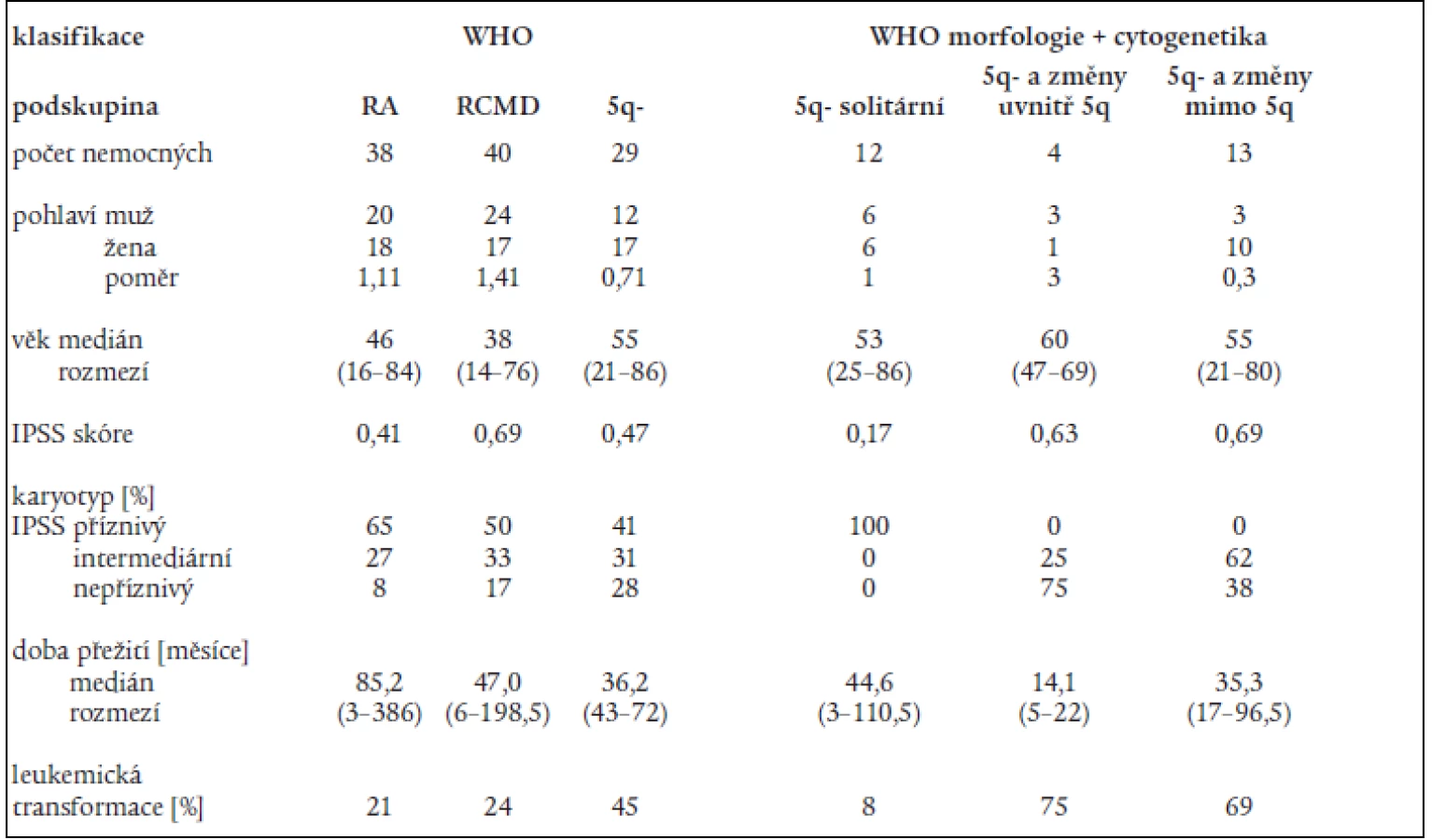
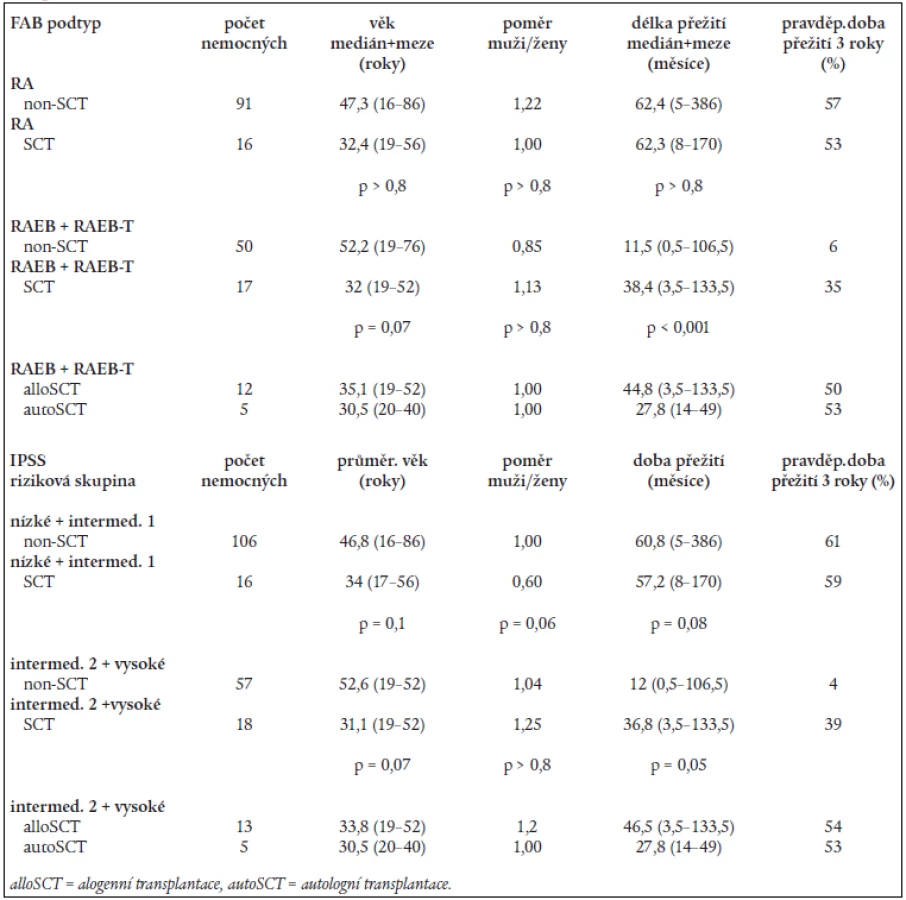
Na základě rozboru nálezů v periferní krvi a kostní dřeni bylo 197 nemocných rozděleno následovně do jednotlivých podskupin dle FAB klasifikace: RA - 107 nemocných, RARS - 13 nemocných, RAEB – 33 nemocných, RAEB-T - 34 nemocných, CMML - 10 nemocných. 107 nemocných s RA a 13 nemocných s RARS pak bylo reklasifikováno dle WHO klasifikace následovně: RA - 43 nemocných, RCMD - 60 nemocných, RCMDs - 5 nemocných, 5q- syndrom - 4 nemocní, RARS - 8 nemocných. V tab. 4 je uvedena délka přežití (medián a rozmezí), procento leukemické transformace a pravděpodobnost 3letého přežití v jednotlivých podskupinách dle FAB či WHO klasifikace a dle IPSS. Statisticky významný rozdíl v délce přežití byl nalezen mezi nemocnými s RA a RAEB či RAEB-T (P < 0,001 pro obě skupiny). Obdobný rozdíl byl přítomen i v pravděpodobnosti přežití 3 roky mezi RA a RAEB či RAEB-T (P < 0,001) a RARS a RAEB či RAEB-T (P < 0,001). Při hodnocení nemocných podle WHO klasifikace byl zjištěn statisticky významný rozdíl v délce přežití mezi nemocnými s RA a RCMD (medián 78,2 vs 42,3 měsíce, P = 0,001), stejné rozdíly byly nalezeny i při hodnocení pravděpodobnosti přežití 3 roky. Delece dlouhého raménka 5. chromozomu (5q-) byla bez ohledu na morfologický nález přítomna na základě rutinního vyšetření karyotypu u 29 nemocných s RA dle FAB klasifikace. Pokud byli při hodnocení dle WHO klasifikace s přihlédnutím k cytogenetickému nálezu (tab. 5) bráni tito nemocní jako zvláštní skupina 5q-, byla jejich prognóza významně horší než u nemocných s 5q-syndromem či RA, medián přežití 5q- skupiny byl 36,2 měsíce ve srovnání s nemocnými s RA (85,2 měsíce, P = 0,0002) a RCMD (47,0 měsíce, P > 0,8). Analýza karyotypu pomocí FISH potvrdila deleci v úseku 5q31 jako solitární přestavbu karyotypu pouze u 12 nemocných, 4 nemocní měli deleci 5q31 spojenou s dalšími přestavbami v oblasti dlouhého raménka 5. chromozomu, 13 nemocných pak mělo přestavbu 5q- spojenu s dalšími změnami karyotypu mimo oblast 5q. Nemocní se solitární delecí 5q potvrzenou metodou FISH měli délku přežití srovnatelnou s nemocnými s RA dle WHO klasifikace (85,2 vs. 44,6 měsíce) na rozdíl od nemocných s přídatnými změnami karyotypu lokalizovanými do oblasti 5q (14,1 měsíce) či mimo tuto oblast (35,3 měsíce). Obdobné rozdíly byly přítomny i při hodnocení procenta leukemické transformace (tab. 5).
Analýza přežití nemocných v jednotlivých rizikových skupinách dle IPSS (tab. 4) ukázala statisticky významný rozdíl v délce přežití mezi nemocnými s nízkým a středním II. či vysokým rizikem (P < 0,001 pro obě skupiny) a mezi nemocnými se středním I. A středním II. či vysokým rizikem (P < 0,001 pro obě skupiny). Obdobně pravděpodobnost přežití 3 roky se významně lišila mezi nemocnými s nízkým či středním I. rizikem a nemocnými se středním II. či vysokým rizikem (P <0,001).
Kombinovanou chemoterapií (většinou kombinací antracyklinů se standardní či středně vysokou dávkou cytosin arabinosidu) bylo léčeno 12 nemocných s diagnózou RAEB. Medián délky přežití těchto nemocných činil v průměru 11,0 měsíce a statisticky významně se nelišil od přežití 11 nemocných léčených podpůrnou léčbou (8,8 měsíce; P = 0,8). Obě skupiny se nelišily statisticky významně věkem (medián 53,1 vs 65,4 měsíce, P = 0,8). Medián délky přežití 23 nemocných s RAEB-T léčených chemoterapií byla výrazně delší než u 4 nemocných léčených paliativně (13,7 měsíce vs 2,1 měsíce), rozdíl však nebyl statisticky významný (P = 0,2). Medián věku činil u nemocných léčených chemoterapií 52,1 měsíce (meze 19-71 let), u nemocných léčených paliativně 65,3 měsíce (meze 58-82 let).
Transplantace krvetvorných buněk (SCT) byla po standardním myeloablativním přípravném režimu provedena u 34 nemocných (16 nemocných s RA, 10 nemocných s RAEB, 7 nemocných s RAEB-T a 1 nemocný s CMML). U 18 nemocných byla provedena SCT od HLA shodného příbuzného dárce, u 11 nemocných šlo o HLA shodného nepříbuzného dárce z registru, 5 nemocných s RAEB či RAEB-T bylo transplantováno autologním štěpem odebraným v první kompletní remisi choroby. Při hodnocení efektu SCT byl nalezen statisticky významný rozdíl v délce přežití mezi transplantovanými a netransplantovanými nemocnými s RAEB a RAEB-T (38,4 měsíce vs 11,5 měsíce, P < 0,001), stejně tak jako u nemocných se středním II. a vysokým rizikem (36,8 měsíce vs 12,0 měsíce, P = 0,05), SCT rovněž významně zvýšila pravděpodobnost přežití 3 roky jak u nemocných s RAEB a RAEB-T (P < 0,001), tak u nemocných se středním II. a vysokým rizikem (P < 0,001) (tab. 6). Na druhé straně, u nemocných s RA SCT významně neovlivnila ani délku přežití (medián přežití činil 62,3 měsíce u transplantovaných vs 62,4 měsíce u netransplantovaných, P > 0,8), ani pravděpodobnost přežití 3 roky (63 % u transplantovaných vs 58 % u netransplantovaných). Stejně tak nebyl významný rozdíl v délce přežití a v pravděpodobnosti 3letého přežití mezi transplantovanými a netransplantovanými nemocnými s nízkými a středním I. rizikem (tab. 6). I přes nižší věk transplantovaných nemocných nebyly rozdíly ve věku ani v poměru muži/ženy statisticky významné v žádné z hodnocených skupin. Při podrobnějším rozboru transplantovaných nemocných nebyl přítomen rozdíl ani v peritransplantační mortalitě, ani v délce přežití mezi nemocnými transplantovanými od příbuzného či nepříbuzného dárce (66,2 vs 38,3 měsíce; P = 0,07), horší prognóza nemocných po autologní SCT byla dána vysokou incidencí relapsů po transplantaci.
WHO klasifikace nemocných s RA a RARS neprokázala statisticky významný rozdíl v délce přežití mezi transplantovanými a netransplantovanými nemocnými ani u nemocných s „čistou“ RA (64,0 měsíce u SCT vs 91,0 měsíce u netransplantovaných; P = 0,3) ani u nemocných s RCMD (60,8 měsíce u SCT vs 41,2 měsíce u netransplantovaných; P = 0,2), přestože transplantace vedla k významnému snížení rizika leukemické transformace (tab. 7).
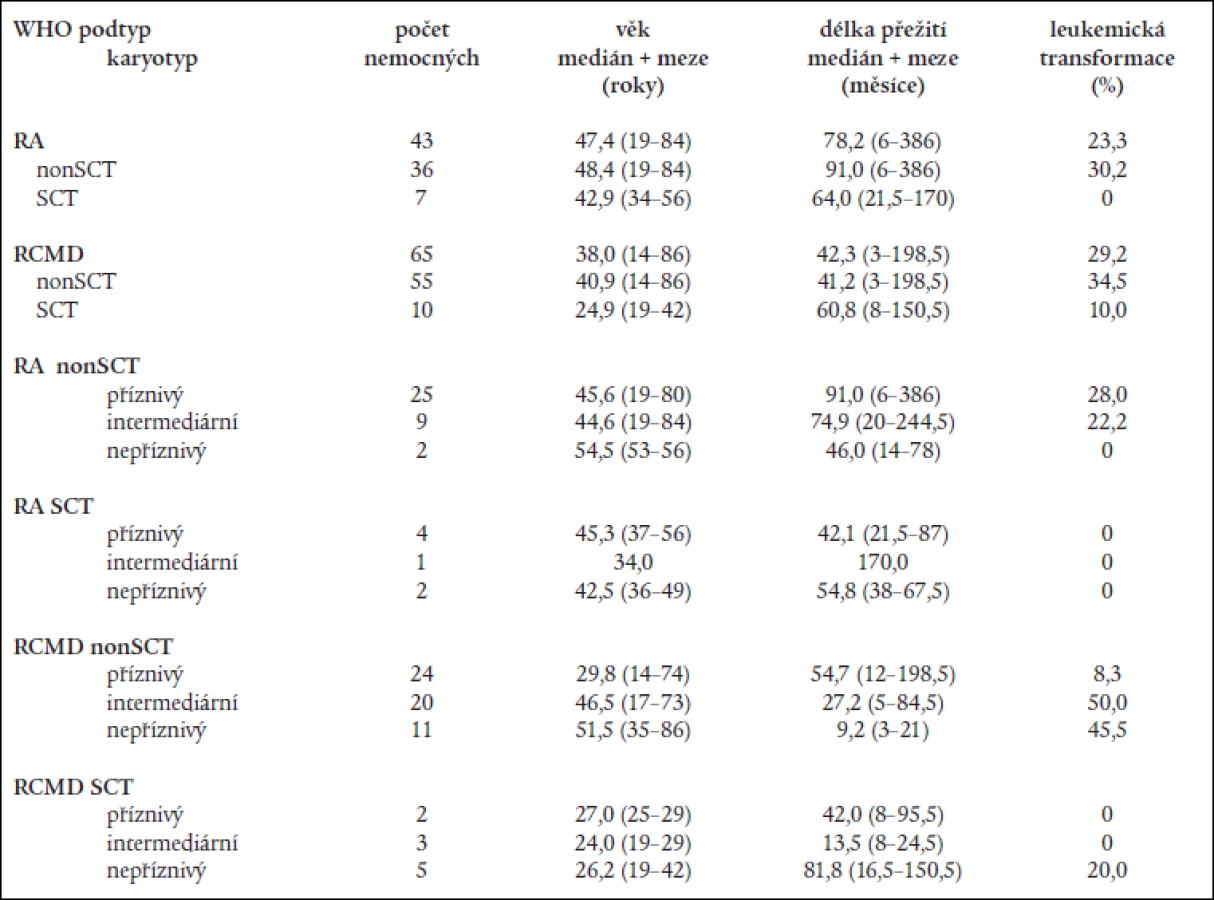
Mortalita spojená s transplantací činila 29 % u nemocných s RA a 60 % u nemocných s RCMD. Podrobnější rozdělení nemocných s RA a RCMD na podkladě rizikových skupin karyotypu dle IPSS neovlivnilo rozdíly mezi transplantovanými a netransplantovanými nemocnými s RA, medián délky přežití transplantovaných nemocných s „příznivým“ karyotypem byl dokonce kratší než u netransplantovaných (42,1 měsíce vs 91,0 měsíce) (tab. 7). Na druhé straně, i když počty nemocných v jednotlivých podskupinách byly poměrně malé a transplantováni byli mladší nemocní, medián délky přežití netransplantovaných nemocných s RCMD a „nepříznivým“ karyotypem činil pouze 9,2 měsíce a 3 roky nepřežil žádný nemocný na rozdíl od transplantovaných nemocných (medián délky přežití: 81,8 měsíce; 3 roky přežívá 80 % nemocných) (tab. 7).
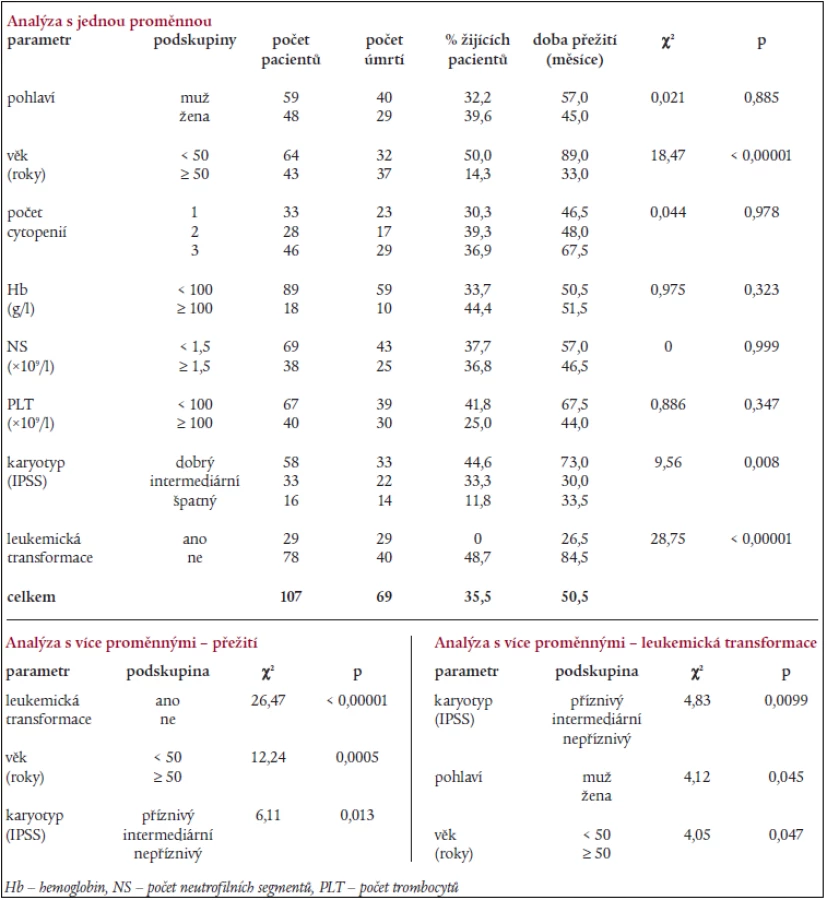
Statistická analýza s jednou proměnnou provedená u nemocných s RA (dle FAB klasifikace) odhalila jako významné proměnné ovlivňující délku přežití věk, karyotyp a přítomnost leukemické transformace (tab. 8). Regresní analýza s více proměnnými ukázala jako nezávislé parametry ovlivňující délku přežití přítomnost „intermediárního“ a „nepříznivého“ karyotypu dle IPSS a věk nad 50 let, karyotyp, mužské pohlaví a věk nad 50 let byly významnými parametry ovlivňujícími leukemickou transformaci (tab. 8).
Diskuse
V současné době představují u nemocných s MDS léčebný problém zejména dvě skupiny nemocných. Prvou skupinu tvoří starší nemocní s pokročilými stadii choroby, kteří špatně tolerují kombinovanou cytostatickou léčbu a nejsou vzhledem k věku a celkovému stavu indikováni k provedení SCT. Výsledky naší analýzy potvrdily, že u nemocných s RAEB a RAEB-T, stejně jako u nemocných se středním II. a vysokým rizikem (jež je dáno dle IPSS zejména nárůstem počtu blastů), má pro dlouhodobé přežití nemocných zásadní význam provedení SCT. Samotná chemoterapie nepředstavovala vzhledem k vysoké incidenci závažných komplikací a četnosti relapsů signifikantní přínos oproti pouhé podpůrné léčbě, byly však hodnoceny poměrně malé skupiny nemocných. Průměrná doba přežití netransplantovaných nemocných s RAEB a RAEB-T činila necelý rok a 3 roky přežívala pouze 4 %, resp. 3 % nemocných, podobně jako ve studii Greenberga et al [8], kde nemocní s RAEB a RAEB-T přežívali 0,6 resp. 1,5 roku. Tato nepříznivá prognóza vyvažuje poměrně vysokou peritransplantační mortalitu i u mladších nemocných s RAEB či RAEB-T, jež činí v průměru 40 %. Pro starší nemocné s pokročilými stadii MDS mohou určitou naději znamenat látky s diferenciačním účinkem, u nichž byl prokázán efekt u 50 % nemocných, možný přínos SCT provedené u starších nemocných po méně toxickém přípravném režimu [12] je třeba zhodnotit v rozsáhlejších studiích, na efektivitu autologní SCT u MDS nejsou názory jednoznačné [7,15]. V naší studii měli nemocní po autologní SCT horší prognózu než nemocní po alogenní SCT zejména díky vysoké incidenci relapsů.
Druhý okruh tvoří nemocní s časnějšími a prognosticky relativně méně závažnými stadii MDS, u nichž SCT může přinést vyléčení, ale na druhé straně je stále spojena s nezanedbatelným rizikem. Ve všech rozsáhlých studiích zabývajících se přínosem SCT k léčbě MDS byli nejlepšími kandidáty pro provedení úspěšné alogenní SCT mladší nemocní s méně pokročilými stadii choroby [1,6,14,15,16]. Pravděpodobnost dlouhodobého přežití činí u těchto nemocných při transplantaci časně po stanovení diagnózy 60-75 %, nicméně i u nich se pohybuje mortalita spojená se SCT mezi 20-50 % [1,6,14,16]. Na druhé straně, jak již bylo zmíněno v úvodu, rozsáhlá multicentrická analýza neléčených nemocných s MDS [9] zjistila, že nemocní mladší 60 let s nízkým rizikem (dle IPSS) přežívají v průměru 11,8 let i bez léčby a riziko leukemické transformace u nich představuje pouze 22 % během 10 let trvání choroby, obdobně jako u nemocných s RA (dle FAB klasifikace). U nemocných se středním I. rizikem činila průměrná doba přežití 5,2 roku a u 28 % nemocných došlo během této doby k rozvoji AL. V naší studii nebyl pozorován statisticky významný rozdíl v délce přežití mezi transplantovanými a netransplantovanými nemocnými s RA či nízkým a středním I. rizikem, nelišila se ani pravděpodobnost přežití 3 roky. Tyto nálezy ukazují na nutnost hledání dalších kritérií, která by pomohla vyčlenit nemocné s méně pokročilými stadii choroby, pro něž představuje časná SCT jasný přínos i přes rizika spojená s jejím provedením.
Naše studie prokázala, stejně jako předchozí rozsáhlá německá studie [8], že WHO klasifikace představuje pro nemocné s méně pokročilými stadii MDS přínos zejména tím, že umožňuje vyčlenit prognosticky jednoznačně nepříznivou podskupinu s RCMD ze skupiny refrakterních anémií. Kromě toho bylo prokázáno, že pro klasifikaci a prognózu nemocných s delecí dlouhého raménka 5. chromozomu má spíše než morfologická charakteristika, jak je navrhováno WHO klasifikací [10], význam analýza karyotypu pomocí metod molekulární cytogenetiky, jež v naší práci umožnila odlišit nemocné s přídatnými změnami karyotypu a jednoznačně nepříznivou prognózou od nemocných se solitární delecí 5q. Lze předpokládat, že obdobný přínos pro prognózu nemocných s MDS může mít i analýza dalších chromozomů pomocí FISH či dalších metod molekulární cytogenetiky.
Nicméně ani při hodnocení pouze dle WHO klasifikace nebyl u našich nemocných přítomen statisticky významný rozdíl v délce přežití mezi transplantovanými a netransplantovanými ani u RA, ani u RCMD, přestože SCT významně snížila riziko leukemické transformace. Pokud pomineme poměrně malé počty nemocných v jednotlivých skupinách, zdá se být nejpravděpodobnější příčinou vysoká mortalita spojená s SCT (44 %) na jedné straně, a pomalu probíhající choroba s nízkým rizikem přechodu do AL (24 % pro RA a 0 % pro RARS dle FAB klasifikace 5 let po diagnóze) na straně druhé. Rozdíl mezi počtem přežívajících s RA dle FAB klasifikace se stal významným ve prospěch transplantovaných teprve po 7 letech (48 % vs 33 %). K obdobným závěrům došla i americká pracovní skupina hodnotící riziko spojené se SCT u nemocných s MDS [4]. U nemocných s nízkým či středním I. rizikem dle IPSS byla při časné SCT v době diagnózy pravděpodobnost přežití o 3 roky nižší než u nemocných transplantovaných ve fázi počínající leukemické transformace. U nemocných s nízkým rizikem byla s maximálním efektem na délku přežití spojena SCT 2,5 roku po diagnóze, pokud byla provedena před přechodem do AL. V matematickém modelu pak autoři prokázali, že u nemocných s MDS a nízkým rizikem by měla časná SCT v době diagnózy jednoznačný přínos pouze, pokud by se podařilo snížit mortalitu spojenou s transplantací pod 15 %.
Teprve při kombinované stratifikaci nemocných podle WHO klasifikace a rizikových skupin dle IPSS klasifikace karyotypu byla v naší studii nalezena skupina nemocných s RCMD a „nepříznivým“ karyotypem, u níž byl přínos časné SCT na délku přežití evidentní i přes malý počet hodnocených nemocných v jednotlivých skupinách. Význam karyotypu pro prognózu nemocných s časnými stadii MDS byl podpořen i výsledkem regresní analýzy laboratorních a klinických parametrů, přítomnost nepříznivých změn karotypu byla nejdůležitějším nezávislým parametrem ovlivňujícím jak délku přežití, tak riziko leukemické transformace nemocných s RA dle FAB klasifikace. K obdobnému závěru dospěla i regresní analýza 816 nemocných s primárním MDS při tvorbě IPSS, u nemocných s RA a RARS byly nezávislými parametry ovlivňujícími přežití a rozvoj AML pancytopenie a nepříznivý karyotyp. Naše studie ukázala i význam metody FISH při analýze karyotypu a odhalování přídatných aberací karyotypu, jejichž přítomnost může mít negativní prognostický význam. Užití metod molekulární cytogenetiky v kombinaci s klasickou morfologií může dále zpřesnit identifikaci rizikových nemocných v podskupině refrakterních anémií, indikovaných k časné SCT i přes relativně vysoké riziko komplikací spojených s transplantací.
Práce vznikla v rámci grantového projektu NK/7713-3 Interní grantové agentury Ministerstva zdravotnictví České republiky a výzkumného záměru MZ 00237360001.
MUDr. Jaroslav Čermák, CSc.
Ústav hematologie a krevní transfuze
U nemocnice 1
128 20 Praha 2
Doručeno do redakce: 27. 6. 2003
Přijato po recenzi: 8. 3. 2004
Sources
1. Anderson JE Bone marrow transplantation for myelodysplasia. Blood Reviews 2000; 14: 63-77.
2. Bennett JM, Catovsky D, Daniel MT et al. Proposals for the classification of the myelodysplastic syndromes. Br J Haematol 1982; 51: 189-199.
3. Cox DR. Regression models and life-tables. J R Stat Soc 1972; 34: 187.
4. Cutler C, Lee S, Greenberg P et al. A decision analysis of allogeneic stem cell transplantation for MDS: Delayed transplantation for low risk MDS is associated with improved outcome. Blood 2002; 100: 74a.
5. Čermák J Myelodysplastický syndrom. In: Mayer J, Starý J. Leukémie. Grada 2002: 221-234.
6. De Witte T, Zwaan F, Hermans J et al. Allogeneic bone marrow transplantation for secondary leukemia and myelodysplastic syndrome:a survey by the Leukemia Working Party of the European Bone marrow Transplantation Group (EBMTG). Br J Haematol 1990; 74: 151-155.
7. De Witte T, Suciu S, Verhoef G et al. Intensive chemotherapy followed by allogeneic or autologous stem cell transplantation for patients with myelodysplastic syndromes (MDSs) and acute myeloid leukemia following MDS. Blood 2002; 98: 2326-2331.
8. Germing U, Gattermann N, Strupp C et al. Validation of the WHO proposals for a new classification of primary myelodysplastic syndromes: a retrospective analysis of 1600 patients. Leuk Res 2000; 24: 983-992.
9. Greenberg P, Cox C, Le Beau MM et al. International scoring system for evaluating prognosis in myelodysplastic syndromes. Blood 1997; 89: 2079-2088.
10. Harris NL, Jaffe ES, Diebold J et al. World Health Organization classification of neoplastic diseases of the hematopoietic and lymphoid tissues: report of the Clinical Advisory Committee Meeting - Airlie House, Virginia, November 1997. J Clin Oncol 1999; 17: 3835-3849.
11. Kaplan EL, Meier P. Nonparametric estimation from incomplete observations. J Am Stat Assoc 1958; 53: 457-481.
12. Parker JE, Shafi T, Pagliuca A et al. Allogeneic stem cell transplantation in the myelodysplastic syndromes: interim results of outcome following reduced-intensity conditioning compared with standard preparative regimens. Br J Haematol 2002; 119: 144-154.
13. Raza A, Gregory A, Preisler HD. The myelodysplastic syndromes in 1996: complex stem cell disorders confounded by dual actions of cytokines. Leukemia Res 1996; 20: 881-890.
14. Sierra J, Pérez WS, Rozman C et al. Bone marrow transplantation from HLA-identical siblings as treatment for myelodysplasia. Blood 2002; 100: 1997-2004.
15. Steensma DP, Tefferi A The myelodysplastic syndrome(s): a perspective and review highlighting current controversies. Leuk Res 2003; 27: 95-120.
16. Sutton L, Chastang C, Ribaud P et al. Factors influencing outcome in de novo myelodysplastic syndromes treated by allogeneic bone marrow transplantation: a long term study of 71 patients. Blood 1996; 88: 358-365.
17. Van Den Berghe H, Cassiman JJ, David G et al. Distinct hematological disorder with deletion of long arm of No. 5 chromosome. Nature 1974; 251: 437.
18. Wijermans P, Lubbert M, Verhoef G et al. Low-dose 5-Aza-2,-deoxycytidine, a DNA hypomethylating agent, for the treatment of high-risk myelodysplastic syndrome: a multicenter phase II study in elderly patients. J Clin Oncol 2000; 18: 956-962.
Labels
Diabetology Endocrinology Internal medicineArticle was published in
Internal Medicine

2005 Issue 1
Most read in this issue
- Liver disease at alpha-1-antitrypsin deficiency
- Myelodysplastic syndrome in the new millennium. How to classify and cure patients?
- Hypertension and hyperuricemy
- Extramedullary plasmacytoma of the thyroid gland – a rare vause of a solitary struma nodosa and hyperthyroidism