
Antifosfolipidový syndrom – diagnostika a léčba
Antiphospholipid syndrome – diagnosis and treatment
Antiphospoholid syndrome is the most common form of acquired thrombophilia. It can cause significant morbidity and even mortality. Any organ system and any size of vessel can be affected during clinical course of the disease. Therefore, the antiphospholipid syndrome can manifest itself in a wide variety of clinical features. Despite advances in the understanding of the pathogenesis of antiphospholipid antibodies impact on their target structures, the mainstay of management is still anticoagulation. In this review, we focused on diagnostic procedure in patients with antiphospholipid antibodies and we summarised therapeutic approaches.
Key words:
antiphospholipid syndrome – diagnostic procedure – treatment
:
A. Buliková; M. Penka
:
Oddělení klinické hematologie FN Brno, pracoviště Bohunice, přednosta prof. MUDr. Miroslav Penka, CSc.
:
Vnitř Lék 2005; 91(7 a 8): 809-817
:
128th Internal Medicine Day - 21rd Vanysek's Day Brno 2005
Antifosfolipidový syndrom je nejčastější příčinou získané trombofilie. Může vést k významné mortalitě i morbiditě. Jakýkoli orgánový systém a céva jakékoli velikosti mohou být v průběhu onemocnění postiženy. Z tohoto důvodu se sám antifosfolipidový syndrom může manifestovat širokým spektrem klinických příznaků. Bez ohledu na pokroky v pochopení patogeneze vlivu antifosfolipidových protilátek na jejich cílové struktury, zůstává antikoagulační léčba hlavím způsobem léčby. V tomto přehledu jsme se zaměřili na proces diagnostiky u pacientů s antifosfolipidovými protilátkami a shrnuli terapeutické možnosti.
Klíčová slova:
antifosfolipidový syndrom – proces diagnostiky – terapie
Úvod
Antifosfolipidový syndrom (APS) je klinicko-laboratorní diagnostická jednotka charakterizovaná přítomností antifosfolipidových protilátek a současně klinických projevů, tj. venózní a/nebo arteriální trombózy či mikrotrombotizace v cirkulaci a/nebo reprodukčních ztrát.
Cílovými antigeny antifosfolipidových protilátek, které jsou asociovány s klinickou manifestací APS, jsou makromolekulární struktury vázané na negativně nabité, většinou fosfolipidové povrchy. Je však nutno říci, že tato místa zásahu antifosfolipidových protilátek jsou velmi heterogenní a jejich stručný přehled je shrnut rámcově v tab. 1 [1,2,3,4,5,6].

V posledním desetiletí je zdůrazňována role buněčných elementů v procesech krevního srážení, a tím se také stále častěji potvrzuje jejich úloha v patofyziologii působení antifosfolipidových protilátek. Klíčovou roli zde hrají endoteliální buňky, trombocyty a monocyty [1,2,7,8,9].
Výskyt
Antifosfolipidové protilátky (APA) se mohou prokazovat i u jinak zdravých jedinců jako náhodný nález (tab. 2) [10,11]. Nicméně tyto náhodně detekované protilátky bývají častěji přechodné, většinou po infekcích.

Diagnóza antifosfolipidového syndromu
Diagnóza antifosfolipidového syndromu doznala v průběhu let několik revizí. Poslední ucelené diagnostické schéma vzešlo z workshopu, který navazoval na 8. mezinárodní sympozium o antifosfolipidových protilátkách v Sapporu v roce 1998. Kritéria definitivní diagnózy podle mezinárodního konsenzu shrnuje tab. 3 [12].

Na základě poznatků, že přítomnost inhibitoru lupus antikoagulans a protilátek proti β2-glykoproteinu I je více asociována s trombózou nežli přítomnost antikardiolipinů [13], byla v roce 2002 na jednání standardizačního výboru ISTH (Mezinárodní společnosti pro trombózu a hemostázu) doporučena nová klasifikace laboratorních kritérií pro diagnózu antifosfolipidového syndromu [14].
Doporučuje rozlišovat:
- typ I: pozitivita lupus antikoagulans a současně pozitivita protilátek proti β2-glykoproteinu I
- typ II: izolovaný průkaz lupus antikoagulans
- typ III: izolovaná pozitivita protilátek proti β2-glykoproteinu I
- typ IV: průkaz jiných antifosfolipidových protilátek (proti fosfatidyletanolaminu, proti protrombinu atd).
Nález antikardiolipinových protilátek v tomto doporucení na novou klasifikaci nebyl výslovne zminován s tím, že prukaz protilátek proti kardiolipinu bude nahrazen více specifickým vyšetrením protilátek proti #2-glykoproteinu I.
Primární a sekundární antifosfolipidový syndrom
Stav, u nějž můžeme stanovit diagnózu antifosfolipidového syndromu, může vyplývat z nejrůznějších klinických situací. Obvykle rozlišujeme čtyři kategorie [15]:
- a) antifosfolipidový syndrom spojený se systémovým onemocněním pojiva, obvykle se SLE;
- b) pacienti s antifosfolipidovým syndromem, u nichž nebylo zjištěno žádné doprovodné onemocnění systémového onemocnění pojiva – primární antifosfolipidový syndrom;
- c) pacienti s antifosfolipidovým syndromem, u nichž je podezření na systémové onemocnění pojiva, ale klinická kritéria tohoto onemocnění nebyla s jistotou naplněna – „lupus-like“ choroba;
- d) antifosfolipidový syndrom z jiných příčin, jako jsou léky, nádorová onemocnění, infekce. Řada těchto pacientů má jen antifosfolipidové protilátky bez klinické manifestace onemocnění, obvykle jen v nízkých hladinách pozitivity a často přechodné.
Stavy, které jsou spojeny s výskytem APA, ukazuje tab. 4. Vylučovací kritéria primárního antifosfolipidového syndromu byla stanovena již v roce 1993 Piettem [16] a jsou shrnuta v tab. 5.
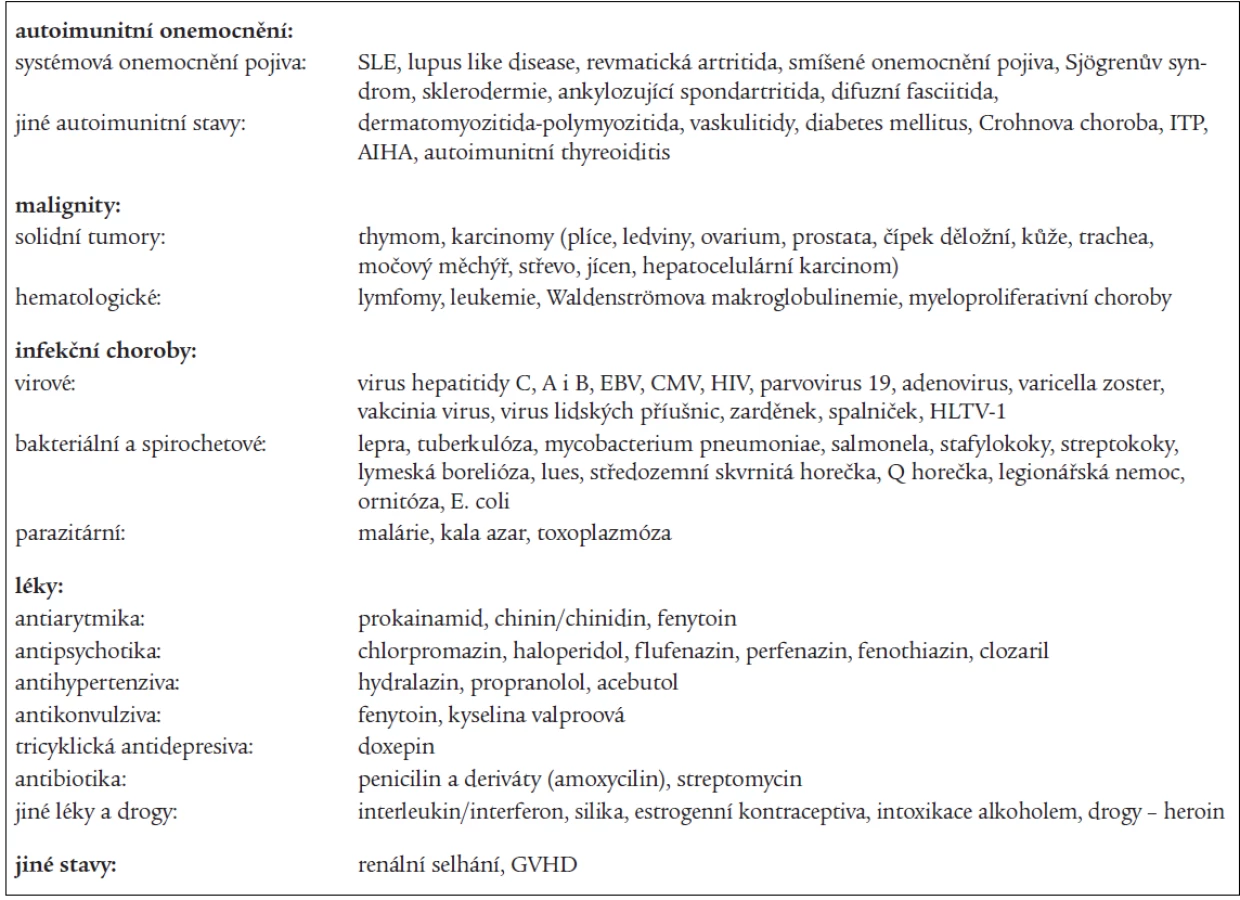

Klinické projevy působení antifosfolipidových protilátek
Klinické projevy APS jsou velmi pestré. Většina z nich je zapříčiněna trombotizací venózní nebo arteriální části cévního řečiště nejrůznějších systémů od velkých cév po mikrocirkulaci. Na změnách se podílí trombotická mikroangiopatie, druhotná ischemie zapříčiněná poruchou přítoku na základě okluze arteriálního řečiště, periferní embolizace z venózních, arteriální či intrakardiálních zdrojů. Teoreticky může být postižen jakýkoli orgán. U některých typů klinické manifestace však nikdy trombóza ani mikrotrombotizace cílového orgánu prokázána nebyla; zde se předpokládá přímé působení APA na cílové struktury.
Venózní trombóza, zejména manifestující se na dolních končetinách, je nejčastější klinickou manifestací antifosfolipidového syndromu, která se objevuje s frekvencí 29–55 % u nemocných s nálezem antifosfolipidových protilátek při průměrné době sledování méně než 6 let [17]. Ve velkém prospektivním sledování italského registru pro antifosfolipidové protilátky se trombotická manifestace objevovala s celkovou incidencí 2,5 % pacientů ročně; u nemocných s anamnézou předchozí trombózy to bylo 5,4 % pacientů ročně, bez této předchozí manifestace 0,95 % pacientů ročně. Zřetelná většina nemocných měla idiopatický typ trombotické manifestace [18].
Arteriální trombóza a/nebo okluze je u antifosfolipidového syndromu o něco méně častým klinickým příznakem. Nejčastějším postižením je ischemická cévní mozková příhoda, která tvoří 50 % arteriálních postižení. Okluze koronární tepny pak tvoří 23 % případů, zatímco zbývajících 27 % znamená arteriální postižení v nejrůznějších oblastech [17].
Přítomnost antifosfolipidových protilátek může být spojena s výskytem celé řady kožních projevů až u 41 % pacientů s antifosfolipidovým syndromem [19]. Patří sem livedo reticularis, kožní ulcerace, nekrotizující a livedoidní vaskulitida, kožní gangréna a nekróza, povrchová tromboflebitida, pseudovaskulitické léze, resp. subunguální hemoragie.
Také neurologické symptomy jsou přítomny u řady pacientů s prokázanými antifosfolipidovými protilátkami, avšak ne vždy je jasná korelace mezi těmito klinickými nálezy a hladinou antifosfolipidových protilátek [20].
Cerebrovaskulární ischemie je nejčastější arteriální trombotickou manifestací u nemocných s antifosfolipidovým syndromem [21]. Průměrný věk postižených pacientů je o několik desetiletí nižší, než je běžná klinická manifestace těchto příhod bez přítomnosti APA [20]. K dalším projevům patří trombóza cerebrálního venózního sinu. Častá je i demence a jiné poruchy kognitivních funkcí jako například zhoršení výkonnosti, verbálního učení a paměti – tyto nálezy mohou indikovat preklinickou fázi neurologického postižení [20,21]. Tyto projevy nemusí být spojeny s žádným nálezem na nukleární magnetické rezonanci (NMR) mozku, a proto se předpokládá nejen snížený průtok krve CNS, ale i přímý efekt antifosfolipidových protilátek na funkci neuronu. Literární data dávají různé výsledky při studiu vztahu epilepsie a antifosfolipidových protilátek. Nicméně pokusy na zvířatech prokazují přímou vazbu APA v mozku a jejich schopnost ovlivňovat receptory kyseliny γ-aminobutyrové (GABA) – tím mohou snižovat prahovou pohotovost ke křečovým stavům. U primárního i sekundárního APS se může vyskytnout také chorea. Vzhledem k časté manifestaci v graviditě, puerperiu či při hormonální kontracepci se předpokládá role estrogenů při rozvoji klinických příznaků. Za použití pozitronové emisní tomografie (PET) byl u těchto nemocných prokázán přechodný hypermetabolizmus v kontralaterálním bazálním gangliu, a proto se zde předpokládá spíše excitační vliv protilátek nežli dříve předpokládaná porucha prokrvení [20]. Obraz transverzální myelopatie a „multiple sclerosis–like“ syndrom jsou dalšími možnými klinickými projevy APS.
U části z nich se předpokládá cévní postižení, u jiných negativní NMR nález a úspěšnost imunosupresivní a nikoli antitrombotické léčby předpokládá spíše přímý vliv autoprotilátek či zánětlivé vlivy.
Přítomnost antifosfolipidových protilátek byla prokázána u některých případů Guillainova-Barrého syndromu, u senzoneurální ztráty sluchu, zejména při jejím náhlém vzniku. Do souvislosti s APA jsou dávány také některé stavy přechodné globální amnézie – náhlé nevysvětlitelné ztráty paměti. K projevům APS patří i postižení optického nervu. Při neurologických symptomech v přítomnosti antifosfolipidových protilátek je indikováno vyšetření mozku resp. i míchy NMR. Nejčastějším nálezem jsou malé fokusy vysokého signálu v subkortikální bílé hmotě rozeseté difuzně po mozku. Tento nález však není specifický. I když je užitečné prokázat strukturální léze, jako jsou infarkty a hemoragie, NMR špatně koreluje s difuzní či globální dysfunkcí. Příčinu potíží nezřídka neodhalí ani PET [20].
Výskyt antifosfolipidových protilátek je dále spojen s nálezem postižení chlopenního aparátu srdce, což je u těchto nemocných nejčastější kardiální manifestací [22]. V patofyziologii se předpokládá APA vyvolaná aktivace endoteliálních buněk na chlopních a následná lokální zánětlivá reakce, která vyústí v chlopenní postižení. Celá situace je navíc podpořena obecnou protrombotickou tendencí. V echokardiografických nálezech je patologie prokázána u třetiny nemocných s primárním antifosfolipidovým syndromem [23].
Přítomnost antifosfolipidových protilátek se zdá se však být nezávislým rizikovým faktorem pro rozvoj infarktu myokardu a náhlou kardiální smrt [24]. Svou roli zde zřejmě hraje proaterogenní efekt protilátek proti β2-glykoproteinu I, a tím nový model imunitně podmíněné aterosklerózy. Přítomnost antifosfolipidových protilátek hraje svou roli při terapii koronární srdeční nemoci – je vyšší incidence okluze štěpu po provedeném bypassu i restenózy po perkutánní koronární angioplastice. Je popisováno vyšší riziko vzniku intrakardiálního trombu, ojediněle i souvislost s akutní kardiomyopatií (na patofyziologickém základě kardiální mikrovaskulopatie), resp. i nález chronické kardiomyopatie [24].
Podobně jako v ostatních parenchymatózních orgánech je přítomnost antifosfolipidových protilátek provázena zvýšenou tendencí k trombotickému procesu v ledvinných cévách, a to na všech úrovních cévního zásobení, počínaje hlavní renální tepnou a jejími větvemi, přes arterioly, glomerulární kapiláry až po ledvinné žíly [25]. Proces v ledvinách je jak z patofyziologického, tak i klinického pohledu komplikován tím, že ledviny jsou orgánem, jež může být postižen přítomností vlastních antifosfolipidových protilátek i základním onemocněním – procesem, který je příčinou indukce těchto protilátek (nejčastěji SLE).
U nemocných s antifosfolipidovými protilátkami mohou vznikat okluze či stenózy ledvinných tepen – velkých i středně velkých. Klinickou manifestací je především těžká renovaskulární hypertenze, bolesti v bederní oblasti, hematurie a zejména v případech primárního antifosfolipidového syndromu i renální selhání [26].
Nálezy provázející intrarenální cévní postižení při APS jsou někdy shrnovány do pojmu „nefropatie antifosfolipidového syndromu“ (APSN). V klinickém obrazu dominuje obvykle těžká systémová hypertenze, akutní nebo chronické ledvinné selhání, proteinurie (lehká až obraz nefrotického syndromu) je přítomna téměř u všech nemocných. Hematurie je nekonstantním nálezem. V histologickém obrazu nacházíme postižení intrarenálních cév s fibrotickou vaskulární okluzí a akutními mikrotromby, u třetiny nemocných je zjišťována trombotická mikroangiopatie.
Jak trombóza hlavní žíly, tak i trombózy menších ledvinných žil jsou taktéž popisovány u primárního i sekundárního antifosfolipidového syndromu. Bilaterální postižení může být příčinou renálního selhání. V těch případech terminální renální choroby („end-stage renal disease“), kdy není příčina renálního postižení jasně určena, je taktéž vyšší prevalence nálezu antifosfolipidových protilátek, zejména lupus antikoagulans [26].
Plicní embolie a infarkt patří k nejčastějším manifestacím antifosfolipidového syndromu [27] – až třetina pacientů s trombózou má tuto komplikaci, u 9 % nemocných jde o první projev choroby [28]. Také plicní parenchym může být v případě antifosfolipidových protilátek postižen mikrotrombotizací – v klinickém obrazu může při chronickém průběhu dojít ke vzniku plicní hypertenze, při akutním či subakutním průběhu dochází ke vzniku syndromu akutního respiračního selhání nebo ke vniku intraalveolární plicní hemoragie [17].
Trombotické postižení očních struktur je jedním z dalších rysů antifosfolipidového syndromu. Amaurosis fugax – epizodická monookulární ztráta vizu – je popisována v souvislosti s přítomností antifosfolipidových protilátek od konce 80. let 20. století [29]. Je charakterizována vznikem v mladém věku a vysokou frekvencí epizod – až 20 denně. Obvykle není prokázána embolizace retiny a nález na echokardiogramu může být normální.
K dalším projevům patří vazookluzivní retinopatie a choroideopatie nebo optická neuropatie.
Při přítomnosti APA může dojít k manifestaci v gastrointestinálním traktu – jaterní postižení je relativně vzácné; APA jsou však udávána jako častá příčina Budd-Chiariho syndromu [30]. Může vzniknout venookluzivní nemoc jater, nodulární regenerativní hyperplazie, vzácně je taktéž popisována jinak nevysvětlitelná portální hypertenze. Trombotizace mezenterických cév či jejich okluze může vést ke klinické manifestaci infarktu střeva či abdominální anginy. Také ischemická kolitida patří do spektra možné, byť poměrně vzácné intestinální manifestace antifosfolipidového syndromu. Popisovány jsou i ezofageální ulcerace [17], vzácně i splenický infarkt, postižení pankreatu případně i s rozvojem akutní pankreatitidy [17,30].
Úloha antifosfolipidových protilátek se zdá být kauzální při vzniku řady případů Addisonovy choroby [31]. Infarkty či mikrotrombotizace mohou postihnout i jiné endokrinní orgány – jsou popisovány pituitární insuficience (zřejmě některé případy Sheehanova syndromu jsou této geneze), infarkty testikulární, epidydimální a infarkty prostaty [31].
Trombocytopenie je poměrně častým nálezem u antifosfolipidového syndromu, je popisována u 20–30 % nemocných [31]. Počet trombocytů bývá obvykle jen lehce snížen, trombocytopenie má chronický charakter a nebývá provázena krvácivými komplikacemi. Počet destiček nižší než 50 × 109/l je vzácný a také v tomto případě jsou spontánní krvácivé projevy spíše neobvyklé. I u závažných poklesů počtu trombocytů však nejsou vyloučeny trombotické komplikace [32]. Nález antifosfolipidových protilátek může provázet idiopatickou trombocytopenickou purpuru až ve třetině případů [32]. Počet trombocytů je zde obvykle nižší než u pouhé přítomnosti antifosfolipidových protilátek (méně než 50 × 109/l), je běžný současný průkaz specifických antitrombocytárních protilátek. V rámci diferenciální diagnostiky trombocytopenie u pacientů s průkazem antifosfolipidových protilátek je nutno brát v úvahu možnost indukce diseminované intravaskulární koagulace nebo trombotické trombocytopenické purpury buď identickým základním onemocněním, nebo jeho terapií. Trombotická mikroangiopatie, někdy provázená i mikroangiopatickou hemolytickou anémií, je však relativně častým nálezem u nemocných s antifosfolipidovým syndromem, a proto trombocytopenie zde zjišťovaná může být částečně i této geneze.
Projevy autoimunitní hemolytické anémie (AIHA) jsou méně časté – u nemocných s primárním výskytem antifosfolipidových protilátek okolo 4 % [32]. Přímá vazba antifosfolipidových protilátek na membránu erytrocytů je pravděpodobná, což podporuje řada nálezů – průkaz antikardiolipinových protilátek v eluátech z erytrocytů u nemocných s AIHA a APA, korelace mezi titrem antifosfolipidových protilátek a intenzitou hemolýzy, antierytrocytární aktivita z eluátů může být inhibována absorpcí na fosfolipidové struktury a antikardiolipinová aktivita přítomná v eluátech může být inhibována absorpcí na fixované erytrocytární membrány. Antigen, který by však mohl být antifosfolipidovými protilátkami vázán na povrhu erytrocytů, však nebyl objeven.
Diagnostický proces u antifosfolipidového syndromu
Při naplnění diagnostických kritérií APS je zapotřebí komplexního vyšetření každého pacienta, které směruje k:
- určení, zda jde o primární či sekundární proces; i v případě, že není nalezeno žádné vyvolávající onemocnění a je stanovena diagnóza primárního antifosfolipidového syndromu, je indikováno dlouhodobé sledování k vyloučení systémového onemocnění pojiva;
- stanovení orgánového postižení pomocí klinického, laboratorního, zobrazovacího vyšetření, v některých případech je nutné i histologické vyšetření (zejména postižení ledvin);
- stanovení dalších rizikových faktorů trombózy;
- stanovení krvácivé tendence.
Protrombotickou tendenci antifosfolipidového syndromu mohou zvyšovat vrozené trombofilní stavy – leidenská mutace F V, deficit antitrombinu. U deficitu proteinu S a proteinu C není při jejich průkazu možné vždy rozhodnout, zda jde v konkrétním případě o vrozený stav, či situaci navozenou přítomností vlastní APA, zejména lupus antikogulans. Nebyl naopak prokázán aditivní vliv mutace protrombinu 20210A ani termolabilní varianty metylentetrahydrofolát–reduktázy na klinickou manifestaci trombózy u pacientů s APA [33]. Při posuzování protrombotické tendence APA je nutno brát v úvahu současnou přítomnost celé řady dalších obecně známých získaných rizikových faktorů trombózy v arteriálním či venózním řečišti.
Krvácivé projevy u APS mohou nastat, je-li přítomna jinak vzácná významná trombocytopenie. Také funkční poruchy destiček jsou popisovány až u 40 % pacientů s APA [34]. Část antifosfolipidových protilátek svou vazbou na trombocyty zřejmě mohou pokrývat vazebná receptorová místa a vést k membránovému typu funkční poruchy destiček. Jednou z nejzávažnějších příčin krvácivých projevů je vznik sekundární hypoprotrombinemie. Imunoglobulin s lupus antikoagulans aktivitou reaguje s molekulou protrombinu mimo jeho funkční místo a nevede přímo k ovlivnění – neutralizaci aktivity faktoru. Vznikají však komplexy antigen-protilátka, které jsou odstraňovány z cirkulace, a tímto způsobem může vznikat získaný deficit protrombinu, výjimečně i závažný spojený i se spontánními krvácivými projevy [35]. Na rozdíl od ostatních lupus antikoagulantů, které běžně neovlivňují hodnotu protrombinového času [36] dle Quicka (fosfolipid je u tohoto testu při běžném provedení dodáván v nadbytku a neutralizuje přítomnou antifosfolipidovou protilátku), jsou lupus antikoagulanty se sekundární hypoprotrombinemií provázeny při klinicky významném snížení faktoru II prodloužením tohoto testu. Proto je patologie v protrombinovém času u lupus antikoagulans významným signálem rizika jinak vzácného krvácení a je nutné stanovit aktivitu protrombinu.
Primární prevence
Léčebný přístup k nemocným, u nichž je průkaz APA doposud klinicky asymptomatický, není s definitivní platností vyřešen. Řada těchto pacientů nikdy tromboembolickou příhodu neprodělá, i když je přítomnost APA dlouhodobá či trvalá. V úvahu přichází následující postupy [37]:
- žádná trvalá léčba: Pacientům je doporučeno vystříhat se dalších rizikových faktorů – orální kontracepce, kouření, obezita, ale také např. léčba COX-2 inhibitory. Je naopak zavedena náležitá léčba hypertenze a hypercholesterolemie. Krátkodobá antitrombotická profylaxe obvykle nízkomolekulárním heparinem je zavedena při dalším navýšení trombofilního rizika – operace, úrazy, imobilizace. Nemocní by měli být pečlivě monitorováni.
- kyselina acetylsalicylová (ASA): Nízké dávky aspirinu (75–100 mg denně) jsou v těchto indikacích nejčastěji používanou léčbou [38]. Jen ojedinělé práce však dokládají, že ASA chrání proti trombóze, a to navíc jen v úzce vymezených situacích – po porodu u žen s APS manifestujícím se pouze ztrátami plodu, nikoli však tromboembolickou příhodou [39]. Většina sdělení neprokazuje žádný přesvědčivý benefit [37,38]. V současné době probíhá prospektivní studie k zhodnocení efektu tohoto léčebného přístupu – APLASA study [40].
- antimalarika: Protidestičkové působení hydrochlorochinu či chlorochinu je známo od 70. let minulého století, kdy byly využívány pro profylaxi žilního tromboembolizmu po ortopedických operacích. Opakovaně byl v retrospektivních studiích i na zvířecích modelech prokázán jejich efekt jak u primárního, tak i sekundárního výskytu APA.
- nízkodávkovaná antikoagulační léčba warfarinem: Efekt tohoto typu léčby s cílovým INR 1,5 v kombinaci s nízkodávkovanou ASA je v současné době porovnáván v prospektivní multicentrické studii ve Velké Británii s účinností samotného aspirinu [37].
Léčba
Iniciální léčba akutní trombotické epizody se u pacienta a APS v ničem neodlišuje od terapie trombózy vzniklé z jiných důvodů [41]. Dle místa lokalizace, rozsahu a klinického nálezu lze využít léčbu nízkomolekulárním heparinem, trombolýzou, trombektomií resp. angioplastikou v případě koronární trombózy. Použití nefrakcionovaného heparinu naráží na problém sledování – aktivovaný parciální tromboplastinový čas je ovlivněn přítomnosti lupus antikoagulans již před zavedením terapie a jeho další prodloužení vlivem heparinu není proporcionální.
Hlavním terapeutickým zásahem pro sekundární profylaxi je antikoagulační léčba. Na základě retrospektivních studií [42,43] bylo stanoveno doporučení dosažení cílového INR 3,0–4,0 a dlouhodobé resp. i doživotní podávání dikumarolových preparátů. V současné době prospektivní studie prokazují, že řada nemocných je úspěšně léčena konvenčním dávkováním kumarinů s cílovým INR 2,0–3,0 [44,45]. Vysokodávkovaná a dlouhodobá resp. doživotní léčba bude zřejmě vyhrazena nemocným s rekurentní idiopatickou tromboembolickou manifestací.
Jasný přínos antikoagulační léčby ve srovnání s antiagregační terapií nebyl prokázán u nemocných s cévní ischemickou mozkovou příhodou [46].
Pacienti na antikoagulační léčbě mohou mít relativní rezistenci na warfarin s potřebou vysokých dávek (denní dávka vyšší než 25 mg není vzácností). Na této rezistenci se mimo jiné může podílet i současná léčba revmatického procesu, zejména azathioprin, který významně ovlivňuje metabolizmus warfarinu indukcí hepatálních enzymů. Náhlé vysazení azathioprinu bez kontrol nastavení antikoagulace může vést k rychlému předávkování a fatálním krvácivým komplikacím [47].
Při selhání antikoagulační léčby warfarinem je zvažováno přidání ASA k vysokodávkované léčbě dikumarolovými preparáty, podání imunomodulační terapie [47] nebo nízkomolekulární heparin [38], a to zejména v případech arteriální manifestace.
Imunosupresivní léčba není pro prevenci rekurence trombózy účinná [41]. Má své přesně vymezené indikace, ať už je to léčba základního onemocnění u sekundární indukce APA systémovým procesem, je jí možno ovlivnit sekundární hypoprotrombinemii s krvácivými projevy [1], klinicky významnou trombocytopenii [38]. Její podání je taktéž zvažováno tam, kde se předpokládá jiný než protrombotický mechanizmus klinické manifestace. Do této indikace patří zejména některé z neurologických symptomů. Postupy ovlivňující imunitní systém jsou taktéž používány u katastrofického průběhu APS [48].
Nové léčebné přístupy
V současné době jsou diskutovány různé další léčebné možnosti u APS. Jednak je to využití léků, které se používají rutinně v jiných indikacích, druhým směrem je pak specifické ovlivnění imunitního systému.
Jako perspektivní přístup se jeví využití statinů. Tyto léky, primárně užívané k ovlivnění nepříznivého lipidového spektra, mají i vícečetný efekt na krevní elementy (trombocyty, monocyty). V současné době byl in vitro prokázán inhibující vliv fluvastatinu na protrombotické a prozánětlivé vlastnosti endoteliálních buněk navozené přítomností APA [49]. Na zhodnocení v této indikaci čekají jak novější typy protidestičkových léků – z nichž širší využití u APS doznal pouze klopidogrel [50], stejně tak i řada dalších nových antitrombotik – např. přímé inhibitory trombinu melagatran/ximelagatran, rekombinantní inhibitor cesty tkáňového faktoru a jiné [38].
Příkladem léčebného přístupu s imunomodulačním zaměřením je syntéza toleragenu (LJP 993) určeného k inaktivaci B-lymfocytů specifických k epitopu β2 glykoproteinu I [38, 41].
Závěr
Antifosfolipidový syndrom je onemocnění s velmi variabilní klinickou manifestací, obtížnou diagnostikou a stále diskutovaným a rozvíjeným přístupem k léčbě těchto nemocných. Z tohoto pohledu je stavem, který vyžaduje multidisciplinární spolupráci v problematice zainteresovaných klinických i laboratorních pracovníků.
MUDr. Alena Buliková
www.fnbrno.cz
e-mail: abulik@fnbrno.cz
Doručeno do redakce: 14. 3. 2005
Přijato k otištění: 14. 3. 2005
Sources
1. Arnout J, Vermylen J. Current status and implications of autoimmune antiphospholipid antibodies in relation to thrombotic disease. J Tromb Haemost 2003; 1 : 931–942.
2. Arnout J, Jankowski M. Antiphospholipid syndrome. Hematology J 2004; 5: S1–S5.
3. Roubey RAS. Mechanisms of autoantibody-mediated thrombosis. Lupus 1998; 7 : 114–119.
4. de Groot PG, Horbach DA, Derksen RHWM. Protein C and other cofactor involved in the binding of antiphospholipid antibodies: relation to the pathogenesis of thrombosis. Lupus 1996; 5 : 488–493.
5. Esmon LN, Smirnov MD, Esmon CHT. Thrombogenic mechanisms of antiphospholipid antibodies. Thromb Haemost 1997; 78 : 79–82.
6. Adams MJ, Donohoe S, Mackie IJ et al. Anti–tissue factor pathway activity in patients with primary antiphospholipid syndrome. British J Haematol 2001; 114 : 375–379.
7. Joseph EJ, Harrison P, Mackie IJ et al. Increased circulation platelet-leukocyte and platelet activation in patients with antiphospholipid syndrome, systemic lupus erythematosus and rheumatoid arthritis. British J Hematol 2001; 115 : 451–459.
8. Hill MB, Phipps JL, Hughes P et al. Anti-endothelial cell antibodies in primary antiphospholipid syndrome and SLE: patterns of reactivity with membrane antigens on microvascular and umbilical venous cell membranes. British J Haematol 1998; 103 : 416–421.
9. Cuadrado MJ, López–Pedrera Ch, Khamashta MA et al. Thrombosis in primary antiphospholipid syndrome: a pivotal role for monocyte tissue factor expression. Arthritis Rheum 1997; 40 : 834–841.
10. Petri M. Epidemiology of the antiphospholipid syndrome. J Autoimmunity 2000; 15 : 145–151.
11. Buliková A, Matýšková M, Zavřelová J et al. Antiphosholipid antibodies in blood donors. Haemostasis 1998; Suppl 2 : 460.
12. Wilson WA, Gharavi AE, Koike T et al. International consensus statement on preliminary classification criteria for definite antiphospholipid syndrome. Arthritis Rheumat 1999; 42 : 1309–1311.
13. Galli M, Luciani D, Bertolini G et al. Anti-β2-glykoprotein I, antiprothrombin antibodies, and the risk of thrombosis in the antiphospholipid syndrome. Blood 2003; 102 : 2717–2723.
14. Arnout J. ISTH scientific standardisation subcommittee lupus anticoagulants/phospholipid dependent antibodies. International society of thrombosis and haemostasis. Boston: July 2002.
15. Vincent T, Mackworht-Young C. The primary antiphospholipid syndrome. In Khamashta MA. Hughes Syndrome. London: Springer Verlag 2000 : 111–126.
16. Piette JC, Wechsler B, Frances C et al. Exclusion criteria for primary antiphospholipid syndrome. J Rheumatol 1993; 20 : 1802–1804.
17. Levine JS, Branch DW, Rauch J. The antiphospholipid syndrome. N Engl J Med. 2002; 346 : 752–763.
18. Finazzi G, Brancaccio V, Moia M et al. Natural history risk and risk factors in 360 patients with antiphospholipid antibodies. A four year prospect study forms the Italian registry. Am J Med 1996; 100 : 530–536.
19. Battagliotti CA. Skin Manifestation of the antiphospholipid antibody syndrome. In Khamashta MA. Hughes Syndrome. London: Springer Verlag 2000 : 59–69.
20. Navarrete MG, Brey RL, Levine SR Cerebral disease in the antiphospholipid syndrome. In: Khamashta MA. Hughes Syndrome. London: Springer Verlag 2000 : 43–58.
21. Bray RL. Differential diagnosis of central nervous system manifestation of the antiphospholipid antibody syndrome: J Autoimmunity 2000; 15 : 133–138.
22. Cervera R. Recent advances in antiphospholipid antibody-related valvulopathies. J Autoimmunity 2000; 15 : 123–125.
23. Levy RA. Clinical manifestations of the aPL syndrome. Lupus 1996; 5 : 393–397.
24. Font J, Cervera R. Cardiac manifestation in the antiphospholipid syndrome. In: Khamashta MA. Hughes Syndrome. London: Springer Verlag 2000 : 32–42.
25. Nochy D, Daugas E, Huong DLT et al. Kidney involvement in the antiphospholipid syndrome. J Autoimmunity 2000; 15, 127–132.
26. Amigo MC, García-Torres R. Kidney disease in antiphosphopipid syndrome. In Khamashta MA. Huges Syndrome 2000; London: Springer Verlag 2000 : 70–81.
27. Cervera R, Garcia-Carrasco M, Asherson RA. Pulmonary manifestation in the antiphospholipid syndrome. In Asherson RA, Cervera R, Piette JCh et al. The Antiphospholipid Syndrome. Boca
Raton: CRC Press 1996 : 161–167.
28. Cervera R, Piette JCh, Font J at al. Euro-phospholipid project group. Antiphospholipid syndrome: Clinical and immunologic manifestation and patterns of disease expression in a cohort of 1000 patients. Arthritis Rheum 2002; 46 : 1019.
29. Levine SR, Crofts JW, Lesser GR et al. Visual symptoms associated with the presence of a lupus anticoagulant. Ophthalmology 1988; 95 : 686–692.
30. Asherson RA. Adrenal, hepatic, and other intraabdominal manifestation in the antiphospholipid syndrome. In: Asherson RA, Cervera R, Piette JCh et al. The Antiphospholipid Syndrome. Boca Raton: CRC Press 1996 : 183–191.
31. Hughes GRV. Of the beaten tracks: A clinician’s view. In: Khamashta MA. Huges Syndrome. London: Springer-Verlag 2000 : 105–110.
32. Montecucco C, Caporali R. Hemocytopenias in antiphospholipid syndrome. In: Khamashta MA. Huges Syndrome. London: Springer–Verlag 2000 : 20–31.
33. Galli M, Finazzi G, Duca F et al. The G1691→A mutation of factor V, but not the G20210→A mutation of factor II or the C677→T mutation of methylentetrahydrofolate reductase genes, is associated with venous thrombosis in patients with lupus anticoagulants. British J Haematol 2000; 108 : 865–870.
34. Galli M, Barbui T. Management of thrombocytopenia in Hughes Syndrome. In Khamashta MA. Hughes Syndrome. London: Springer-Verlag 2000 : 408–418.
35. Triplett DA. Use of the dilute Russell viper venom time (dRVVT): its importance and pitfalls. J Autoimmunity 2000; 15 : 173–178.
36. Mackie IJ, Donohoe S, Machin SJ. Lupus anticoagulant measurement. In Khamashta MA. Hughes Syndrome. London: Springer-Verlag 2000 : 214–224.
37. Khamashta MA. Primary prevention of thrombosis in subjects with positive antiphospholipid antibodies. J Autoimmunity 2000; 15 : 249–253.
38. Öztürk MA, Haznedaroglu IC, Turgut M et al. Current debates in antiphospholipid syndrome: The acquired antibody-mediated thrombophilia. Clin Appl Tromb/Hemost 2004; 10 : 89–126.
39. Erkan D, Merril JT, Yazici Y et al. High thrombosis rate after fetal loss in antiphospholipid syndrome: Effective prophylaxis with aspirin. Arthritis Rheumat 2001; 44 : 1466–1467.
40. Erkan D, Yazici Y, Harrison M et al. APLASA study: primary thrombosis prevention in asymptomatic antiphospholipid antibody (aPL) patients with low-dose aspirin. Lupus 2004; 11 : 573.
41. Roubey R. Treatment of the antiphospholipid syndrome. Curr Opin Rheumatol 2002; 14 : 238–242.
42. Khamashta MA, Cuadrado MJ, Mujic F et al. The management of thrombosis in the antiphospholipid-antibody syndrome. N Engl J Med 1995; 332 : 993–997.
43. Ruiz-Irastorza G, Khamashta MA, Hunt BJ et al. Bleeding and recurrent thrombosis in definite antiphospholipid syndrome: analysis of a series of 66 patients treated with oral anticoagulation to a target international normalized ratio of 3,5. Arch Intern Med 2002; 162 : 1164–1169.
44. Crowther MA, Ginsberg JS, Julian J et al. A comparison of two intensities of warfarin for the prevention of recurrent thrombosis in patients with the antiphospholipid antibody syndrome. N Engl J Med 2003; 349 : 1133–1138.
45. Finazzi G, Marchioli R, Barbui I. A randomized clinical trial of oral anticoagulant therapy in patients with the antiphospholipid syndrome. The WAPS study. J Tromb Haemost 2003; Suppl 1: Abstract OC 365.
46. APASS Investigators. Antiphospholipid antibodies and subsequent trombo-occlusive events in patients with ischemic stroke. JAMA 2004; 291 : 576–584.
47. Khamashta MA. Management of thrombosis in the antiphospholipid syndrome. In Khamashta MA. Hughes Syndrome. London: Springer-Verlag 2000 : 391–396.
48. Asherson RA, Cervera R, de Groot PG et al. Catastrophic antiphospholipid syndrome Registry Project Groop: Catastrophic antiphospholipid syndrome: International consensus statement on classification criteria and treatment guidelines. Lupus 2003; 12 : 530.
49. Ferrari DE, Swerlic R, Casper K et al. Fluvastatin inhibits up-regulation of tissue factor expression by antiphospholipid antibodies on endothelial cells. J Tromb Haemost 2004; 2 : 1558–1563.
50. Bick RL. Antiphospholipid thrombosis syndromes. Clin Appl Tromb Haemost 2001; 7 : 241–258.
Labels
Diabetology Endocrinology Internal medicineArticle was published in
Internal Medicine

2005 Issue 7 a 8
Most read in this issue
- Post transfusion reactions
- Thrombocytosis and thrombocythemia
- Antiphospholipid syndrome – diagnosis and treatment
- Antiaggregant therapy