
Molekulární metody v diagnostice trombofilních stavů
Molecular methods in thrombophilic states diagnostics
Molecular genetic methods passed into the field of investigation of thrombophilic states in 90th years of last century, along with the first discoveries of coagulation inhibitors (AT III, protein C and protein S). They have acquired a widespread use above all with the detection of the molecular basis of activated protein C (APC) resistance in 1994 by prof. Bertina. At the present time, a wide range of molecular genetic markers, linked with a clearly documented increased risk of thrombophilia are adapted. They include mutations of factor V Leiden 506R/Q, of protrombin 20210G/A, MTHFR 677C/T in homozygous form, mutation of PAI‑1 4G/5G, mutations of different coagulation inhibitors and finally a range of polymorphisms with still not precisely defined increased risk for thrombophilia (F XIII Val34leu, platelets glycopeproteins, endothelial protein C receptor and trombomodulin). From the methodological viewpoint, all these techniques are based on the principle polymerase chain reaction (PCR). In the last period of time, however there was a rapid evolution, allowing a significant improvement in their laboriousness. Nowadays, splitting with the aid of restriction endonucleases, real time PCR or allel specific primers for PCR. The second, where molecular genetic methods are currently under use, is pathophysiological investigation of the single coagulation processes. Here, in a fact, most significant progress has been in the field of APC resistance made elucidation. Although still in the 90th years of the past century the genetical cause of these coagulation disturbance was unequivocally documented its clinically heterozygous appears not yet fully understood at the moment. Similarly, in prothrombin mutation, only the latest investigations have outlined the probable mechanism of expression. Concerning the future evolution of molecular genetic methods, there can be observed a clear cut tendency to better understanding the pathophysiologic cause of thrombophilia in comparison with the searching for new coagulation defects which consecutively bear lesser a relative risk of thrombosis.
Key words:
thrombophilia – genetic markers of thrombophilia – F V Leiden – protrombin mutation
Authors:
L. Slavík; V. Krčová; A. Hluší; J. Procházková; J. Úlehlová
Authors‘ workplace:
Hemato-onkologická klinika Lékařské fakulty UP a FN Olomouc, přednosta prof. MUDr. Karel Indrák, DrSc.
Published in:
Vnitř Lék 2009; 55(3): 302-309
Category:
15th Parizek's Days
Overview
Molekulárně genetické metody pronikly do oblasti vyšetřování trombofilních stavů v 90. letech minulého století s prvními objevy inhibitorů koagulace – antitrombinu III (AT III), proteinu C (PC) a proteinu S (PS). Velkého rozšíření dosáhly s objevem molekulární příčiny APC rezistence v roce 1994 prof. Bertinou. V současné době je využívána celá skupina molekulárně genetických markerů s jasně prokázaným rizikem trombofilie – mutace F V Leiden 506R/Q, mutace protrombinu (F II) 20210G/A, mutace metylentetrahydrofolát reduktázy (MTHFR) 677C/T v homozygotní formě, mutace inhibitoru plazminogen aktivátoru (PAI‑1) 4G/5G, mutace jednotlivých inhibitorů koagulace a pak řada polymorfizmů, jejichž riziko je diskutováno – F XIII Val34leu, destičkové glykoproteiny, endoteliální protein C receptor a trombomodulin. Z metodologického hlediska jsou všechny metody postaveny na bázi polymerázové řetězové reakce (PCR), nicméně v poslední době prošly překotným vývojem, který umožnil značné snížení jejich pracnosti. V současné době se používá štěpení produktů PCR pomocí restrikčních endonukleáz, real time PCR a také multiplex PCR metod. Druhou oblastí, kde se využívá molekulárně genetických metod, je studium patofyziologie jednotlivých procesů. Zde byl v poslední době učiněn největší pokrok na poli rezistence na aktivovaný protein C (APC rezistence). Ačkoli se v 90. letech minulého století podařilo jednoznačně prokázat genetickou příčinu APC rezistence, stále se nezdařilo vysvětlit klinickou různorodost exprese tohoto stavu. Obdobná situace byla u protrombinové mutace, kde až poslední výzkumy nastínily možný mechanizmus exprese genetických změn do samotného koagulačního mechanizmu. Z hlediska dalšího vývoje molekulárně genetických metod je jasný příklon ke komplexnímu porozumění patofyziologických stavů a odklon od hledání jednotlivých defektů se stále menším relativním rizikem vzniku trombózy.
Klíčová slova:
trombofilie – genetické příčiny trombofilie – F V Leiden – protrombinová mutace
Úvod
Molekulárně genetické metody dovolují objasnit genetické příčiny v tomto případě trombofilních stavů. Již od samotného počátku diagnostiky trombofilních stavů, který vycházel ze studia rodin s vysokým výskytem trombotických komplikací, bylo zřejmé, že se jedná v řadě případů o geneticky podmíněné hereditární dominantně dědičné stavy. Již u prvních rodin s výskytem deficitu AT III byly popsány genetické příčiny tohoto defektu. Takto bylo u AT III popsáno na 150 příčinných mutací.
Molekulárně genetické metody pronikly do oblasti screeningového vyšetřování trombofilních stavů v 90. letech minulého století s prvními objevy inhibitorů koagulace (AT III, proteinu C a proteinu S). Velkého rozšíření dosáhly s objevem molekulární příčiny APC rezistence v roce 1994 prof. Bertinou.
Přes veškeré rozšíření těchto metod byly v následujících letech objevovány defekty na molekulární úrovni se stále menším rizikem pro vznik trombózy, což je patrné z obr. 1. Přesto až do konce minulého století existovala jasná představa, že pouze zjištění veškerých genetických příčin trombofilie povede k celkovému objasnění genetického rizika trombofilie.
![Grafické vyjádření prevalence a rizika jednotlivých trombofilních defektů s časovým vyjádřením doby objevení [1].](https://www.prelekara.sk/media/cache/resolve/media_object_image_large/media/image/ddb8b616beda546c3fd0d97e33f5979c.jpeg)
V současné době je patrný odklon od této představy a stále větší pozornost je věnována objasnění komplexní patofyziologie koagulace na molekulární úrovni.
F V Leiden
Mezi bílou populací je faktor V Leiden (faktor V 1691G-A) nejčastější genetický defekt způsobující trombózu [1–6]. Mutace faktoru V byla poprvé popsána v roce 1994 Bertinou a spolupracovníky na univerzitním pracovišti v Leidenu [7], na základě nálezu rezistence na aktivovaný protein C (APC), která byla poprvé popsána v roce 1993 [8]. Frekvence výskytu kolísá v bílé populaci mezi 2 a 15 % [1]. Heterozygotní forma faktoru V Leiden zvyšuje riziko trombózy 3–8krát [1,9,10], naproti tomu homozygotní postižení představuje až 80násobné riziko [11] žilní trombózy.
Faktor V Leiden se vyskytuje u 20 % pacientů s žilní trombózou [2,9] a u více než 1/2 probandů u vybraných rodin s trombofilií. To znamená, že představuje nejčastější genetickou abnormalitu u pacientů s trombózou.
Protrombin 20210A
Mutace v 3’-nepřepisované oblasti protrombinového genu v pozici 20210 G-A je spojena se zvýšenou hladinou protrombinu a současně představuje zvýšené riziko vzniku trombózy [12]. Tato mutace byla popsána ve vysoké prevalenci (18 %) v rodinách s trombózou a v 6,2 % u pacientů s první trombózou [12].
Mechanizmus působení mutace v pozici 20210 G-A protrombinového genu je vysvětlován tím, že tato mutace podmiňuje zvýšení hladiny protrombinu, což vede ke zvýšení rizika trombózy [12]. Výskyt mutace v bílé populaci kolísá okolo 2 % s mnoha geografickými variacemi, přičemž výskyt mutace je vyšší v jižní než v severní Evropě [13]. To způsobuje značné rozdíly mezi pracemi udávajícími výskyt této mutace v populaci a relativní riziko mutace pro vznik trombózy. Např. v severní Evropě je výskyt 2 % (obdobně jako v české populaci) [14] s relativním rizikem okolo 2, zatímco v katalánské oblasti Španělska je prevalence 6,5 % [15] a relativní riziko 2, což představuje více než 6 % všech trombotických příhod.
Hyperhomocysteinemie
Lehce zvýšené hladiny homocysteinu jsou spojeny se zvýšeným rizikem trombózy [16–18]. Dvě studie prováděné na nerozdělené skupině pacientů ukázaly, že hladina 18,5 μmol/l u 5 % (resp. 10 %) jedinců vede k dvojnásobnému zvýšení rizika trombózy [17–19]. Z toho plyne, že hyperhomocysteinemie představuje 5–10 % všech trombotických příhod.
Hyperhomocysteinemie může vzniknout na základě genetických nebo získaných dispozic [20]. Mezi získané dispozice patří zejména nízký příjem vitaminů (B6, B12, kyseliny listové), který vede ke zvýšení hladiny homocysteinu [21,22]. Mezi genetické můžeme počítat velmi raritní deficit cystathion-b-syntetázy, jejíž homozygotní forma představuje klasickou hyperhomocysteinemii s velmi vysokými hladinami [23], a naopak velmi častou variantu genu metyltetrahydrofolátreduktázy (MTHFR), která vede k termolabilní variantě enzymu s lehce zvýšenou hladinou homocysteinu [24–26]. V současné době není jednoznačně stanovena přímá spojitost mezi variantou MTHFR 677TT, zvýšenou hladinou homocysteinu a rizikem vzniku trombózy [27].
Antitrombin III
Familiární deficit antitrombinu III po-prvé popsal Egeberg v roce 1965 [28–30]. Již tato první práce ukázala, že defekt antitrombinu III je podstatně závažnější rizikový faktor pro vznik trombózy než defekty proteinů C a S, přičemž většina pacientů má manifestaci klinických projevů do 25 let věku [29,31]. To se netýká změny heparinového vazebného místa, která se vyskytuje často, ale v heterozygotní formě nepředstavuje riziko [31].
Na základě rozsáhlé studie bylo stanoveno trombotické riziko deficitu antitrombinu III jako 5násobné, což vychází z 1,1% výskytu deficitu ve skupině pacientů s žilní trombózou oproti 0,2% výskytu v kontrolní skupině [32]. Další práce uvádí výskyt deficitu antitrombinu III v rozmezí 1–0,5 % [28]. Mezi 4 000 zdravými dárci byl deficit antitrombinu III nalezen v 0,02 % [33]. Tato data ukazují, že defekt antitrombinu III je velmi raritním postižením, nicméně je možné konstatovat, že se jedná o vysoce rizikový faktor pro vznik trombózy, což je patrné ze srovnání prevalence tohoto defektu u pacientů s žilní trombózou a u zdravých dárců (1 % oproti 0,02 %), což ukazuje na 50krát vyšší riziko u pacientů s deficitem antitrombinu III. I přes toto vysoké riziko představuje deficit antitrombinu III pouze 1 % všech trombotických stavů.
Z hlediska příčin mohou být deficity antitrombinu III klasifikovány do 2 skupin: vrozené a získané. Vrozené deficity jsou charakterizovány sníženou hladinou antigenu antitrombinu III, sníženou aktivitou antitrombinu III nebo kombinací obou těchto parametrů. Tyto deficity mají příčinu v dědičné poruše syntézy antitrombinu III nebo v syntéze mutantní molekuly antitrombinu. Poruchy jsou autozomálně dominantně dědičné.
Vrozené deficity antitrombinu III jsou rozděleny do dvou základních typů. Typ I, kvantitativní nedostatek antitrombinu III, je charakterizován paralelním snížením aktivity i antigenu antitrombinu III. Porucha je tedy ve snížené rychlosti syntézy antitrombinu III. Tento defekt představuje 80–90 % všech defektů antitrombinu III, přičemž hladiny antitrombinu III jsou sníženy na 50–70 % normálních hodnot. Typ II, funkční defekt antitrombinu III, je charakterizován nesouladem mezi koncentrací antigenu a funkční aktivitou antitrombinu III. Hladina antigenu je tedy normální, ale je porušena funkčnost molekul. Zde můžeme zařadit typy antitrombinu se sníženou afinitou k heparinu (AT III „Budapešť“, AT III „Milano“, AT III „Tokyo“), dále typy se sníženou schopností inaktivace serinových proteáz (AT III „Northwick Park“, AT III „Glasgow“) a také typy s porušenou schopností vazby na heparin (AT III „Tours“, AT III „Rouen“). Typ III je charakterizován syntézou abnormálních molekul antitrombinu III, kdy je snížena hladina pod fyziologickou mez (AT III „Johanesburg“, AT III „Budapest 2“, AT III „Utah“).
Do dnešní doby bylo popsáno okolo stovky mutantních forem molekul AT III [34,35]. Zavádění podrobnější klasifikace deficitů antitrombinu III naráží na velmi nízkou frekvenci tohoto postižení.
Získané deficity se mohou vyskytovat jednak u fyziologických stavů (např. u novorozenců, v pokročilém těhotenství) a dále u řady onemocnění a představují závažný problém. Příčinou deficitu může být snížená syntéza antitrombinu III (u jaterní cirhózy, léčby asparaginázou), zvýšené ztráty AT III (gastrointestinální choroby, nefrotický syndrom) a v neposlední řadě zvýšená konzumpce AT III (disseminovaná intravaskulární koagulace u traumat, sepsí, popálenin, úžehu a pooperačních stavů).
Protein C
V roce 1981 upozornily první práce na zvýšené riziko vzniku trombózy u pacientů s heterozygotním deficitem proteinu C [36,37]. Nebyl stanoven rozdíl mezi pacienty s různým typem deficitu (I nebo II) a základní mutací. Tyto studie upozornily, že velká většina pacientů má manifestaci klinických projevů trombózy již v mladém věku [38]. Současně je pozoruhodné, že u některých nemocných se vyskytuje i APC resistence jako další faktor zvyšující riziko trombózy [39].
Výskyt deficitu proteinu C u pacientů s trombózou analyzovaly 3 velké studie [40–42]. Ve všech byl stanoven výskyt deficitu proteinu C 3 %. Při srovnání s kontrolní skupinou bylo stanoveno také relativní trombotické riziko deficitu proteinu C, které činí 6,5 [42]. Prevalence 0,2% výskytu ve zdravé populaci byla stanovena na základě obsáhlé studie [36]. Tato prevalence v populaci, kombinovaná však s vysokým rizikem trombogenicity, znamená 1–2% podíl deficitů proteinu C na všech trombofilních stavech.
Gen způsobující deficit proteinu C je děděn autozomálně dominantně. Defekt se klinicky projevuje tromboembolickou nemocí typu superfaciální tromboflebitidy a juvenilní žilní trombózy.
Z hlediska diferenciace jsou známy dva typy deficitů proteinu C: kvantitativní (snížení antigenu proteinu C) a funkční. Funkční deficity proteinu Cmohou být způsobeny řadou mutací. Do dnešní doby bylo popsáno na 160 rozdílných mutací [44]. Heterozygotní postižení defektem proteinu C je spojeno se 7násobně zvýšeným rizikem trombózy.
Protein S
Protein S je důležitým antikoagulačním proteinem, který působí jako neenzymatický kofaktor APC při inaktivaci faktorů Va a VIIIa. Laboratorní screening deficitů proteinu S je komplikován faktem, že protein S cirkuluje v krevním oběhu ve dvou formách [45]. Volný protein S, který působí jako kofaktor APC, představuje asi 40 % celkového množství proteinu S.
Deficity proteinu S můžeme rozdělit na 3 základní formy. Typ I je charakterizován snížením celkového proteinu S nejčastěji v důsledku snížené syntézy, typ II je charakteristický sníženým aktivity proteinu S a III typ se sníženou hladinou volného proteinu S s normální aktivitou celkového proteinu S.
Většinu deficitů představuje typ I nebo směs typů I a III. Do současné doby bylo popsáno velmi málo případů deficitů proteinu S typu II. Prevalence deficitů proteinu S představuje 1–2 % pacientů s hlubokou žilní trombózou a 6 % rodin s trombofilií. Z genetického hlediska bylo dosud popsáno 70 odlišných mutací v genu pro protein S.
Patofyziologie působení mutace F V Leiden
Z hlediska patofyziologie koagulačních reakcí hraje F V klíčovou roli jak v prokoagulační, tak antikoagulační kaskádě procesů. V aktivované formě slouží jako kofaktor F Xa v protrombinázovém komplexu, který takto katalyzuje přeměnu protrombinu na trombin. V neaktivované formě slouží F V jako kofaktor APC v regulaci aktivity F VIIIa. Tato dvojí role umožňuje F V při genetických a získaných defektech ovlivňovat manifestaci těchto poruch do hemoragické nebo trombotické formy [46,47]. Pro objasnění této manifestace je nutné přesně poznat prokoagulační a antikoagulační formu F V.
Gen kódující lidský faktor V má velikost přibližně 80 kb a je lokalizován na dlouhém rameni chromozomu I. Jak je patrné z obr. 2, gen sestává z 25 exonů a 24 intronů [46], které se transkribují do 6,8 kB mRNA, jež kóduje řetězec 2 224 aminokyselinových zbytků s 28 aminokyselinami signálního peptidu. Tento peptid je pak odstraněn při přechodu do endoplazmatického retikula.
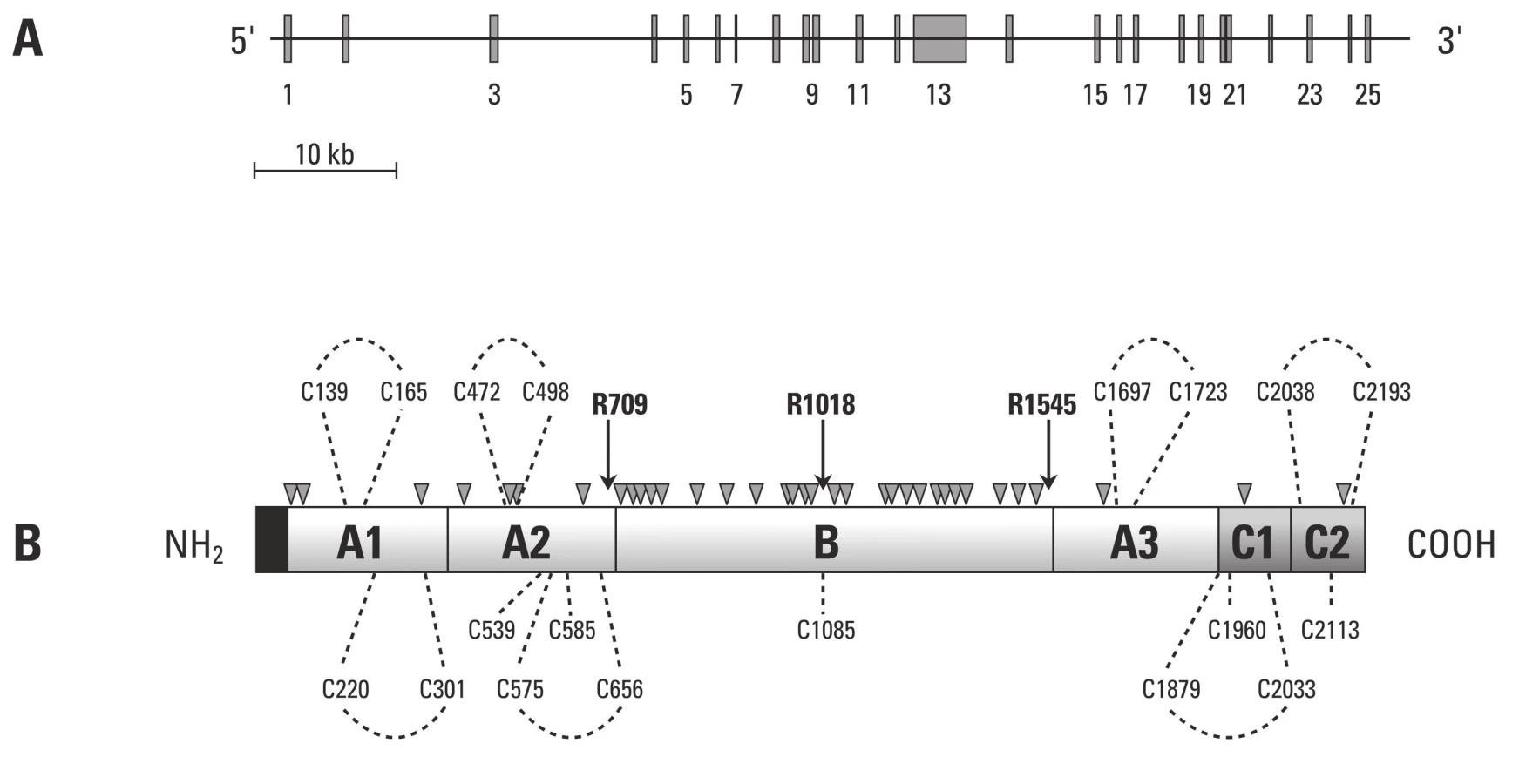
Takto vzniklé molekuly F V jsou přítomné v plazmě jako jednořetězcový glykoprotein v koncentraci 21 nM [48] (20–25 % F V je dále skladováno v a granulích trombocytů) [49]. F V sestává z A1–A2–B–A3–C1–C2 domén s totožnou organizační strukturou, jako má F VIII. A domény F V a F VIII vykazují značnou homologii a mají přibližně ve 40 % totožné sekvence.
Maturace F V obsahuje extenzivní post‑ribozomální modulaci. V poslední době se studie zaměřily zejména na N-glykosylaci. Bylo studováno 37 glykosylačních míst a jejich vliv na strukturu a funkci kofaktorů. Rozdílná glykosylace C2 domény vede ke tvorbě dvou rozdílných variant F V, nazvaných FV1 a FV2. Obě varianty cirkulují v plazmě v poměru 33 : 67 (FV1/FV2). Tyto dvě varianty disponují rozdílnými vlastnostmi (pro ‑ a antikoagulačními) a v modelových případech se také chovají rozdílně [50].
Prokoagulační funkce F Va
Neaktivovaný F V má malou prokoagulační aktivitu až do doby aktivace malým množstvím trombinu nebo F Xa prostřednictvím štěpení v místech Arg709, Arg1018 a Arg1545. Aktivací vzniklá molekula F Va je heterodimer [51].
Na rozdíl od F V, F Va zvyšuje F Xa aktivovanou přeměnu protrombinu v protrombinázovém komplexu. Lze předpokládat, že vyštěpená B-doména alostericky inhibuje vazbu F V v aktivním místě F Xa [52].
Mechanizmus, jakým F Va působí na F Xa, není plně znám. Na základě posledních prací lze předpokládat, že F Va zvyšuje vazebnou afinitu F Xa k fosfolipidům přibližně 100krát. Současně se podařilo prokázat, že F Va v protrombinázovém komplexu nemění vazebné místo F Xa, ale že zvyšuje afinitu protrombinázového komplexu k protrombinu a zajišťuje nárůst vazebných míst.
F Va kofaktorová aktivita je balancována prostřednictvím APC, který F Va proteolyticky štěpí v místech Arg306, Arg506 a Arg679 těžkého řetězce, přičemž nejslabší inaktivace je prostřednictvím štěpení v místě Arg679. Dle posledních poznatků jsou navrženy 2 patofyziologické modely štěpení F Va. První model předpokládá preferenční štěpení v místě Arg506 následované štěpením v místě Arg306. Alternativně může být F Va inaktivován přímo štěpením v místě Arg306. Štěpení v místě Arg506 též snižuje afinitu F Va k F Xa, zatímco štěpení v místě Arg306 způsobuje plnou inaktivaci F Va [53].
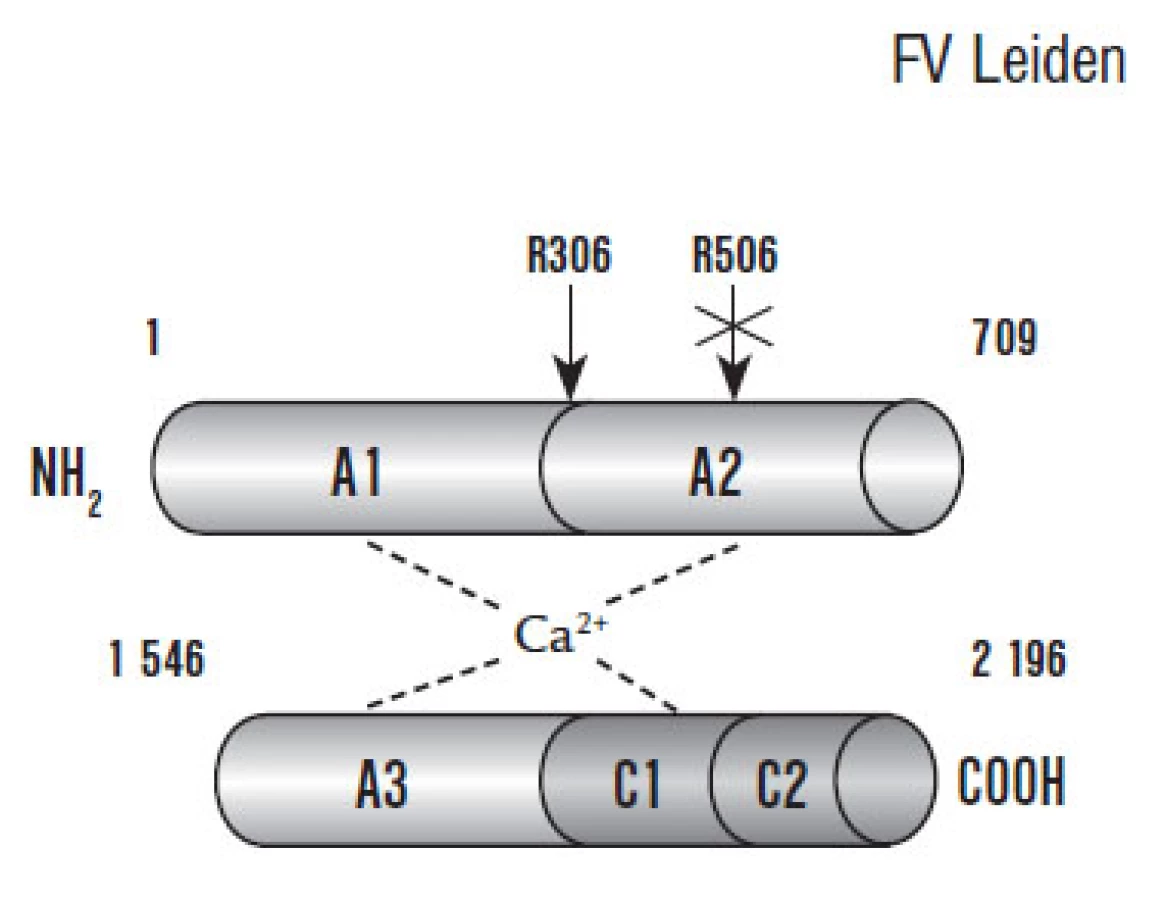
Antikoagulační funkce F Va
Kromě prokoagulační funkce disponuje F Va i antikoagulační funkcí, a to v případě aktivace APC. V tomto případě stimuluje proteolytické štěpení F VIII [54]. Tento model dokazují poslední experimentální práce, kdy byla přídavkem čištěného F V obnovena funkce APC jak u zdravých pacientů, tak u rodin s Leidenskou mutací. Experimenty využívaly měření generace trombinu.
Pokud tedy požadujeme po F V antikoagulační funkci, musí být štěpen v pozici Arg506. Na rozdíl od APC zprostředkovaného štěpení v místech Arg306 a Arg679, které nevykazuje antikoagulační aktivitu.
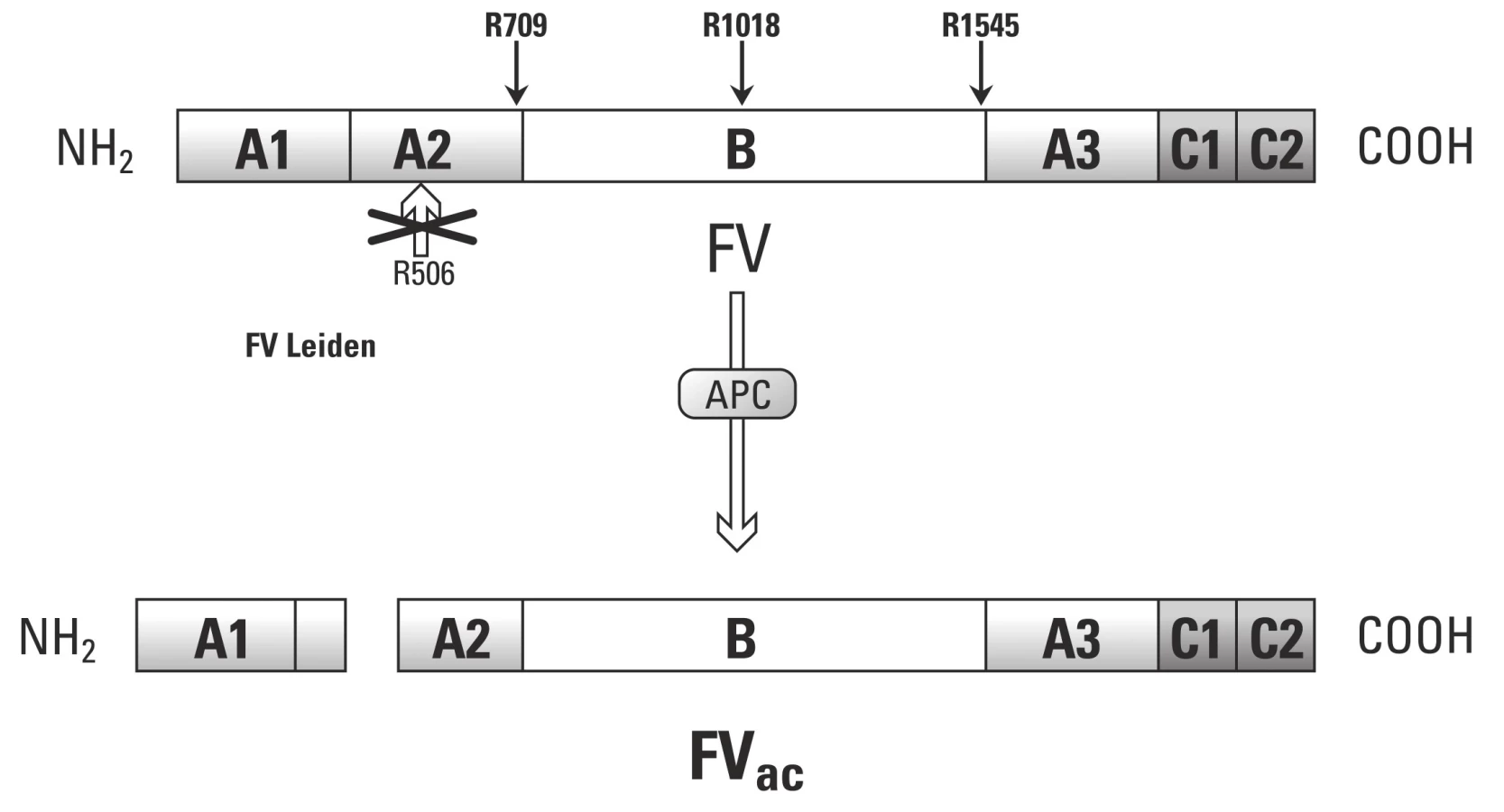
F V a trombofilie
Takto popsané cesty aktivace F V nám ukazují na balancování pro antikoagulační aktivity faktoru V a její význam pro udržení hemostatické rovnováhy.
Vliv mutace F V Leiden na prokoagulační a antikoagulační formu F V
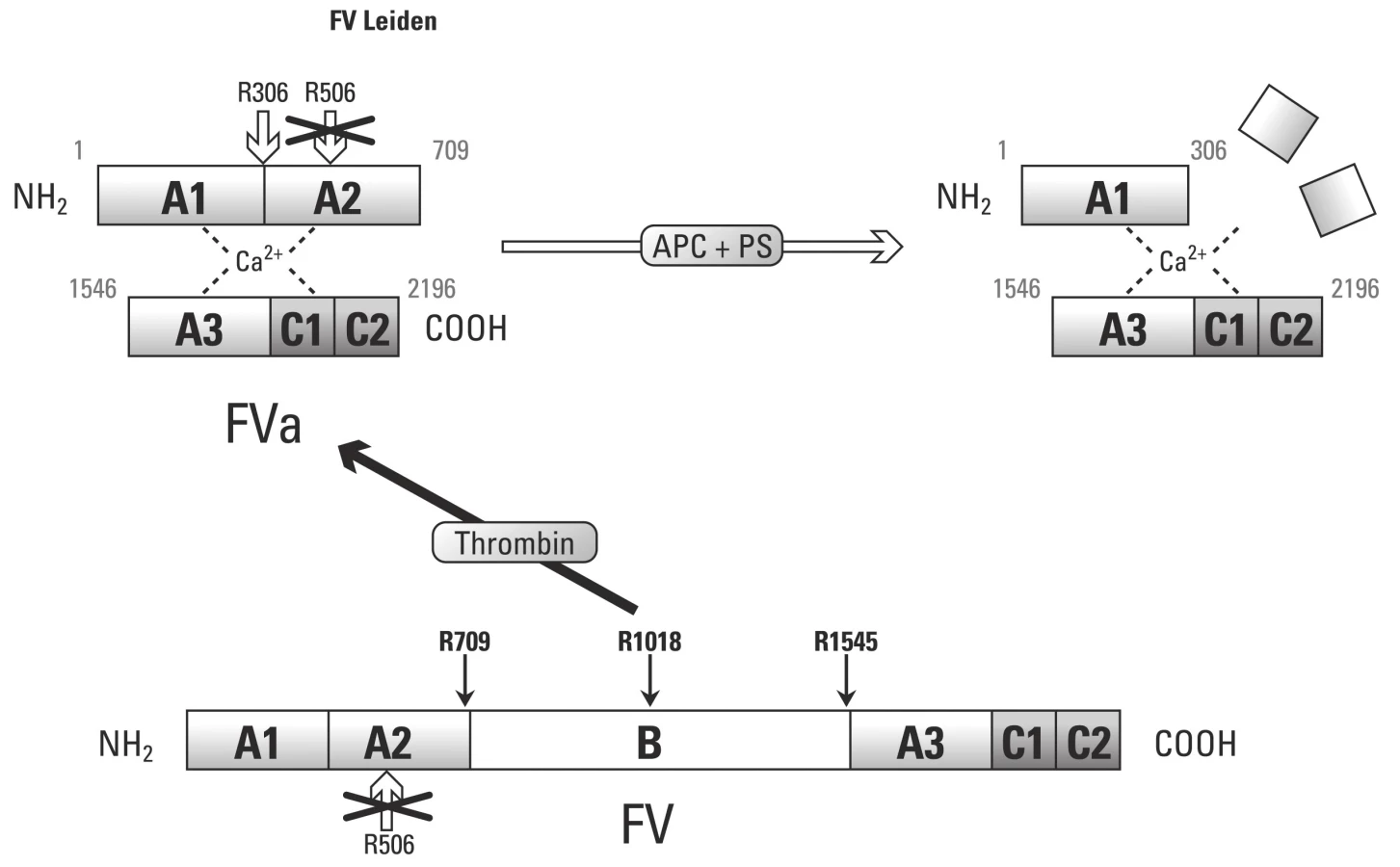
APC rezistence je in vitro popsaný fenomén, který je charakterizován slabou antikoagulační odpovědí na APC v plazmě. Takto snížená citlivost k APC vede k nedostatečné regulaci tvorby trombinu. Takto je APC rezistence asociována se zvýšeným rizikem vzniku trombózy. APC rezistence je až v 90 % případů spojována s mutací F V Leiden.
Mutace F V Leiden zásadně ovlivňuje pro ‑ a antikoagulační rovnováhu F Va. Leidenská mutace, která je příčinou zániku štěpného místa Arg506, nám způsobuje nedostatečné snížení prokoagulační aktivity F Va, což nám vysvětluje prokoagulační stavy u nositelů Leidenské mutace [55]. Nicméně poslední práce ukázaly, že PS a F Xa mohou kompletně eliminovat vlivy APC zprostředkované inkativace F Va a F VIIIa. Takto lze uspokojivě vysvětlit účinek nízkomolekulárních heparinů v prevenci tromboembolické choroby u nosičů mutace F V Leiden.
Objevení druhé cesty působení F Va, kdy prostřednictvím APC slouží jako kofaktor při proteolytickém štěpení F VIII, nám pomůže vysvětlit vliv Leidenské mutace. Jelikož F V s leidenskou mutací nedisponuje štěpným místem v pozici Arg506, nemůže vznikat antikoagulační forma F Vac a takto je vykazována slabá kofaktorová aktivita APC při proteolýze F VIII.
Avšak nejen genetické defekty mohou vysvětlovat patologické působení APC rezistence v některých případech. V poslední době byl popsán také výskyt inhibitorů proti F V, což také může zvyšovat expresi APC rezistence. K vývoji inhibitorů proti FV stačí malé množství bovinního trombinu, který je přidáván do derivátů fibrinogenu získaných z kryoprecipitátu [56].
Objasnění podrobné patogeneze působení F V Leiden vede k závěru, že pro vysvětlení klinické exprese trombofilního stavu je nejdůležitější ozřejmění interakce alespoň jedné geneticky podmíněné trombofilie s jedním a více získanými stavy.
F II 20210G/A
Trombin hraje klíčovou roli v regulaci koagulační kaskády, jelikož interaguje jako prokoagulační enzym a interaguje s aktivovanými faktory XII, XI, VIII, V a také trombinem aktivovaným inhibitorem fibrinolýzy (TAFI) a hlavně štěpí fibrinogen na fibrin.
Rozhodující role trombinu v regulaci hemostázy je ilustrována právě na případu protrombinové mutace 20210 G/A. Tato mutace je příčinou zvýšené koncentrace protrombinu v plazmě a takto predisponuje nosiče ke vzniku trombózy.
F II 20210A mutace je lokalizovaná v 3’netranslatované oblasti (3’UTR) do protrombinové mRNA, kde je většina těchto pozic pre‑mRNA endonukleolyticky štěpená a polyadenylovaná. Molekulární mechanizmus mutace byl dosud vysvětlován zlepšením funkce 3’ukončování, což zvyšuje rozpoznávání štěpného místa protrombinu a akumulaci 3’korektně ukončené mRNA v cytoplazmě a takto podpořenou syntézu proteinu [57,58].
Nicméně poslední práce zabývající se mutacemi v blízkosti místa 20210 ukázaly, že tento předpoklad ne zcela vysvětluje celou situaci. Danckwardtovi et al se totiž podařilo prokázat u 2 pacientů, kteří prodělali trombotickou komplikaci v mladém věku, mutaci F II 20221T. Při podrobném studiu modelů působení vlivu této mutace na štěpné místo v pozici 20210 autoři nejdříve prokázali, že mutace F II 20210G má největší pozitivní vliv na mRNA expresi (2,15krát násobek normálního stavu), dále mají pozitivní vliv i záměny F II 20210C a F II 20210T (1,4krát a 1,5krát). Z tohoto důvodu autoři provedli řadu experimentů k objasnění patofyziologické exprese těchto změn.
Po řadě experimentů se podařilo prokázat, že mutace F II 20221T je zajímavá, jelikož leží 11 párů bazí od místa 20210 v lemující oblasti genu (FS). Specifita a efektivita prodlužování 3’ konce mRNA je závislá na vazbě multiproteinového komplexu. Proto je důležité poznat funkci v těsné oblasti kolem štěpného místa. V kompetitivní studii byl porovnáván vliv mutací F II 20210C a F II 20210T s protrombogenní mutací F II 20210G a vliv mutace F II 20221T v oblasti blízké tomuto štěpnému místu.
Bylo prokázáno, že mutace F II 20221T (právě na modelech štěpného místa 20210 s mutací C a T) zvyšuje efektivitu prodlužování 3’ konce mRNA stejně jako mutace F II 20210A, což demonstruje, že mutace v FS oblasti může ovlivnit prodlužování 3’ konce F II.
Systematická analýza 3’ prodlužování F II odhalila neobvyklou strukturu pozměněné sekvence, která zajišťuje vyváženost tvorby F II. Autoři takto demonstrují, že poruchy F II jak ve štěpném místě, tak v blízké oblasti štěpného místa predisponují ke klinické manifestaci mutace. Takto ozřejměná funkce mutace F II 20221T představuje první mutaci spojenou s trombofilií v uridin bohaté oblasti, která není přepisována do mRNA. Tudíž pro její diagnostiku není možné použít RT PCR metod, ale musí být prováděna diagnostika na bázi genomické DNA.
Práce byla podpořena z grantu IGA MZ NR-9282/2007.
Doručeno do redakce: 5. 2. 2009
Mgr. Luděk Slavík, Ph.D.
www.fnol.cz
e‑mail: ludek.slavik@fnol.cz
Sources
1. Rees DC, Cox M, Clegg JB. World distribution of factor V Leiden. Lancet 1995; 346 : 1133–1134.
2. Rosendaal FR, Koster T, Vandenbroucke JP et al. High risk of thrombosis in patients homozygous for factor V Leiden (activated protein C resistance). Blood 1995; 85 : 1504–1508.
3. Ridker PM, Miletich JP, Hennekens CH et al. Ethnic distribution of factor V Leiden in 4047 men and women. JAMA 1997; 277 : 1305–1307.
4. Vorlova Z, Hrachovinova I, Matyskova M. Probability of thrombosis in patients with factor V Leiden. Thromb Haemost 1997; 78 : 309.
5. Chrobák L, Dulíček P. Resistance to activated protein C as pathogenic factor of venous thromboembolism. Hradec Králové: Acta Medica 1996; 39 : 55–62.
6. Dulíček P, Šafářová M, Chrobák L. Mutace FV Leiden – nejčastější rizikový faktor pro vznik žilní trombózy. Hematológia a transfuziológia 1997; 4 : 6–9.
7. Bertina RM, Koeleman RPC, Koster T et al. Mutation in blood coagulation factor V associated with resistance to activated protein C. Nature 1994; 369 : 64–67.
8. Dahlbäck B, Carlsson M, Svensson PJ. Familial thrombophilia due to a previously unrecognised mechanism characterized by poor anticoagulant response to activated protein C: prediction of a cofactor to activated protein C. Proc Natl Acad Sci USA 1993; 90 : 1004–1008.
9. Koster T, Rosendaal FR, De Ronde H et al. Venous thrombosis due to a poor anticoagulant response to activated protein C: Leiden Thrombophilia Study. Lancet 1993; 342 : 1503–1506.
10. Ridker PM, Hennekens CH, Lindpainter K et al. Mutation in the gene coding for coagulation factor V and the risk of myocardial infarction, stroke, and venous thrombosis in apparently healthy men. N Engl J Med 1995; 332 : 912–917.
11. Anderson FA, Wheeler HB, Goldberg RJ et al. A population based perspective of the hospital incidence and case-fatality rates of deep vein thrombosis and pulmonary embolism. The Worcester DVT study. Arch Intern Med 1991; 151 : 933–938.
12. Poort SR, Rosendaal FR, Reitsma PH et al. A commongenetic variation in the 3’-untranslated region of the prothrombin gene is associated with elevated plasma prothrombin levels and an increase in venous thrombosis. Blood 1996; 88 : 3698–3703.
13. Rosendaal FR, Doggen CJM, Zivelin A et al. Geographic distribution of the 20210 G to A prothrombin variant. Thromb Haemost 1998; 79 : 706–708.
14. Matýšková M, Buliková A, Šlechtová M et al. The prevalence of the prothrombin mutation 20210A in Brno. XV meeting of the ISH – African and European division. Final programme and abstracts, 124.
15. Souto JC, Coll I, Llobet D et al. The prothrombin 20210A allele is the most present genetic risk factor for venous thromboembolism in the Spanish population. Thromb Haemost 1998; 80 : 366–369.
16. Falcon CR, Cattaneo M, Panzeri D et al. High prevalence of hyperhomocyst(e)inemia in patients with juvenile venous thrombosis. Arterioscler Thromb 1994; 14 : 1080–1083.
17. Den Heijer M, Koster T, Blom HJ et al. Hyperhomocysteinemia as a risk factor for deep-vein thrombosis. N Engl J Med 1996; 334 : 759–762.
18. Simioni P, Prandoni P, Burlina A et al. Hyperhomocysteinemia and deep-vein thrombosis: a case-control study. Thromb Haemost 1996; 76 : 883–886.
19. Hyánek J, Hoffman R. Hyperhomocysteinémie a její diagnostický význam u cévních onemocnění. Praktická flebologie 1997; 2 : 61–71.
20. D’Angelo A, Selhub J. Homocysteine and thrombotic disease. Blood 1997; 90 : 1–11.
21. Kang SS, Wong PWK, Norusis M. Homocysteinemia due to folate deficiency. Metabolism 1987; 36 : 458–462.
22. Kang SS, Wong PWK, Norusis M. Homocysteinemia due to folate deficiency. Metabolism 1987; 36 : 458–462.
23. Rees MM, Rodgers GM. Homocysteinemia: association of a metabolic disorder with vascular disease and thrombosis. Thrombosis Research 1993; 71 : 337–359.
24. Ubbink JB, Vermaak WJ, Van der Merwe A et al. Vitamin B12, vitamin B6, and folate nutritional status in men with hyperhomocysteinemia. Am J Clin Nutr 1993; 57 : 47–53.
25. Mudd SH, Skovby F, Levy HL et al. The natural history of homocystinuria due to cystathionine beta‑synthase deficiency. Am J Hum Genet 1985; 37 : 1–31.
26. Engbertsen AMT, Franken DG, Boers GHJ et al. Thermolabile 5,10-methylenetetrahydrofolate reductase as a cause of mild hyperhomocysteinemia. Am J Hum Genet 1995; 56 : 142–150.
27. Frosst P, Blom HJ, Milos R et al. A candidate genetic risk factor for vascular disease: a common mutation in methylenetetrahydrofolate reductase. Nat Genet 1995; 10 : 111–113.
28. Egeberg O. Inherited antithrombin deficiency causing thrombophilia. Thromb Diath Haemorrh 1965; 13 : 516–530.
29. Thaler E, Lechner K. Antithrombin III deficiency and thromboembolism. Clin Haematol 1981; 10 : 369–390.
30. Demers C, Ginsberg JS, Hirsh J et al. Thrombosis in antithrombin III deficient persons: report of a large kindred and literature review. Ann Intern Med 1992; 116 : 754–761.
31. Hirsh J, Piovella F, Pini M. Congenital antithrombin III deficiency: incidence and clinical features. Am J Med 1989; 87 : 34–38.
32. Lane DA, Mannucci PM, Bauer KA et al. Inherited Thrombophilia: Part 1. Thromb Haemost 1996; 76 : 651–662.
33. Tait RC, Walker ID, Perry DJ et al. Prevalence of antithrombin deficiency in the healthy population. Br J Haematol 1994; 87 : 106–112.
34. Bock SC et al. Assignement of the human antithrombin III structural gene to chromosome 1q23–25. Cytogenet Cell Genet 1985; 39 : 67–69.
35. Lane DA et al. Antithrombin III: A database of mutations. Thrombos Haemostasis 1991; 66 : 657–661.
36. Griffin JH, Evatt B, Zimmerman TS et al. Deficiency of protein C in congenital thrombotic disease. J Clin Invest 1981; 68 : 1370–1373.
37. Allaart CF, Poort SR, Rosendaal FR et al. Increased risk of venous thrombosis in carriers of protein C deficiency defect. Lancet 1993; 341 : 134–138.
38. Broekmans AW, Veltkamp JJ, Bertina RM. Congenital protein C deficiency and venous thromboembolism: a study of three Dutchfamilies. N Engl J Med 1983; 309 : 340–344.
39. Allaart CF, Poort SR, Rosendaal FR et al. Increased risk of venous thrombosis in carriers of protein C deficiency defect. Lancet 1993; 341 : 134–138.
40. Koeleman BPC, Reitsma PH, Allaart CF et al. APC‑resistance as an additional risk factor for thrombosis in protein C deficient families. Blood 1994; 84 : 1031–1035.
41. Heijboer H, Brandjes DPM, Büller HR et al. Deficiencies of coagulation‑inhibiting and fibrinolytic proteins in outpatients with deep-vein thrombosis. N Engl J Med 1990; 323 : 1512–1516.
42. Mateo J, Oliver A, Borrell M et al. The EMET Group. Laboratory evaluation and clinical characteristics of 2,132 consecutive unselected patients with venous thromboembolism-results of the Spanish multicentric study on thrombophilia (EMET-study). Thromb Haemost 1997; 77 : 444–451.
43. Koster T, Rosendaal FR, Briët E et al. Protein C deficiency in a controlled series of unselected outpatients: an infrequent but clear risk factor for venous thrombosis (Leiden Thrombophilia Study). Blood 1995; 85 : 2756–2761.
44. Reitsma PH, Bernardi F, Doig RG et al. Protein C deficiency a database of mutations. 1995 Update. Thromb Haemost 1995; 73 : 876–889.
45. Walker ID et al. Guidelines on the Investigation and Management of Thrombophilia. J Clin Path 1990; 43 : 703–709.
46. Cripe LD, Moore KD, Kane WH. Structure of the gene for human coagulation factor V. Biochemistry 1992; 31 : 3777–3785.
47. Jenny RJ, Pittman DD, Toole JJ et al. Complete cDNA and derived amino acid sequence of human factor V. Proc Natl Acad Sci USA 1987; 84 : 4846–4850.
48. Tracy PB, Eide LL, Bowie EJ et al. Radioimmunoassay of factor V in human plasma and platelets. Blood 1982; 60 : 59–63.
49. Suehiro Y, Veljkovic DK, Fuller N et al. Endocytosis and storage of plasma factor V by human megakaryocytes. Thromb Haemost 2005; 94 : 585–592.
50. Hoekema L, Nicolaes GAF, Hemker HC et al. Human factor Va1 and factor Va2: properties in the procoagulant and anticoagulant pathways. Biochemistry 1997; 36 : 3331–3335.
51. Esmon CT. The subunit structure of thrombin‑activated factor V. Isolation of activated factor V, separation of subunits, and reconstitution of biological activity. J Biol Chem 1979; 254 : 964–973.
52. Husten EJ, Esmon CT, Johnson AE. The active site of blood coagulation factor Xa. Its distance from the phospholipid surface and its conformational sensitivity to components of the prothrombinase complex. J Biol Chem 1987; 262 : 12953–12961.
53. Yegneswaran S, Fernandez JA, Griffin JH et al. Factor Va increases the affinity of factor Xa for prothrombin: a binding study using a novel photoactivable thiol-specific fluorescent probe. Chem Biol 2002; 9 : 485–494.
54. Dahlbäck B, Carlsson M, Svensson PJ. Familial thrombophilia due to a previously unrecognized mechanism characterized by poor anticoagulant response to activated protein C: prediction of a cofactor to activated protein C. Proc Natl Acad Sci USA 1993; 90 : 1004–1008.
55. Bertina RM, Koeleman BP, Koster T et al. Mutation in blood coagulation factor V associated with resistance to activated protein C. Nature 1994; 369 : 64–67.
56. Leroy-Matheron C, Mallat A, Duvoux C et al. Inhibitor against coagulation factor V after liver transplantation. Transplantation 1999; 68 : 1054–1056.
57. Gehring NH, Frede U, Neu-Yilik G et al. Increased efficiency of mRNA 3 end formation: a new genetic mechanism contributing to hereditary thrombophilia. Nat Genet 2001; 28 : 389–392.
58. Ceelie H, Spaargaren-van Riel CC, Bertina RM et al. G20210A is a functional mutation in the prothrombin gene; effect on protein levels and 3-end formation. J Thromb Haemost 2004; 2 : 119–127.
59. Danckwardt S, Gehring N, Neu-Yilik G et al. The prothrombin 3 end formation signal reveals a unique architecture that is sensitive to thrombophilic gain‑of-function mutations. Blood 2004; 104 : 428–435.
Labels
Diabetology Endocrinology Internal medicineArticle was published in
Internal Medicine

2009 Issue 3
Most read in this issue
- Laboratorní postup při nálezu trombocytopenie
- Péče o pacienty s nemocí chladových protilátek, kryoglobulinemií a kryofibrinogenemií před kardiochirurgickými zákroky
- Prevence žilní tromboembolické nemoci v ortopedii a traumatologii
- Antitrombotická profylaxe v těhotenství