
Indikace k vyšetření rizikových faktorů žilní trombózy
Indicati on for examinati on of risk factors for veno us thrombosis
Pathogenesis of thrombosis includes many interacting factors, both inherited and environmental. We are able to detect a lot of thrombotic risk factors. Surveying these sho uld be indicated only in cases where knowledge of the defect can influence pati ent’s tre atment. Wide range screening of defects in which there is no definite proven relati on to clinical picture is not recommended.
Key words:
thrombophili a – veno us thrombosis – laboratory examinati on
Authors:
M. Matýšková; M. Šlechtová; J. Zavřelová; M. Penka
Authors‘ workplace:
Oddělení klinické hematologie FN Brno, pracoviště Bohunice, přednosta prof. MUDr. Miroslav Penka, CSc.
Published in:
Vnitř Lék 2009; 55(Suppl 1)(Supplementum 1): 41-47
Overview
Patogeneze trombózy zahrnuje mnoho vrozených a získaných faktorů, které se navzájem ovlivňují. V so učasnosti jsme schopni detekovat řadu faktorů, které zvyšují riziko trombózy. Jejich vyšetřování by mělo být cílené po uze na případy, kdy znalost defektu může ovlivnit postup léčby či profylaxi u daného jedince. Plošné vyšetřování a vyšetřování faktorů, u kterých není jednoznačně prokázána závislost na klinice, není doporučováno.
Klíčová slova:
trombofili e – žilní trombóza – laboratorní vyšetření
Úvod
Tromboembolická nemoc (TEN) je hlavní příčinou mortality a morbidity všude ve světě. Výskyt žilních trombóz (VT) je udáván 1 – 3 na 1 000 obyvatel za rok a je úzce závislý na věku a s věkem so uvisejících faktorech (v mládí těhotenství, HAK, později malignity, imobilizace a další) [1,2]. VT je třetí nejčastější kardiovaskulární chorobou po infarktu myokardu a mozkové příhodě.
Trombofili e je vrozený nebo získaný defekt hemostázy, který je s největší pravděpodobností příčinou zvýšeného sklonu k trombóze [3,4]. Tato široce po užívaná definice má však svá úskalí. Je řada osob, které mají tzv. „trombofilní defekt“, ale nikdy trombózu neměli. A naopak, jso u rodiny s těžkými trombotickými projevy, u kterých dosud žádný defekt nebyl nalezen. V posledních letech se podařilo zjistit definovatelno u eti ologii u více než 75 % jedinců s především VT hlubokých žil (DVT), z toho asi u 30 – 50 % byl nalezen vrozený defekt v systému hemostázy. Většina vrozených defektů je a utozomálně dominantně dědičná.
Někdy je trombofili e vymezována nálezem vrozeného rizikového stavu a jinak se mluví o hyperkoagulačním stavu. Naopak někdy je za hyperkoagulační stav označován laboratorní nález defektu a o trombofilii se mluví, pokud dojde ke klinické manifestaci TEN. Zdá se, že je vhodnější po užívat klinicko u definici trombofilie (tab. 1), která nám současně i indikuje osoby, které jsou vhodné k vyšetření trombofilních defektů.

Rizikové faktory
Trombóza je vždy komplexní multifaktori ální geneze, nikdy není vyvolána po uze jedním faktorem, tj. ani vrozeným defektem. Trombóza jako symptom je závažno u komplikací mnoha vrozených a získaných stavů. Ukázalo se, že je řada faktorů, které se u jedinců s trombózo u vyskytují častěji, a můžeme je nazývat rizikové. Stále platí Virchowova hypotéza vyslovená před více než 150 lety: příčino u trombózy moho u být změny cévní stěny, krevního toku a chemických látek v krvi přítomných. Dosud veškeré odchylky či stavy, které moho u k trombóze vést, můžeme zařadit do těchto tří kategori í. Rizikové faktory trombózy většino u dělíme na obecné (běžné), klinické (spíše lze mluvit o chorobných stavech, při kterých se trombózy častěji vyskytují (tab. 2), vrozené či získané poruchy plazmatických proteinů [5].

Mezi „obecné“ rizikové faktory trombózy bývá řazen věk nad 45 let (v rozmezí 40 – 50 let), obezita, operace, úraz, imobilizace, cestování, těhotenství, šestinedělí, hormonální kontracepce i substituce [6]. Každý tento faktor zvyšuje riziko o 4 – 18 % [7].
Je známo, že jedinci s krevní skupino u A, B či AB (ne 0) mají 2krát vyšší riziko DVT oproti osobám s krevní skupino u 0. Tento vliv je pozorován i u arteri álních trombóz. Tento fenomén popsali poprvé Jick et al v roce 1969 [8], když si všimli, že ve skupině jedinců s trombózo u je krevní skupina 0 zasto upena méně, než je v dané populaci. Toto pozorování bylo opakovaně potvrzeno [9 – 11].
V so učasnosti je uznáváno vedle velmi vzácné dysfibrinogenemi e 5 genetických rizikových faktorů pro VT (mluvíme o vrozené trombofilii) – defekt antitrombinu, proteinu C či S (PC, PS), mutace F V Leiden (FVL) a PT G20210A (tab. 3). Přestože asoci ace těchto defektů s VT je jasně prokázána, setkáváme se u jednotlivých defektů s velko u heterogenito u projevů [12]. Toto i nadále vyvolává otázku, zda jso u tyto defekty příčino u, následkem, či ko incidencí VT. Mezi rizikové faktory smíšené (tj. kombinace vrozené dispozice a získaných faktorů) jso u řazeny zvýšené hladiny ko agulačních faktorů, především F VIII [13], fibrinogenu a hyperhomocysteinemi e [14]. Ze získaných rizikových faktorů je nejčastější přítomnost antifosfolipidových protilátek, které však pro svo u obsáhlost nejso u předmětem tohoto sdělení.
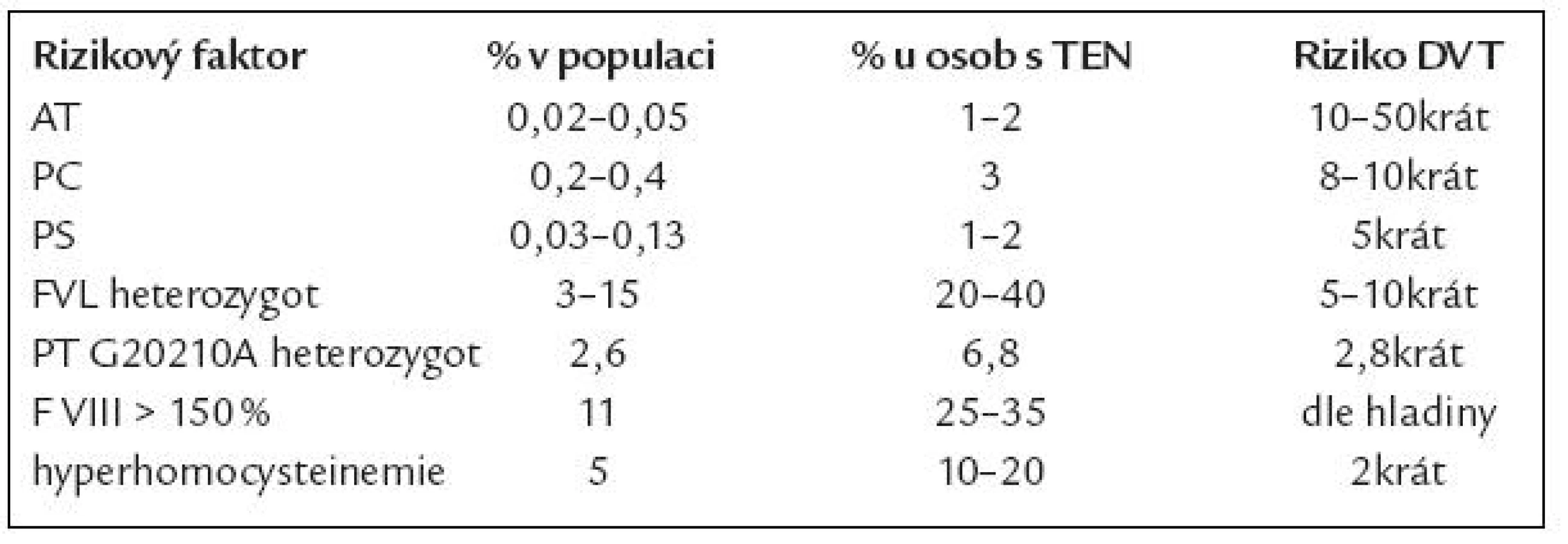
V evropské populaci je nejčastějším trombofilním defektem Leidenská mutace F V (FVL), v naší zdravé populaci je prevalence asi 7 % [15,16]. Tzv. rezistence na aktivovaný protein C (APCR) byla popsána skupino u prof. Dahlbäcka v roce 1993 [17] a prakticky vzápětí byla identifikovaná příčina většiny APCR (asi 90 %) – mutace faktoru Vnazvaná F V Leiden [18].
Klinicky se defekt projevuje většino u distální žilní trombózo u (hlavně hluboko u, ale je popsána i povrchová). Riziko trombózy je zvýšené u heterozygotní formy 5 – 10krát, u homozygotů 50 – 100krát. So učasně je však nižší riziko emboli e plicní. Průměrný věk první manifestace je u heterozygotů 44 let, homozygotů potom 31 let [19 – 22]. V našem so uboru nositelů FVL v homozygotní formě máme klinická data od 109 žen (83,21 % ze 131). Dle retrospektivní analýzy anamnézy se trombóza vyskytla u 76 žen (69,72 % z této podskupiny). Průměrný věk první trombózy byl 34,26 let (ve skupině mužů 42,60 let), ale rozsah věku byl od 9 do 82 let [23].
V graviditě a šestinedělí riziko TEN významně sto upá [24 – 26]. U nositelek tohoto polymorfizmu je popsáno i zvýšené riziko potratů v II. trimestru gravidity [27] a patologi í v graviditě. Naopak se zdá, že v I. trimestru je tento polymorfizmus výhodo u [28,29].
Stran zvýšeného rizika recidivy trombózy u heterozygotních jedinců se stále vedo u debaty: některé práce vyšší riziko nepotvrzují [30], jiné zvýšené riziko nalézají [31], ale nepovažují je za důvod delšího podávání antiko agulační léčby [32]. Podobně jso u nejednoznačné údaje i u PT G20210A [31 – 33].
FVL je „balanční“ polymorfizmus s výhodami pro heterozygoty s to uto mutací, jako např. prokazatelně signifikantně nižší krevní ztráty po porodu [34], snižuje riziko intrakrani álního krvácení. Předpokládá se, že mutace se objevila před 21 000 – 34 000 lety, a so učasný výskyt odpovídá ge ografické distribuci [35] podobně jako polymorfizmus PT G20210A [36].
Protrombin G20210A je druhým nejčastějším defektem [37], riziko trombózy se zvyšuje u heterozygotů asi 3krát (3 – 11krát). Klinicky se opět projevuje hlavně VT, zvyšuje i riziko PE a komplikací v graviditě [25]. Není rizikovým faktorem u mužů pro infarkt myokardu ani mozkovo u příhodu [26,38 – 40].
Prevalence vrozeného defektu antitrombinu v populaci je udávána mezi 0,05 a 0,2 na 1 000 obyvatel, paci enti jso u většino u heterozygoti s aktivito u AT 40 – 50 %. Defekt antitrombinu byl popsán jako první trombofilní defekt v roce 1965 Egebergem [41]. V bývalém Československu popsal první rodinu s defektem antitombinu doc. Hule v roce 1977 [42].
Klinická manifestace defektu antitrombinu závisí na typu defektu [43]. Nejtěžší postižení bývá u typu I, což je klasický deficit (snížení aktivity i antigenu). Riziko trombózy je nižší u typu II, u kterého jso u přítomny vari antní formy AT. Tento funkční defekt postihuje buď re akční místo, místo vazby heparinu (po uze u tohoto defektu je popsán výskyt homozygotů), nebo jde o plei otropní defekt. Porucha je primárně sdružena s žilními trombózami, zvyšuje i riziko PE. Jso u známy i trombózy v atypických oblastech. První manifestace se objevují okolo puberty, u žen často v těhotenství a šestinedělí. Asi u 67 % postižených se defekt klinicky projeví mezi 10 a 35 lety věku. Vzhledem ke konzumpci AT při trombotických příhodách je nutné stanovení zopakovat mimo akutní stav [39,43 – 46].
Nedostatek proteinu C nebo S zvyšuje riziko trombózy asi 3 – 10krát. Klinicky se může projevit již v dětství, setkáváme se s žilními trombózami hlavně dolních končetin, ale moho u se objevit i trombózy v netypických místech (horní končetiny), včetně žil povrchových, ojediněle jso u popsány i tepenné trombózy. I riziko trombózy u proteinu S závisí na typu defektu a opět není rizikový faktor pro arteri ální trombózy. U homozygotních jedinců se v raném dětství může manifestovat jako „purpura fulminans“, která končí často smrtelně. Za příčinu se považují mikrotrombózy kapilár, které poškodí cévní stěnu s následným prokrvácením. U dospělých se moho u objevit kožní nekrózy po kumarinech. I zde je za příčinu považován pokles PC, který má kratší poločas než většina ostatních faktorů na K vitaminu závislých. Opět v důsledku poklesu PC se tvoří mikrotrombózy cév kůže s následno u nekrózo u v těchto místech.
Prevalence mutace PC v populaci je udávána 2/ 1 000 obyvatel, prevalence defektu PS 0,3 – 1,3/ 1 000 obyvatel [47 – 49].
Zvýšené riziko trombózy představuje i zvýšená hladina ko agulačních faktorů VIII, IX, X, XI. F VIII zvyšuje i riziko recidivy TEN po první trombotické příhodě. F IX nad 129 % zvyšuje 2 – 3krát riziko trombózy, riziko je větší u žen než u mužů [13,50 – 52]. Byl popsán i rodinný výskyt kombinace zvýšené hladi ny F VIII, IX a XI u 6 rodin s významným zvýšením rizika trombózy [53]. Klinický dopad snížení F XII zůstává stále předmětem diskuze (první paci ent, u kterého byl homozygotní defekt F XII popsán, zemřel na plicní embolii).
Za nejzávažnější rizikové faktory trombózy jso u považovány defekty v homozygotní formě (např. FVL), dále potom kombinace defektů [54]. Ale např. i u závažné kombinace, jako je nedostatek antitrombinu a FVL, závisí riziko trombózy na typu defektu a projevy jso u v rodinách různé [55]. Podobně přítomnost vrozené či smíšené trombofili e a získaného faktoru může riziko trombózy neúměrně navýšit. Jedná se např. o těhotné ženy (včetně šestinedělí) či užívání hormonální léčby [56 – 59].
V posledních letech se významně zvyšuje počet prací, které popisují nálezy zejména vrozených faktorů, které by mohly být zařazeny mezi rizikové – polymorfizmy F XIII, polymorfizmy destičkových glykoproteinů, fibrinogenu, mutace heparin kofaktor II, trombomodulinu, TFPI a řada dalších. Velká část z nich je i běžně dostupná k vyšetření. Mezi jednoznačně prokázané rizikové faktory však zatím zařazeny nejso u. Otázko u tak zůstává, zda má význam je vyšetřovat, a pokud ano, tak které a za jakých situ ací.
Indikace vyšetření trombofilie
Problematice vyšetřování vrozených trombofilních dispozic je v literatuře věnována řada publikací, část z nich se přiklání k indikaci vyšetření, pokud jsou splněna klinická kritéri a, zčásti je vyšetření velmi zpochybňováno [60 – 64].
Kdy je vyšetření vrozené trombofili e indikované? Kdo a kdy by měl být vyšetřován? Co vyšetřením nemocný získá? Bude lepší prevence v zátěžových situ acích? Bude déle podávána odpovídající antiko agulační léčba? Pomůže nám vyšetření v případě zvažování indikace hormonální antikoncepce či substituce? Při zvažování těchto otázek je vždy nejdůležitější a na prvním místě řádně odebraná rodinná i osobní anamnéza! Pokud bychom se měli řídit po uze nalezeným laboratorním defektem, můžeme přehlédno ut těch 50 % nemocných, u kterých se dosud žádný defekt neprokázal.
Ovlivní detekce léčebný postup?
Detekce defektu v žádném případě ne ovlivní postup při léčbě akutní VTE příhody [65,66]. Rezistence na heparin u defektů AT je vzácná a můžeme se s ní setkat i u získaných defektů. Pokud řádně překrýváme heparinem zahájení warfarinizace, můžeme se vyhno ut i kumariny indukované nekróze.
Má přítomnost defektu význam pro určení délky (event. síly) trvání antikoagulace? Ani zde se přístup u nositelů běžných rizikových faktorů nebude lišit od ostatních. Délka se vždy bude řídit tíží příhody, tj. klinickým nálezem, souběžnými chorobami či přídatnými rizikovými faktory (budeme jinak postupovat při trombóze po operaci a jinak u tzv. „idiopatické“).
V poslední době se v odborném tisku také opakovaně probírá délka antikoagulační léčby. Většinou se po ukazuje na zvyšující se riziko krvácivých komplikací u osob dlouhodobě antikoagulovaných, které významně převažuje nad rizikem recidivy trombózy [66]. Trvání léčby vždy primárně závisí na riziku recidivy. Delší profylaxe se doporučuje pouze, pokud první příhoda byla život ohrožující, idiopatická nebo je přítomen vysoce rizikový vrozený defekt (včetně kombinací). Opět je ale nutné mít na paměti, že to, že nebyl nalezen defekt, neznamená, že daná osoba nemá zvýšené riziko recidivy TEN. Riziko recidivy trombózy je vyšší u osob s laboratorní trombofilií v případě vysazení warfarinizace po 3 měsících. V případě vysazení po jednom roce nehraje vrozená trombofilie žádnou roli v případě recidivy [67]. Při zvažování délky léčby musíme vždy zohlednit klinický stav (souběžné choroby) včetně cévního vyšetření, komplikace léčby, anamnézu, rizikovost situace, za které TE vznikla (idiopatická?!), aktuální laboratorní výsledky (stanovení D‑dimerů, ProC® Global a další) [66,68,69], nalezené vrozené rizikové faktory a v neposlední řadě spolupráci nemocného. Po jedné trombotické příhodě, byť u nemocného s nalezeným vrozeným rizikovým faktorem, většinou není indikována doživotní antikoagulační léčba. A už vůbec není důvod pro nasazení antikoagulační léčby u bezpříznakových osob, když je diagnostikován vrozený rizikový faktor [70]. Sám vrozený defekt nezvyšuje riziko recidivy trombózy při řádně vedené warfarinizaci [71].
Může nález trombofilního faktoru ovlivnit tromboprofylaxe v zátěžové situaci?
Ano, v tomto případě může nositel defektu z vyšetření jednoznačně profitovat, pokud bude mladší 40 – 45 let. Víme ale, že obecné rizikové faktory mají při stratifikaci rizika minimálně stejno u váhu jako vrozené. Vždy musíme zhodnotit rizikové situace (operace, úrazy, imobilizace, těhotenství, dlouhé lety nad 8 hod). Profylaxe se vždy na prvním místě řídí anamnézou a stavem, pro který je indikována. Stanovení defektů může ovlivnit tromboprofylaxi pouze asi do 40 let, potom se profylaxe již běžně podává. Toto ale neplatí pro zdravé děti. Prospektivní studie 143 dětí (věk 1 – 14 let) z trombofilních rodin, z nichž 81 (56,6 %) bylo nositeli defektu, ukázala, že přestože se vyskytlo celkem 31 rizikových situ ací (20 ve druhé skupině), nebyla pozorována žádná trombóza v období sledovaných let (průměr 5, rozmezí 1 – 8), tj. 395, resp. 296 sledovaných let. Screening trombofilie není u zdravých jedinců do 15 let zdůvodnitelný [72], je ale vhodný v rizikových situacích [73].
Kdo by měl být vyšetřován?
V podstatě je možné říct, že vyšetření je indikováno, pokud jso u přítomny alespoň 2 znaky definující klinickou trombofilii (tab. 1). Vyšetření je vhodné u osoby do asi 45 let (většino u udávané rozmezí 40 – 50), hlavně pokud se jedná o tzv. idiopaticko u trombózu nebo o recidivující stavy. Podobně u jedinců jak s arteriální, tak venózní trombózou bez známé jiné choroby v anamnéze, nebo u ženy s trombózou či opakovanými potraty, zejména pokud budo u mít i pozitivní rodinnou anamnézu [74]. Studie prokázaly, že není např. ekonomické vyšetřování F V Leiden u všech žen před nasazením hormonální antikoncepce či v graviditě [75,76]. Vždy je nutné mít na paměti, že sama hormonální antikoncepce je nejčastější rizikový faktor VTE u žen ve fertilním věku [77], podobně jako významně stoupá riziko trombózy v těhotenství a hlavně šestinedělí [78]. Vyšetření je však zdůvodnitelné v případě pozitivní rodinné či osobní anamnézy [79].
Není na místě vyšetřovat bezpříznakové členy rodin především vyššího věku. Pokud už vyšetřovat, tak např. u mladších rodinných příslušníků by vyšetření mělo být vždy cílené a ne celé spektrum trombofilních faktorů, zejména u bezpříznakových jedinců.
Je nutné si také ujasnit, zda vyšetřujeme rodinu, kde jsme nalezli vysoce rizikové faktory (např. FVL v homozygotní formě, defekt antitrombinu I. typu nebo kombinace vrozených faktorů) nebo rodinu s nálezem lehčího rizikového faktoru, jako je např. heterozygotní forma Leidenské mutace F V nebo vari antní protrombin G20210A, kde je riziko tak malé, že vyšetření příbuzných ne odůvodní [38]. Už sama pozitivní rodinná anamnéza by měla kliniky varovat a těmto osobám by měla být věnována v případě nutnosti tromboprofylaxe vyšší pozornost [60].
Jako zbytečné se ukazuje vyšetření vrozených rizikových faktorů u trombóz, které vznikly při nádorovém onemocnění, a uto imunitních onemocněních apod. nebo ve vysokém věku.
Co vyšetřovat?
Při zvažování indikace vyšetření bychom také měli, dle typu trombózy, rozlišovat, které faktory vyšetřit. Tj. např. nedělat celé spektrum rizikových faktorů pro žilní trombózy u jedinců po infarktu myokardu. Studie publikovaná v roce 2003, která testovala 9 protrombotických polymorfizmů u 1 210 osob po prvním MI ve věku < 45 let, nenalezla asociaci s žádným testovaným faktorem. Jednalo se o polymorfizmus G - 455A β-fibrinogenu, F V Leiden, PT20210A, faktor VII G10976A, faktor XIII G185T, PAI 4G/ 5G, glykoproteid GP Ia C807T a GP IIIa C1565T, MTHFR C677T [80].
Práce z poslední doby ukazují na vhodnost identifikace mutace JAK2 (Janus kinase 2) V617F v případě výskytu trombóz v oblasti splanchniku či mozkových. Tato mutace se vyskytuje u chronické myeloproliferativní nemoci a může na ni upozornit i v časném stadiu [81,82].
V neposlední řadě si musíme být vědomi limitací vyšetření a znát řádno u interpretaci výsledků [6]. S výjimkou molekulárně biologických vyšetření jsou testy ovlivněny akutním stavem (např. pokles antitrombinu, zvýšení F VIII, zkrácení PCG), warfarinizací (K - dependentní faktory), těhotenstvím (významný pokles proteinu S).
Závěr
Patogeneze trombózy zahrnuje řadu vrozených a získaných faktorů. Incidence trombózy i u nositelů známých vrozených defektů je velmi variabilní a řada z nich nikdy trombózu neměla a mít nebude. Jiní naopak mají těžké trombotické příhody již v mladém věku. Potvrzuje to multifaktori ální patogenezi trombózy a vzájemného ovlivňování řady získaných a vrozených faktorů (interakce genové, geny a prostředí, ovlivnění věkem). Vyšetření vrozených či smíšených rizikových faktorů by mělo být prováděno pouze u mladších jedinců, kteří splňují minimálně dvě kritéria klinické trombofilie. Neměli bychom zapomínat, že už sama anamnéza trombotických příhod je varováním a indikací k důslednější prevenci v zátěžových situacích i tehdy, kdy o žádném vrozeném defektu nevíme [60]. V případě vyšetření musíme mít zcela jasno v tom, jak případná přítomnost trombofilních defektů ovlivní léčbu či profylaxi u nositelů [83]. Neměli bychom provádět vyšetření pro vyšetření, kdy závěrem je pouze sdělení, že polymorfizmus/ mutace je či není přítomen/ na, a v podstatě není jasné, proč vyšetření bylo děláno.
V každém případě je nezbytné poskytno ut pacientům řádnou informaci o typu defektu, o rizicích, o možné prevenci/ profylaxi. I u bezpříznakových „pacientů“ (jedná se o zdravé jedince) bychom si vždy měli najít čas a nálezy řádně vysvětlit a informovat je o daném defektu, neměli bychom je bezpředmětně vystrašit.
MUDr. Miloslava Matýšková, CSc.
www.fnbrno.cz
e-mail: mmatys@fnbrno.cz
Doručeno do redakce: 16. 4. 2009
Sources
1. Naess IA, Christi ansen SC, Romundstad P. Incidence and mortality of veno us thrombosis: a populati on‑based study. J Thromb Haemost 2007; 5 : 692 – 699.
2. Rosendaal FR. Risk factors for veno us thrombotic dise ase. Thromb Haemost 1999; 82 : 610 – 619.
3. Lane DA, Mannucci PM, Ba uer KA et al. Inherited thrombophili a: Part 1. Thromb Haemost 1996; 76 : 651 – 662.
4. Walker ID, Gre aves M, Preston FE, on behalf of the Haemostasis and Thrombosis Task Force British Committee for Standards in Hematology. Investigati on and management of heritable thrombophili a. Br J Haematol 2001; 114 : 512 – 528.
5. Rosendaal FR. Veno us thrombosis: the role of genes, environment, and behavi or. Hematology Am Soc Hematol Educ Program 2005; 47 : 1 – 12.
6. Dulíček P. Trombofilní stavy. Vnitř Lék 2005; 51 : 819 – 824.
7. Gre aves M, Baglin T. Laboratory testing for heritable thrombophili a: impact on clinical management of thrombotic dise ase annotati on. Br J Haematol 2000; 109 : 699 – 703.
8. Jick H, Slone D, Westerholm B et al. Veno us thromboembolic dise ase and ABO blo od type. A co operative study. Lancet 1969; 1 : 539 – 542.
9. Wa utrecht JC, Galle C, Motte S et al. The role of ABO Blo od gro ups in the incidence of deep vein thrombosis. Thromb Haemost 1998; 79 : 688 – 689.
10. González Ordóñez AJ, Medina Rodriguez JM, Martín L et al. The 0 blo od gro up protects against veno us thromboembolism in individu als with the factor V Leiden but not the prothrombin (factor II G20210A) mutati on. Blo od Co agul Fibrinolysis 1999; 10 : 303 – 307.
11. Matýšková M, Zavřelová J, Pejchalová A et al. Krevní skupiny AB0/ H a faktor V Leiden. Čas lék čes 2002; 141 : 146 – 151.
12. Martinelli I, Mannucci PM, De Stefano V et al. Different risks of thrombosis in fo ur co agulati on defects associ ated with inherited thrombophili a: A study of 150 famili es. Blo od 1998; 92 : 2353 – 2358.
13. Kraaijenhagen RA, in’t Anker PS, Ko opman MM et al. High plasma concentrati on of factor VIIIc is a major risk factor for veno us thromboembolism. Thromb Haemost 2000; 83 : 5 – 9.
14. Verstraete M. Hyperhomocysteinemi a as a risk factor for arteri al and veno us thrombosis. Hämostase ol 1998; 18: S14 – S18.
15. Vorlová Z, Hrachovinová I, Matýšková M. APC - Resistance in Pati ents with a Thromboembolism. In: Hermann FH (ed). Molekulargenetik hereditärer Hämostasedefekte. Greifswalder Hämophili e - Tagung: Pabst Sci ence Publishers 1996 : 113 – 116.
16. Matýšková M, Paseka J, Vorlová Z et al. Prevalence of factor V Leiden mutati on in he althy women. In: Scharrer I, Schramm W (eds). 29. Hämophili e – Symposi on Hamburg 1998.Berlin Heidelberg: Springer 1999 : 309 – 311.
17. Dahlbäck B, Carlsson M, Svensson PJ. Famili al thrombophili a due to a previ o usly unrecognized mechanism characterized by po or antico agulant response to activated protein C: Predicti on of a cofactor to activated protein C. Proc Natl Acad Sci USA 1993; 90 : 1004 – 1008.
18. Bertina RM, Koeleman BP, Koster T et al. Mutati on in blo od co agulati on factor V associ ated with resistance to activated protein C. Nature 1994; 369 : 64 – 67.
19. Bo uname a ux H. Factor V Leiden paradox: risk of deep - vein thrombosis but not of pulmonary embolism. Lancet 2000; 356 : 182 – 183.
20. Rosendaal FR, Koster T, Vandenbro ucke JP et al. High risk of thrombosis in pati ents homozygo us for factor V Leiden (activated protein C resistance). Blo od 1995; 85 : 1504 – 1508.
21. Matýšková M, Vorlová Z, Hrachovinová I et al. Klinické nálezy u jedinců s Leidensko u mutací faktoru V. Vnitř Lék 1997; 43 : 298 – 301.
22. Dulícek P, Malý J, Safárová M. Risk of thrombosis in pati ents homozygo us and heterozygo us for factor V Leiden in the East Bohemi an regi on. Clin Appl Thromb Hemost 2000; 6 : 87 – 89.
23. Matýšková M, Šlechtová M, Čech Z et al. Factor V Leiden homozygosity in women. Thromb Res 2009; 123 (Suppl 2): S155. Abstract P54.
24. Gerhardt A, Scharf RE, Zotz RB. Effect of hemostatic risk factors on the individu al probability of thrombosis during pregnancy and the puerperi um. Thromb Haemost 2003; 90 : 77 – 85.
25. Dulíček P, Malý J, Kalo usek I et al. Výskyt venózního tromboembolizmu u žen s Leidensko u mutací v so uvislosti s gravidito u a šestinedělím. Čes Gynekol 2005; 70 : 133 – 138.
26. Pomp ER, Lenselink AM, Rosendaal FR et al. Pregnancy, the postpartum peri od and prothrombotic defects: risk of veno us thrombosis in the MEGA study. J Thromb Haemost 2008; 6 : 632 – 637.
27. Kist WJ, Janssen NG, Kalk JJ et al. Thrombophili as and adverse pregnancy o utcome – A confo und problem! Thromb Haemost 2008; 99 : 77 – 85.
28. van Dunné FM, Doggen CJ, Heemskerk M. Factor V Leiden mutati on in relati on to fecundity and miscarri age in women with veno us thrombosis. Hum Reprod 2005; 20 : 802 – 806.
29. Simur A, Özdemir S, Acar H et al. Repe ated in vitro fertilizati on failure and its relati on with thrombophili a. Gynecol Obstet Invest 2009; 67 : 109 – 112.
30. Eichinger S, Weltermann A, Mannhalter C et al. The risk of recurrent veno us thromboembolism in heterozygo us carri ers of factor V Leiden and a first spontane o us veno us thromboembolism. Arch Intern Med 2002; 162 : 2357 – 2360.
31. Marchi ori A, Mosena L, Prins MH et al. The risk of recurrent veno us thromboembolism among heterozygo us carri ers of factor V Leiden or prothrombin G20210A mutati on. A systematic revi ew of prospective studi es. Haematologica 2007; 47 : 1107 – 1114.
32. Ho WK, Hankey GJ, Quinlan DJ et al. Risk of recurrent veno us thromboembolism in pati ents with common thrombophili a: a systematic revi ew. Arch Intern Med 2006; 166 : 729 – 736.
33. De Stefano V, Martinelli I, Mannucci PM et al. The risk of recurrent veno us thromboembolism among heterozygo us carri ers of the G20210A prothrombin gene mutati on. Br J Hame atol 2001; 113 : 630 – 635.
34. Lindqvist PG, Svensson PJ, Dahlbäck B et al. Factor V Q506 mutati on (activated protein C resistance) associ ated with reduced intrapartum blo od loss - a possible evoluti onary selecti on mechanism. Thromb Haemost 1998; 79 : 69 – 73.
35. Zivelin A, Griffin JH, Xu X et al. A single genetic origin for a common ca ucasi an risk factor for veno us thrombosis. Blo od 1997; 89 : 397 – 402.
36. Zivelin A, Rosenberg N, Fai er S et al. A single genetic origin for the common prothrombotic G20210A polymorphism in the prothrombin gene. Blo od 1998; 92 : 1119 – 1124.
37. Hrachovinová I, Vorlová Z, Matýšková M et al. Thrombotic risk of the prothrombin gene G20210A mutati on and clinical fe atures of thrombophili a in 50 carri ers of the mutati on. Thromb Haemost 1999; Suppl. Abstracts XVII. Congress of the ISTH, Washington: 652, No 2060.
38. Coppens M, van de Poel MH, Bank I et al. A prospective cohort study on the absolute incidence of veno us thromboembolism and arteri al cardi ovascular dise ase in asymptomatic carri ers of the prothrombin 20210A mutati on. Blo od 2006; 108 : 2604 – 2607.
39. Rossi E, Za T, Ciminello A et al. The risk of symptomatic pulmonary embolism due to proximal deep veno us thrombosis differs in pati ents with different types of inherited thrombophili a. Thromb Haemost 2008; 99 : 1030 – 1034.
40. Ridker PM, Hennekens CH, Miletich JP. G20210A stati on in prothrombin gene and risk of myocardi al infarcti on, stroke, and veno us thrombosis in a large kohort of US men. Circulati on 1999; 99 : 999 – 1004.
41. Egeberg O. Inherited antithrombin defici ency ca using thrombophili a. Thromb Di ath Haemorrh 1965; 13 : 516 – 530.
42. Hule V. Vrozené rodinné snížení antitrombinu III. Vnitř Lék 1977; 23 : 887 – 892.
43. Hrachovinová I, Habart D, Salaj P et al. Molekulární podstata vrozeného defektu antitrombinu u deseti českých rodin. Čas Lék Čes 2000; 139 : 595 – 597.
44. van Boven HH, Lane DA. Antithrombin and its inherited defici ency state. Semin Hematol 1997; 34 : 188 – 204.
45. van Boven HH, Vandenbro ucke JP, Briët E et al. Gene - gene and gene - enviromental interacti ons determine risk of thrombosis in famili es with inherited antithrombin defici ency. Blo od 1999; 94 : 2590 – 2594.
46. Ra u JC, Be a uli e u LM, Huntington JA et al. Serpins in thrombosis, hemostasis and fibrinolysis. J Thromb Haemost 2007; 5 (Suppl 1): 102 – 115.
47. Koster T, Rosendaal FR, Briët E et al. Protein C defici ency in a controlled seri es of unselected o utpati ents: an infrequent but cle ar risk factor for veno us thrombosis (Leiden Thrombophili a Study). Blo od 1995; 85 : 2756 – 2761.
48. Dykes AC, Walker ID, McMahon AD et al. A study of Protein S antigen levels in 3788 he althy volunteers: influence of age, sex and hormone use, and estimate for prevalence of defici ency state. Br J Haematol 2001; 113 : 636 – 641.
49. Bro uwer JLP, Veeger NJGM, van der Schaaf W et al. Difference in absolute risk of veno us and arteri al thrombosis between famili al protein S defici ency type I and type III. Results from a family cohort study to assess the clinical impact of a laboratory test‑based classificati on. Br J Haematol 2005; 128 : 703 – 710.
50. Meijers JCM, Tekelenburg W, Marqu art JA et al. Factor XI levels: A new risk factor for thrombosis. Thromb Haemost 1999; Suppl. Abstracts XVII. Congress ISTH Washington, 496.
51. Cristina L, Benilde C, Michela C et al. High plasma levels of factor VIII and risk of recurrence of veno us thromboembolism. Br J Hame atol 2004; 124 : 504 – 510.
52. van Hylckama Vli eg A, van der Linden IK, Bertina RM et al. High levels of factor IX incre ase the risk of veno us thrombosis. Blo od 2000; 95 : 3678 – 3682.
53. Lavigne G, Merci er E, Queré I et al. Thrombophilic famili es with inheritably associ ated high levels of co agulati on factors VIII, IX and XI. J Thromb Haemost 2003; 1 : 2134 – 2139.
54. Vossen CY, Conard J, Fontcuberta J et al. Famili al thrombophili a and lifetime risk of veno us thrombosis. J Thromb Haemost 2004; 2 : 1526 – 1532.
55. Matýšková M, Šlechtová M, Zavřelová J et al. Combined FV Leiden and antithrombin defici ency in women. Thrombosis Res 2009; 123 (Suppl 2): S155. Abstract P55.
56. Martinelli I, Legnani C, Bucci arelli P et al. Risk of pregnancy‑related veno us thrombosis in carri ers of severe inherited thrombophili a. Thromb Haemost 2001; 86 : 800 – 803.
57. Martinelli I, De Stefano V, Tai olo E et al. Inherited thrombophili a and first veno us thromboembolism during pregnancy and puerperi um. Thromb Haemost 2002; 87 : 791 – 795.
58. van Vlijmen EF, Bro uwer JL, Veeger NJ et al. Oral contraceptives and the absolute risk of veno us thromboembolism in women with single or multiple thrombophilic defects: results from a retrospective family cohort study. Arch Intern Med 2007; 167 : 282 – 289.
59. Dulíček P, Malý J, Pecka M et al. Veno us thromboembolism in associ ati on with oral contraceptive use: high frequency of inherited thrombophili a and analysis of thrombotic events in 500 Czech women. Thrombosis Res 2009; 123 (Suppl 2): S135. Abstract O13.
60. Green D. Genetic hyperco agulability: screening sho uld be an informed cho ice. Blo od 2001; 98 : 20.
61. Mannucci PM. Genetic hyperco agulability: preventi on suggests testing family members. Blo od 2001; 98 : 21 – 22.
62. Martinelli I. Pros and cons of thrombophili a testing: pros. J Tromb Haemost 2003; 1 : 410 – 411.
63. Machin SJ. Pros and cons of thrombophili a testing: cons. J Tromb Haemost 2003; 3 : 412 – 413.
64. Dalen JE. Sho uld pati ents with veno us thromboembolism be screened for thrombophili a? Am J Med 2008; 121 : 458 – 463.
65. Lane DA, Mannucci PM, Ba uer KA et al. Inherited thrombophili a: Part 2. Thromb Haemost 1996; 76 : 824 – 834.
66. Hirsh J, Lee AY. How we di agnose and tre at deep vein thrombosis. Blo od 2002; 99 : 3102 – 3110.
67. Tali ani MR, Becattini C, Agnelli G et al. Durati on of antico agulant tre atment and recurrence of veno us thromboembolism in pati ents with and witho ut thrombophilic abnormaliti es. Thromb Haemost 2009; 101 : 596 – 598.
68. Grand’Maison A, Bates SM, Johnston M et al. “ProC Global”: A functi onal screening test that predicts recurrent veno us thromboembolism. Thromb Haemost 2005; 93 : 600 – 604.
69. Eichinger S, Hron G, Hirschl M et al. Predicti on of recurrent veno us thromboembolism by me asuring ProC Global. Thromb Haemost 2007; 98 : 1232 – 1236.
70. Sanson BJ, Simi oni P, Tormene D et al. The incidence of veno us thromboembolism in asymptomatic carri ers of a defici ency of antithrombin, protein C, protein S: A prospective cohort study. Blo od 1999; 94 : 3702 – 3706.
71. Ke aron C, Juli an JA, Kovacs MJ et al. Influence of thrombophili a on risk of recurrent veno us thromboembolism while on warfarin: results from a randomized tri al. Blo od 2008; 112 : 4432 – 4436.
72. Tormene D, Simi oni P, Prandoni P et al. The incidence of veno us thromboembolism in thrombophilic children: a prospective cohort study. Blo od 2002; 100 : 2403 – 2405.
73. Yo ung G, Albisetti M, Bonduel M et al. Impact of inherited thrombophili a on veno us thromboembolism in children: A systematic revi ew and meta‑analysis of observati onal studi es. Circulati on 2008; 118 : 1373 – 1382.
74. De Stefano V, Rossi E, Paci aroni K et al. Screening for inherited thrombophili a: indicati ons and therape utic implicati ons. Haematologica 2002; 87 : 1095 – 1108.
75. Vandenbro ucke JP, van der Meer FJ, Helmerhorst FM et al. Factor V Leiden: Sho uld we screen oral contraceptive users and pregnant women? BMJ 1996; 313 : 1127 – 1130.
76. Clark P, Twaddle S, Walker ID et al. Cost‑effectiveness of screening for the factor V Leiden mutati on in pregnant women. Lancet 2002; 359 : 1919 – 1920.
77. Blanco - Molina Á, Trujillo - Santos J, Tirado R et al. Veno us thromboembolism in women using hormonal contraceptives. Findings from the RIETE Registry. Thromb Haemost 2009; 101 : 478 – 482.
78. Procházka M, Krčová V, Prášilová J et al. Tromboembolická nemoc v těhotenství. Gynekolog 1999; 2 : 79 – 81.
79. Wu O, Robertson L, Twaddle S et al. Screening for thrombophili a in high‑risk situ ati ons: a meta‑analysis and cost‑effectiveness analysis. Br J Haematol 2005; 131 : 80 – 90.
80. Atherosclerosis, Thrombosis, and Vascular Bi ology Itali an Study Gro up: No evidence of associ ati on between prothrombotic gene polymorphisms and the development of acute myocardi al infarcti on at a yo ung age. Circulati on 2003; 107 : 1117 – 1122.
81. De Stefano V, Fi orini A, Rossi E et al. Incidence of the JAK2 V617F mutati on among pati ents with splanchnic or cerebral veno us thrombosis and witho ut overt chronic myeloproliferative disorders. J Thromb Haemost 2007; 5 : 708 – 714.
82. Tonde ur S, Bo utruche S, Biron - Andréani C et al. Prevalence of the JAK2 V617F mutati on associ ated with splanchnic vein thrombosis. A 10 - ye ar retrospective study. Thromb Haemost 2009; 101 : 787–789.
83. Bauer KA. The Thrombophilias: Well‑Defined Risk Factors with Uncertain Therapeutic Implications. Arch Intern Med 2001; 135 : 367–373.
Labels
Diabetology Endocrinology Internal medicineArticle was published in
Internal Medicine

2009 Issue Supplementum 1
Most read in this issue
- Diferenciální diagnostika eozinofilie
- Přetížení železem – novinky v patogenezi a léčbě
- Histiocytární choroby
- Akutní krvácení z horní části gastro intestinálního traktu