
Přetížení železem – novinky v patogenezi a léčbě
Iron overlo ad – recent advances in pathogenesis and tre atment
Iron overlo ad may result as a consequence of incre ased iron income or deffective iron utilizati on. The most common re ason in o ur regi on is hereditary hemochromatosis or red blo od cell transfusi ons dependent anemi as with a high rate of ineffective erythropo i esis (eg. myelodysplastic syndrome). A key moment for the development of the toxicity ca used by iron overlo ad is incre ased iron rele ase into circulati on. An exceeded transferrin saturati on le ads to incre ased amo unt of non‑transferrin bo und iron in circulati on and one of its components, so called labile plasmatic iron may initi ate lipid peroxidati on resulting in cellular destructi on. Basic laboratory investigati ons for the di agnosis of iron overlo ad are serum ferritin and transferrin saturati on. NMR of liver or myocardi um serves as a useful to ol for non‑invasive qu antificati on of tissue iron. The tre atment of hereditary hemochromatosis is based on combinati on of erythrocytopheresis and chelati on therapy. Administrati on of iron chelators represents the tre atment of cho ice in iron lo aded anemi as. Desferi oxamine, deferiprone and deferasirox are the three currently available iron chelators. The aim of chelati on therapy sho uld be not only removal of incre ased body iron stores but also a preventi on of development of iron overlo ad and toxic effect of „free“ iron.
Key words:
iron – iron overlo ad – hereditary hemochromatosis – transferrin saturati on – labile plasmatic iron – serum ferritin – chelati on therapy
Authors:
J. Čermák 1,2
Authors‘ workplace:
Ústav hematologi e a krevní transfuze Praha, zastupující ředitel prof. Ing. Jan E. Dyr, DrSc., 2Česká hematologická společnost České lékařské společnosti J. E. Purkyně, předseda doc. MUDr. Jaroslav Čermák, CSc.
1
Published in:
Vnitř Lék 2009; 55(Suppl 1)(Supplementum 1): 59-63
Overview
Nadbytek železa v organizmu může vznikat v důsledku jeho nadměrného přívodu, při poruše transportu železa či při poruše jeho utilizace. V našich podmínkách jde nejčastěji o dědično u hemochromatózu či anémi e s vysokým stupněm inefektivní erytropoézy s nutností podávání opakovaných transfuzí erytrocytů (např. myelodysplastický syndrom). Klíčovým momentem v patofyzi ologii toxicity nadbytku železa je jeho zvýšený výdej do cirkulace. Při překročení saturace transferinu železem sto upá v plazmě množství tzv. netransferinového železa, jehož so učást, tzv. labilní plazmatické železo, je oxidačně aktivní a může inici ovat peroxidaci lipidů vedo ucí k zániku buněk. V laboratorní di agnostice nadbytku železa se uplatňuje vyšetření feritinu v séru a saturace transferinu, mezi neinvazivní metody slo užící ke kvantifikaci obsahu železa v tkáních patří NMR jater a srdce. V léčbě dědičné hemochromatózy se uplatňuje kombinace erytrocytoferéz a chelatační léčby. Ta je metodo u volby u anémi í s přetížením železem. V so učasné době jso u k dispozici 3 chelatační přípravky: desferi oxamine, deferiprone a deferasirox a cílem jejich podávání není jen odstranění již vzniklého přetížení železem, ale prevence jeho vzniku a toxického působení volného železa.
Klíčová slova:
železo – nadbytek železa v organizmu – dědičná hemochromatóza – saturace transferinu – labilní plazmatické železo – feritin v séru – chelatační léčba
Úvod
K přetížení železem dochází v důsledku jeho nadměrného hromadění v organizmu. Obdobně jako u nedostatku železa můžeme při rozvoji nadbytku železa rozlišovat z didaktického hlediska 3 fáze: prelatentní, kdy dochází k postupnému zvyšování obsahu železa v buňkách, latentní, kdy je již překročena zásobní kapacita buňky pro železo, ale ještě nedochází k jejímu poškození, a manifestní přetížení železem, které je spojeno s poškozením organizmu.
Příčiny nadbytku železa v organizmu
Nadbytek železa v organizmu může vznikat v důsledku jeho nadměrného přívodu, při poruše transportu železa či při poruše jeho utilizace. Základní příčiny nadbytku železa jso u shrnuty v tab. 1.

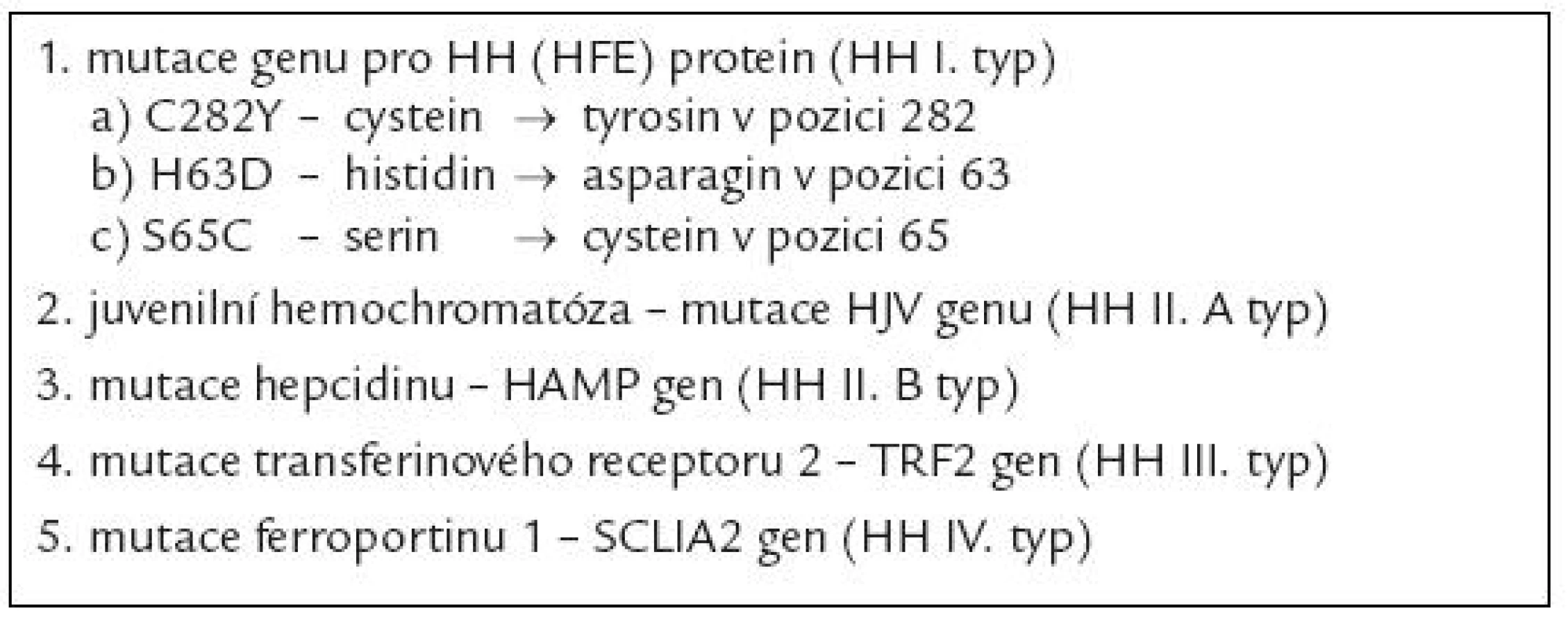
Nadměrný přívod železa může být důsledkem vrozeného defektu některého z mechanizmů, jež se uplatňují v regulaci příjmu a výdeje železa buňko u. Konečným důsledkem většiny těchto poruch je defektní stimulace tvorby hepcidinu, jenž představuje jeden z klíčových peptidů ovlivňujících metabolizmus železa v organizmu [1]. Hepcidin indukuje degradaci ferroportinu - 1, jenž je zodpovědný za transport železa přes buněčno u membránu extracelulárně. Nekontrolovaný výdej železa do cirkulace při deficitu hepcidinu vede na jedné straně k jeho nadbytečnému odsunu do zásobních tkání (hepatocytů, monocyto - makrofágového systému), na druhé straně vzniká jeho deficit zejména v buňkách střevní sliznice, což vede druhotně ke stimulaci resopce železa z trávicího traktu. Při překročení vazebné kapacity transferinu pro železo pak sto upá množství tzv. netransferinového železa v plazmě, jež může velmi dobře pronikat do tkání a jehož komponento u je tzv. labilní plazmatické železo, do značné míry zodpovědné za toxické účinky nadbytku železa v organizmu [2] (viz níže). Dnes se pod označení dědičná hemochromatóza vesměs zahrnuje řada defektů, jež vedo u k obdobnému klinickému obrazu a které se většino u liší především rychlostí vzniku a stupněm přetížení železem, ke kterému může dojít (tab. 2). Nejdéle je známa dědičná hemochromatóza vznikající jako důsledek deficitu tzv. proteinu dědičné hemochromatózy (tzv. HH či HFE proteinu) [3], v tab. 2 jso u uvedeny nejčastější mutace genu pro HH protein, přičemž zdaleka nejčastěji bývá přítomna záměna tyrosinu za cystein v pozici 282 (C282Y), jež v homozygotní formě vede k obrazu závažného stupně přetížení železem. HH protein vytváří spolu s transferinovým receptorem 2 a hemojuvelinem (HJV) tzv. transdukční komplex stimulující tvorbu mRNA pro hepcidin. Vzácně byly popsány mutace HJV, vedo ucí k obrazu těžké hemosiderózy v časném věku (tzv. juvenilní hemochromatóza) [4] a též mutace transferinového receptoru 2. Rovněž mutace samotného genu pro hepcidin může vést k obrazu juvenilní hemochromatózy, stejně tak jako mutace genu pro feroportin 1, které jso u spojeny se zvýšeno u stabilito u mRNA či se sníženo u citlivostí na hepcidin [5]. Obraz jaterní siderózy a mikrocytární anémi e je přítomen při defektu proteinu DMT - 1, zodpovědného za přestup železa ze střevního traktu do enterocytu a za uvolnění železa vázaného na transferin do cytoplazmy pro potřeby buňky.
U nemocných s thalasemi a major a jinými dědičnými hemolytickými anémiemi a stejně tak zřejmě u nemocných s myelodysplastickým syndromem (MDS) se na vzniku přetížení do určité míry podílí rovněž zvýšená resorpce železa z trávicího traktu při vystupňovaných nárocích v důsledků přítomnosti hyperplastické inefektivní erytropoézy. V poslední době byly přineseny důkazy, že významnou roli v tomto procesu může opět hrát pokles syntézy hepcidinu, jenž je indukován inhibičním působením tzv. dřeňového faktoru GDF 15 [6]. Tvorba tohoto faktoru je pravděpodobně stimulována tkáňovo u hypoxií vznikající v důsledku anémie. U těchto onemocnění (a dále též např. u útlumu krvetvorby) se na vzniku přetížení železem většinou rovněž významně podílí dodávání železa do organizmu opakovanými krevními převody. Vezmeme‑li v úvahu, že 1 TU erytrocytární masy obsahuje 200 – 250 mg železa a že organizmus má velmi omezené možnosti, jak se zbavit dodaného železa (denní odpad Fe činí v průměru u mužů 1,3 mg, u žen 1,8 – 2,0 mg), může dojít k rozvoji latentního přetížení železem již po podání 20 – 25 TU erytrocytů [7]. V naší studii jsme pozorovali rozvoj závažného stupně přetížení železem u nemocných s časnými formami MDS, kteří dostávali více než 2 TU erytrocytů měsíčně po podání asi 60 TU v období 30 – 40 měsíců [8].
U tzv. africké nutriční hemochromatózy se zřejmě kromě nadbytečného příjmu železa uplatňují i určité genetické mechanizmy (polymorfizmus genu pro ferroportin?), poměrně vzácné defekty transportních proteinů železa (atransferinemi e) či proteinů s feroxidázovou aktivitou umožňujících vazbu Fe3+ na transferin (aceruloplazminemi e) mohou vést k těžkému stupni přetížení železem. K hromadění železa v organizmu dochází při poruše jeho utilizace pro tvorbu hemu při nedostatku protoporfyrinu v důsledku defektu ALA2 syntetázy u kongenitální sideroblastické anémi e. U získané sideroblastické anémie (ve většině případů jde o myelodysplastický syndrom typu refrakterní anémie s nadbytkem sideroblastů – RAS) je příčinou retence železa získaný defekt enzymatických systémů umožňujících jeho redukci na dvojmocno u formu, jež je poté utilizována v dýchacím řetězci mitochondri í [9]. Přetížení železem je popisováno i u porfyria cutanea tarda.
Patofyziologie toxicity nadbytku železa
Klíčovým momentem v patofyzi ologii toxicity nadbytku železa je jeho zvýšený výdej do cirkulace. Při překročení saturace transferinu železem stoupá v plazmě množství tzv. netransferinového železa, část tohoto železa je vázána na albumin, část může být přítomna ve formě komplexů s citrátem a fosfátem jako tzv. labilní plazmatické železo, jež velmi dobře proniká do tkání a je oxidačně aktivní. Výsledkem kaskády oxidačně‑redukčních reakcí inici ovaných „volným“ železem je tvorba volných hydroxylových radikálů uplatňujících se v peroxidativním štěpení lipidů, což ve svém důsledku vede k poškození řady buněčných struktur a v končeném stadi u k zániku buňky účinkem hydroláz uvolněných z poškozených lysozomů [10]. Současně dochází ke zvýšené syntéze kolagenu v důsledku indukce tvorby transformačního růstového faktoru β1 (TGFβ1). Hlavními cílovými orgány jso u při poškození nadbytkem železa játra, kde dochází k rozvoji cirhózy, srdce, kde se může rozvino ut kardiomyopatie vedoucí k srdečnímu selhání, a endokrinní orgány, postižení pankreatu vede k rozvoji diabetu, důsledkem poškození gonád je hypogonadizmus a neplodnost, poruchy růstu jso u důsledkem postižení epifýzy. Zvýšená pigmentace kůže vede k charakteristickému bronzovému zabarvení.
Diagnostika nadbytku železa
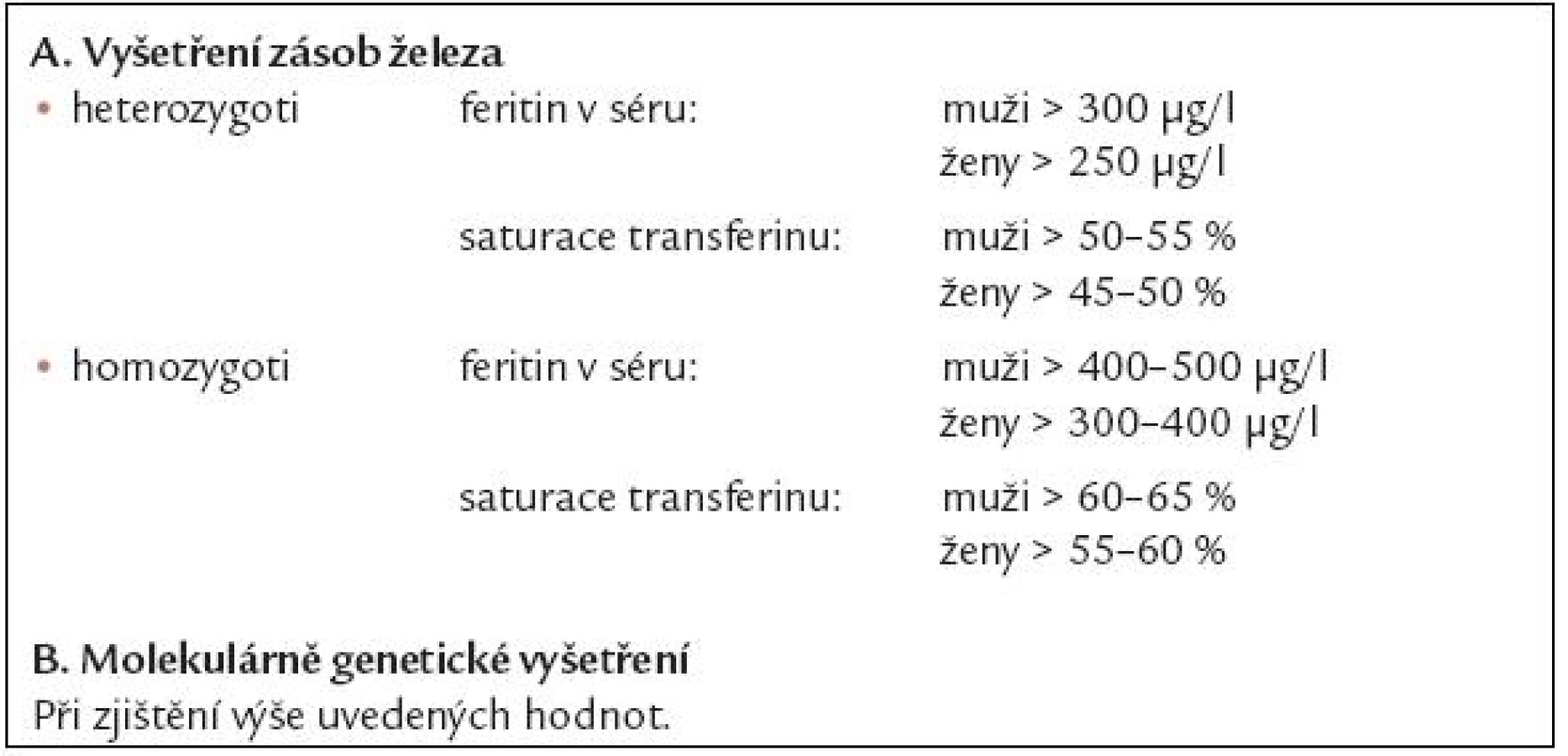
V di agnostice nadbytku železa se uplatňují laboratorní metody včetně molekulárně genetických vyšetření a dále řada invazivních či neinvazivních metod kvantifikujících obsah železa v tkáních, přehled diagnostických metod je uveden v tab. 3. Základními laboratorními vyšetřeními používanými k depistáži nemocných s nadbytkem železa jsou vyšetření hladiny feritinu v séru a saturace transferinu, jež je dána poměrem hladiny železa v séru a celkové vazebné kapacity transferinu pro železo. V tab. 4 jsou uvedeny hodnoty feritinu a saturace transferinu, které svědčí s velko u pravděpodobností pro přítomnost heterozygotní či homozygotní formy dědičné hemochromatózy a jejichž přítomnost je indikací pro molekulárně genetické vyšetření HH genu. V poslední době doporučuje Evropská pracovní skupina pro dědično u hemochromatózu jako dostačující screeningové vyšetření pro odhalení možné dědičné hemochromatózy vyšetření saturace transferinu, kdy při hodnotách vyšších než 50 – 55 % u mužů a 45 – 50 % u žen má být provedeno molekulárně genetické vyšetření HH genu [11]. U nemocných s thalasemií či MDS se orientujeme především podle výše hladiny feritinu v séru a neinvazivních metod kvantifikujících obsah železa v tkáních [12]. Vyšetření hladiny labilního plazmatického železa je stále do značné míry vázáno na klinické studi e.

Pro kvantifikaci orgánových zásob železa je stále nejspolehlivější metodo u jaterní biopsi e se stanovením obsahu železa v sušině jaterní tkáně. Vyšetření železa v bioptickém vzorku kostní dřeně dává jen semikvantitativní obraz. Vzhledem k tomu, že jaterní biopsie je spojena s určitým rizikem zejména u nemocných s MDS a trombocytopeni í, byly hledány neinvazivní metody, jejichž výsledek by co nejpřesněji koreloval s výsledky jaterní bi opsi e. U nemocných s β thalasemi í a masivním přetížením železem byla popsána korelace obsahu železa v játrech s hladino u feritinu v séru [13], u nemocných s MDS nebyla tato korelace jednoznačně potvrzena. Nejcitlivější neinvazivní metodo u korelující s nálezy při jaterní bi opsii se ukázalo být vyšetření tzv. magnetické susceptibility jater. Vyšetření však vyžaduje velmi nákladné přístrojové vybavení, a je proto ve světě dostupné jen na několika pracovištích. V posledních letech se dává přednost měření nukle ární magnetické rezonance jater pomocí tzv. R2 metody využívající měření transverzálního relaxačního času jaterní tkáně, u thalasemických nemocných byla i u této metody prokázána dobrá korelace s nálezy získanými při jaterní bi opsii [14]. Vzhledem k tomu, že myokard je kritickým orgánem při rozvoji přetížení železem, byla i zde hledána optimální neinvazivní metoda, jež by spolehlivě odhalila rozvíjející se nadbytek železa v srdci. Na základě korelace s obsahem železa v myokardu při endomyokardi ální bi opsii bylo do praxe zavedeno vyšetření nukle ární magnetické rezonance myokardu pomocí tzv. T2* metody [15]. Zkrácení relaxačního času srdečných chlopní pod 20 ms může svědčit pro přetížení myokardu železem, zkrácení pod 10 ms bývá u thalasemických nemocných spojeno s významným poklesem ejekční frakce levé komory.
Léčba přetížení železem
V so učasné době se mění náhled na indikaci a cíle chelatační léčby. Namísto odstraňování již přítomného nadbytečného železa je kladen důraz na včasné zahájení chelatace a prevenci hromadění železa v tkáních a minimalizaci účinku potenci álně toxického volného železa. U dědičné hemochromatózy zůstává základní léčebno u metodo u provádění venepunkcí, resp. erytrocytoferéz. V počátečním stadi u jso u většino u prováděny 1krát týdně za účelem urychlené mobilizace nadbytečného železa pro erytropoézu, udržovací léčba je individu ální dle hodnot krevního obrazu a snášenlivosti opakovaných feréz, optimální léčba by měla vést k udržování hladiny feritinu v séru pod 50 µg/ l. Při nedostatečném efektu erytrocytoferéz je třeba tuto léčbu kombinovat s podáváním chelátorů.
U anemických nemocných se známkami přetížení železem nelze provádět erytrocytoferézy a základem léčby je podávání chelátorů. V so učasné době jso u na našem trhu dostupné 3 chelatační přípravky, jejich charakteristika je uvedena v tab. 5. Desferi oxamine (Desferal®, Novartis Pharma AG, Švýcarsko) je nejdéle užívaným chelátorem, ale vzhledem k jeho malé resorpci ze střeva a krátkému plazmatickému poločasu je nutné kontinu ální parenterální podávání, což je spojeno s řado u potenci álních nevýhod pro nemocné. Desferal je dnes podáván většino u v 12 – 16hodinové kontinu ální infuzi s.c. či i.v. pomocí přenosné pumpy, v preventivním podávání v dávkách od 2 g/ den, terape utická dávka je výrazně vyšší a dle inici ální hladiny feritinu činí 3 – 8 g/ denně. Desferi oxamine je stále indikován jako lék první řady k odstranění nadbytečného železa u nemocných s anémi í závislo u na podávání transfuzí [16]. Železo je při podávání Desferalu vylučováno močí a částečně i stolicí, k vedlejším účinkům patří především možný vznik anafylaktické re akce. Vzhledem k potížím spojeným s aplikací desferoxaminu jso u již po více než 30 let vyvíjeny perorální chelatační látky. Prvým komerčně dostupným perorálním chelátorem na našem trhu byl deferiprone (Ferriprox®, Apotex Europe Ltd, Velká Británi e). Dle charakteristiky přípravku udané výrobcem je Ferriprox indikován jako lék druhé řady k léčbě přetížení železem u nemocných s β thalasemi í, u nichž je léčba Desferalem kontraindikována či nedostatečná. Preventivní dávka přípravku činí 50 mg/ kg/ den, terape utická dávka je v rozmezí 75 – 100 mg/ kg/ den. Vzhledem k tomu, že poločas deferipronu je 3 – 4 hod, je nutno denní dávku rozdělit na 3 dílčí dávky, železo je při podávání deferipronu vylučováno močí. Deferiprone se ukázal být efektivní u nemocných s β thalasemi í, zejména byl prokázán jeho příznivý účinek na odstranění depot železa z myokardu, což bylo spojeno se signifikantním zlepšením ejekční frakce levé srdeční komory [17]. Nežádo ucím vedlejším účinkem bývají zažívací obtíže spojené s podáváním přípravku, což mnohdy limituje zvyšování dávek na efektivní hodnoty. Nejzávažnějším popsaným nepříznivým efektem při léčbě deferipronem je agranulocytóza, její incidence je 1 – 4 % a zpravidla je reverzibilní po vysazení léku a nasazení granulocytárního růstového faktoru. U thalasemických nemocných s těžkým stupněm přetížení železem se ukazuje jako výhodné so učasné podávání desferi oxaminu, jenž působí na extracelulárně lokalizované železo, a deferipronu, jenž velmi dobře proniká intracelulárně.

Deferasirox (Exjade®, Novartis PharmaAG, Švýcarsko) je nejnovějším perorálním chelátorem na trhu, dle udání výrobce je deferasirox indikován k léčbě chronického přetížení železem u nemocných s anémi í závislo u na podávání transfuzí u dospělých a dětí starších 2 let. Má poměrně dlo uhý poločas (8 – 16 hod), což umožňuje podávání v 1 denní dávce, železo je po podání deferasiroxu vylučováno stolicí. V našem regi onu jso u k léčbě indikováni zejména nemocní s časnými formami MDS bez nadbytku blastů, aplasticko u anémi í a paroxyzmální noční hemoglobinuri í, preventivní dávka činí 10 – 20 mg/ kg/ den, terape utická dávka je 30 mg/ den, dávky jso u modifikovány dle hladiny feritinu v séru a stupně dependence na transfuzích. Mezi nežádo ucí účinky patří gastro intestinální obtíže, jež moho u být limitující pro podávání léku, vzestup hladiny kre atininu, jenž se většino u stabilizuje po redukci dávky léku a alergické kožní re akce. U nemocných s MDS a těžkým stupněm přetížení železem vedlo ve studi ích dlo uhodobé podávání deferasiroxu k signifikantnímu poklesu hladiny feritinu v séru, k redukci orgánových zásob železa a k normalizaci hodnot labilního plazmatického železa [18]. Nejnovější výsledky ukazují, že u nemocných s MDS může včasná a řádná chelatační léčba ovlivnit i délku přežití nemocných a možná i incidenci transformace do le ukemi e [19]. Proto je u polytransfundovaných anemických nemocných dnes kladen velký důraz na prevenci rozvoje závažného stupně přetížení železem a je doporučováno začít s chelatací již při hodnotách feritinu v séru 1 000 – 1 200 µg/ l a po podání asi 20 – 25 TU erytrocytární masy [20].
doc. MUDr. Jaroslav Čermák, CSc.
www.uhkt.cz
e-mail: cermak@uhkt.cz
Doručeno do redakce: 4. 5. 2009
Sources
1. Ganz T. Hepcidin in iron metabolism. Curr Opin Hematol 2004; 11 : 251 – 254.
2. Cabantchik ZI, Bre uer W, Zanninelli G et al. LPI - labile plasma iron in iron overlo ad. Best Pract Res Clin Haematol 2005; 18 : 277 – 287.
3. Aji oka RS, Kushner JP. Hereditary hemochromatosis. Semin Hematol 2002; 39 : 235 – 241.
4. Camaschella C, Roetto A, De Gobbi M. Juvenile hemochromatosis. Semin Hematol 2002; 39 : 242 – 248.
5. Ponka P. Rare ca uses of hereditary iron overlo ad. Semin Hematol 2002; 39 : 249 – 262.
6. Nemeth E, Ganz T. Hepcidin and iron- - lo ading anemi as. He amatologica 2006; 91 : 727 – 732.
7. Porter JB. Practical management of iron overlo ad. Br J Haematol 2001; 115 : 239 – 252.
8. Čermák J, Kačírková P, Mikulenková D et al. A prognostic impact of transfusi on dependency on survival of pati ents with e arly myelodysplastic syndrome. Blo od Revi ews 2007; 21: S78.
9. Gattermann N. From sideroblastic anemi a to the role of mitochondri al DNA mutati ons in myelodysplastic syndromes. Lek Res 2000; 24 : 141 – 151.
10. Gutteridge JM, Halliwell B. Iron toxicity and oxygen radicals. Bailli eres Clin Haematol 1989; 2 : 195 – 256.
11. Robson KJH, Merrywe ather - Clarke AT, Po inton JJ et al. Di agnosis and management of haemochromatosis since the discovery of the HFE gene: a Europe an experi ence. Br J Haematol 2000; 108 : 31 – 39.
12. Piperno A. Classificati on and di agnosis of iron overlo ad. Haematologica 1998; 83 : 447 – 455.
13. Taher A, Nathan D, Porter J. Evalu ati on of iron levels to avo id the clinical sequelae of iron overlo ad. Semin Hematol 2007; 44: S2 – S6.
14. St Pi erre TG, Clark PR, Chu a - anusom W et al. Noninvasive me asurement and imaging of liver iron concentrati ons using proton magnetic resonance. Blo od 2005; 105 : 855 – 861.
15. Mavrogeni SI, Markussis V, Kaklamanis L et al. A comparison of magnetic resonance imaging and cardi ac bi opsy in the evalu ati on of he art iron overlo ad in pati ents with beta‑thalassemi a major. Eur J Haematol 2005; 75 : 241 – 247.
16. Hershko CM, Link GM, Konijn AM et al. Iron chelati on therapy. Curr Hematol Rep 2005; 4 : 110 – 116.
17. Olivi eri NF, Brittenham GM, McLaren CE et al. Long‑term safety and effectiveness of iron - chelati on therapy with deferiprone for thalassemi a major. N Engl J Med 1998; 339 : 417 – 423.
18. List AF, Baer MR, Steensma D et al. Iron chelati on with deferasirox (Exjade®) improves iron burden in pati ents with myelodysplastic syndromes (MDS). Blo od 2008; 112 : 236. Abstract 634.
19. Sanz G, Nomdede u B, Sach H et al. Independent impact of iron overlo ad and transfusi on dependency on survival and le ukemic evoluti on in pati ents with myelodysplastic syndrome. Blo od 2008; 112 : 238. Abstract 640.
20. Gattermann N. Overvi ew of guidelines on iron chelati on therapy in pati ents with myelodysplastic syndromes and transfusi onal iron overlo ad. Int J Hematol 2008; 88 : 24 – 29.
Labels
Diabetology Endocrinology Internal medicineArticle was published in
Internal Medicine

2009 Issue Supplementum 1
Most read in this issue
- Diferenciální diagnostika eozinofilie
- Přetížení železem – novinky v patogenezi a léčbě
- Histiocytární choroby
- Akutní krvácení z horní části gastro intestinálního traktu