
Genetické pozadie nádorov adrenomedulárneho a extraadrenálneho chromafinného tkaniva – aktuality
Genetic background of tumors originating from adrenomedullar and extraadrenal chromaffin tissue – update
It is anticipated that an inherited/ familial forms of pheochromocytomas cause approximately 20% of all pheochromocytomas. Therefore, the classic “rule of 10” axioma used to remember the key features of disorder is invalid. Various mutations in several genes have been identified, which underly syndromes with paragangliomas and/ or pheochromocytomas. The more candidate genes, the less numbers of patients with apparently sporadic forms of the disorder. This review has summarized the current knowledge of the genetic background of tumors orginating from adrenomedullar and extra‑adrenal chromaffin tissue.
Key words:
adrenal gland – inherited tumor – pheochromocytoma – paraganglioma – neuroblastoma – ganglioneuroma – genes
Authors:
P. Vaňuga 1; M. Pura 1; A. Kreze jr. 2
Authors‘ workplace:
Endokrinologické oddelenie Národného endokrinologického a diabetologického ústavu Ľubochňa, Slovenská republika, prednosta prim. MU Dr. Peter Vaňuga, PhD. 2 II. interní oddělení FN Na Bulovce Praha, přednosta prim. MU Dr. Jiří Koskuba
1
Published in:
Vnitř Lék 2010; 56(12): 1296-1302
Category:
Celebration
Overview
Podiel vrodených/ familiárnych foriem feochromocytómov sa dnes odhaduje na približne 20 % všetkých prípadov feochromocytómov. Preto už neplatí klasické „pravidlo 10“, slúžiace na zapamätanie si kľúčovej klinickej charakteristiky ochorenia. Sú identifikované viaceré gény, ktorých mutácie podmieňujú syndrómy s výskytom paragangliómov a/ alebo feochromocytómov. Pribúdaním kandidátnych génov ubúdajú počty pacientov so zdanlivo sporadickými formami ochorenia. V práci je podaný prehľad aktuálnych poznatkov týkajúcich sa problematiky genetického pozadia nádorov adrenomedulárneho alebo extraadrenálneho chromafinného tkaniva.
Kľúčové slová:
nadoblička – vrodený nádor – feochromocytóm – paraganglióm – neuroblastóm – ganglioneuróm – gény
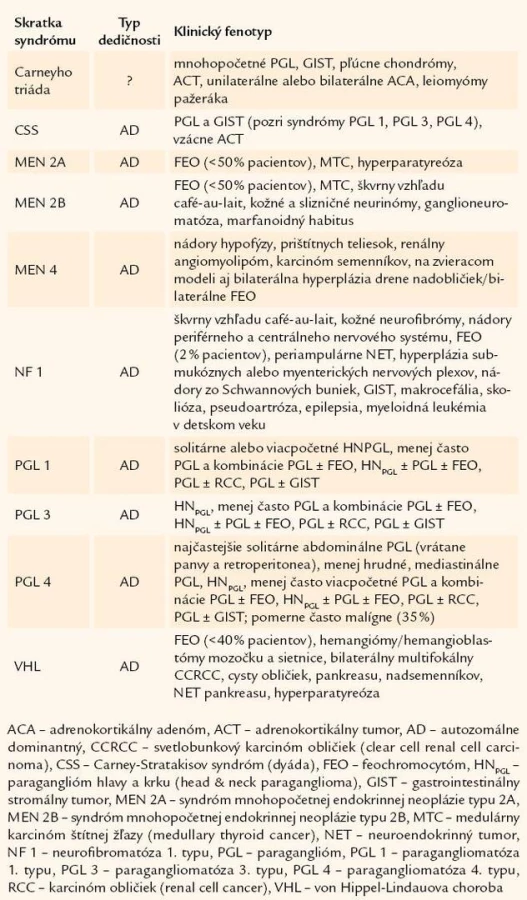
Prvým, všeobecne akceptovaným popisom feochromocytómu (FEO) je prípad 18 - ročnej Minny Rollovej z Wittenweieru z roku 1886 [1]. Hoci v tom čase boli vedomosti o nádoroch vychádzajúcich z drene nadobličiek minimálne, vtedajšia dôkladnosť doktora Felixa Fränkela a jeho spolupracovníkov v Univerzitnej nemocnici vo Freiburgu umožnila aj po 120 rokoch dokázať, že išlo o vrodenú formu FEO v rámci syndrómu mnohopočetnej endokrinnej neoplázie typu 2 [2]. V poslednej dobe sme svedkami prehlbovania poznatkov týkajúcich sa nádorov adrenomedulárneho alebo extraadrenálneho chromafinného tkaniva. Na základe genetických štúdií pomerne veľkých kohort pacientov sa podarilo identifikovať viaceré gény, ktorých mutácie sú podkladom syndrómov s výskytom paragangliómov (PGL) a/ alebo FEO (t.j. PGL-FEO syndrómy) (tab. 1) [3 – 5]. Pribúdaním kandidátnych génov podmieňujúcich vrodené/ familiárne formy nádorov ubúdajú počty pacientov so zdanlivo sporadickými formami ochorenia. Podiel vrodených nádorov sa dnes odhaduje na približne 20 % všetkých prípadov FEO [6 – 8]. Preto už neplatí klasické „pravidlo 10“, slúžiace na zapamätanie si kľúčovej klinickej charakteristiky ochorenia.
Podnetom na napísanie nasledujúceho textu, v ktorom je podaný prehľad aktuálnych poznatkov týkajúcich sa problematiky genetického pozadia nádorov adrenomedulárneho alebo extraadrenálneho chromafinného tkaniva, je významné životné jubileum, ktorého sa v novembri tohto roku dožíva významná osobnosť československej endokrinológie – profesor MU Dr. RNDr. Luboslav Stárka, DrSc. Na rozdiel od FEO pre profesora Stárku „pravidlo 10“ platí. V prvom rade, pre autorov článku a určite aj pre celú endokrinologickú komunitu je pán profesor jedným z „top 10“ morálnych autorít a nedosiahnuteľných vzorov správania. Patrí medzi 10 najuznávanejších odborníkov v odbore endokrinológie a je jedným asi nie z viac ako 10 endokrinológov, ktorí majú steroidogenézu v malíčku. Priemerný počet jeho odborných publikácií je viac ako 10 za rok, pričom veríme, že ešte minimálne 10 rokov bude.
Pán Boh a dehydroepiandrostendión mu v tom pomáhaj.
Feochromocytómy a paragangliómy
FEO a PGL môžu vznikať v rámci viacerých syndrómov, o ktorých sa podrobnejšie zmieňujeme nižšie, v literatúre však existujú aj popisy familiárnych nesyndrómových prípadov FEO. Kandidátnymi lokusmi pre tieto nové formy FEO sú oblasť centroméry 2. chromozómu (2cen) a krátke ramienko 16. chromozómu (16p13) [9,10].
Von Hippel-Lindauova choroba (VHL)
VHL je ochorenie s incidenciou približne 1 : 36 000 a autozomálne dominantným typom dedičnosti, ktoré predisponuje pacientov na viacpočetné vaskularizované nádory (tab. 1 a 2). VHL vzniká ako dôsledok inaktivačnej mutácie tumor-supresorového génu VHL, pričom približne v 20 % prípadov ide o de novo mutácie bez pozitívnej rodinnej anamnézy [11]. VHL sa rozdeľuje na typ 1, pri ktorom môžu mať postihnutí jedinci hemangioblastómy a adenokarcinóm obličky, ale nie FEO, a typ 2, kde minimálne 1 postihnutý jedinec má FEO. Pri VHL koreluje genotyp s fenotypom, najmä čo sa týka prítomnosti FEO [11]. V rodinách s VHL typu 1 dominujú veľké delécie a bodové mutácie génu, ktoré vedú ku skráteniu polypeptidového reťazca, väčšina pacientov s VHL typu 2 má bodové missense mutácie [11,12]. VHL je najčastejšou príčinou familiárneho FEO, ktorý v závislosti od špecifickej mutácie postihuje 10 – 25 % pacientov a diagnostikuje sa veku asi 20 rokov [7,11]. Pre vznik FEO pri VHL je okrem germinatívnej mutácie jednej alely VHL génu nutná strata aktivity druhej alely na somatickej úrovni. Produkty génu VHL (súhrnne nazývané pVHL) sú súčasťou ubiquitínového komplexu, ktorý viaže ubiquitín na substráty, čím ich vystavuje degradácii v proteazómoch [11,13]. Jeden z cieľových substrátov pre ubiquitín ligázový komplex E3 obsahujúci pVHL je HIF-α (transkripčný faktor aktivujúci hypoxiou-indukovateľné gény), pričom práve dysregulácia HIF-α zohráva kľúčovú úlohu v patogenéze FEO a/ alebo PGL pri mutáciách génov VHL a SDHx (pozri nižšie) [4,13].
![Prehľad hereditárnych foriem feochromocytómov a paragangliómov. Podľa [3].](https://www.prelekara.sk/media/cache/resolve/media_object_image_large/media/image/77564d5114e18ce0e7d22f6b5a220849.jpg)
Mnohopočetná endokrinná neoplázia typ 1 (MEN 1)
Syndróm MEN 1 je autozomálne dominantným ochorením s výskytom nádorov endokrinných a neendokrinných orgánov. Napriek tomu, že u pacientov s MEN 1 sa najčastejšie vyskytujú hormonálne inaktívne benígne adrenokortikálne adenómy, zriedkavo prípady periférneho hyperkortizolizmu a primárneho aldosteronizmu, boli popísané aj prípady pacientov s unilaterálnym FEO [6,14].
Mnohopočetná endokrinná neoplázie typ 2 (MEN 2)
Syndróm MEN 2 je ochorením s autozomálne dominantným typom dedičnosti a s incidenciou 1 : 40 000. Aktivačné mutácie RET-protoonkogénu spôsobujú konštitučnú aktiváciu tyrozínkinázového receptoru [15]. So vznikom FEO v rámci MEN 2A sa spájajú germinatívne mutácie v kodónoch 611, 618, 620, 634. Riziko vzniku FEO je vyššie pri mutáciách v kodóne 634. Vznik FEO pri MEN 2B podmieňujú mutácie v kodónoch 883 a 918 [6]. RET ovplyvňuje proliferáciu a diferenciáciu buniek odvodených od neuroektodermy. Pri aktivačných mutáciách RET génu dochádza v dôsledku jeho tkanivo-špecifickej expresie k hyperplázii parafolikulárnych C-buniek v štítnej žľaze a chromafinných buniek v dreni nadobličiek, s vysokým rizikom ich následnej nádorovej transformácie. V určitom počte prípadov sa nepodarí odhaliť mutácie RET, pri oboch podtypoch MEN 2 syndrómu navyše existujú značné fenotypové rozdiely v rámci rodín a medzi rodinami s identickou mutáciou génu RET, čo svedčí pre existenciu genetických modifikátorov a/ alebo ďalších lokusov, resp. kandidátnych génov [15 – 19].
RET je receptorom pre ligandy zo skupiny neurotropných faktorov odvodených od gliovej bunkovej línie (GFL – glial cell-derived neurotrophic factor family of ligands), menovite GDNF (glial cell-derived neurotrophic factor), neurturín, persefín a artemín. Ligandy najskôr vytvoria komplex s ich koreceptorom zo skupiny GFRα1 až GFRα4, následne GFL-GFRα komplex aktivuje RET [15 – 19]. Analýzy génov GDNF, resp. GFRα, zatiaľ neviedli k identifikácii germinatívnych mutácií, preto sa zdá, že mutácie GDNF, resp. GFRα nemajú veľkú úlohu v patogenéze familiárnych alebo sporadických FEO, alelické variácie lokusu GDNF však môžu modifikovať riziko vzniku FEO [15]. Rovnako sa doteraz nedokázali ani somatické mutácie žiadneho z génov kódujúcich GFL/ GFRA, hoci gény GFL/ GFRA sa nachádzajú v oblastiach, v ktorých sa zaznamenali alelické inbalancie napr. pri medulárnom karcinóme štítnej žľazy [19]. Boli publikované práce o polymorfizmoch génov GFL a ich koreceptorov, najmä génu GFRA1 (kódujúceho GFRα1) pri familiárnych a génu GFRA4 (kódujúceho α4) pri sporadických prípadoch MTC [16,18,19]. Inou cestou skúmania vzťahu genotyp-fenotyp pri syndróme MEN 2 je štúdium rodín so zriedkavými dvojitými germinatívnymi mutáciami RET-protoonkogénu. Príkladom je dvojitá mutácia C634Y/ Y791F, pri ktorej sa dokázala vysoká penetrancia FEO (u 11 z 13 nositeľov mutácie), frekventný výskyt bilaterálnych nádorov (5 z 11) a najmä náchylnosť na vznik nádorov veľkých rozmerov (5 – 14 cm). Pri in vitro štúdii bol RET proteín s dvojitou mutáciou C634Y/ Y791F fosforylovaný signifikantne viac ako aktivovaný nemutovaný receptor, resp. receptory so samostatnými mutáciami C634Y a Y791F [20].
Syndróm mnohopočetnej endokrinnej neoplázie typ 4 (MEN 4)
Syndróm MEN 4 je novým syndrómom s autozomálne dominantným typom dedičnosti a prekrývajúcim sa fenotypom MEN 1 a MEN 2 syndrómov (tab. 1). U pacientov s MEN 4 boli identifikované germinatívne mutácie génov kódujúcich inhibítory cyklín-dependentných kináz (CDKi) s predpokladanou tumor-supresorovou aktivitou. Zo 7 známych CDKi sa zatiaľ dokázali mutácie génov CDKN2B (p15Ink4b), CDKN2C (p18Ink4c), CDKN1A (p21Cip1) a CDKN1B (p27Kip1) [21 – 25]. Bilaterálna hyperplázia drene a/ alebo bilaterálne FEO sú súčasťou fenotypu iba na zvieracom modeli s mutáciami génu Cdkn1b (kódujúceho proteín p27Kip1), u doteraz identifikovaných pacientov s mutáciami CDKi sa nádory vychádzajúce z chromafinných buniek zatiaľ nedokázali [22,26,27]. Je zaujímavé, že regulátormi expresie p27Kip1 sú menín (podmieňujúci syndróm MEN 1), ako aj AIP (aryl hydrocarbon receptor-interacting protein) (predisponujúci na familiárny výskyt adenómov hypofýzy, navyše s novoozrejmenou úlohou v tumorigenéze adrenokortikálnych nádorov) [22,23,28,29].
Neurofibromatóza 1. typu
NF1 je ochorenie s autozomálne dominantným typom dedičnosti, s incidenciou asi 1 : 3 000 – 4 000. Podkladom sú mutácie génu NF1, pričom vysoké percento (približne 50 %) mutácií vzniká de novo. Génový produkt, neurofibromín, pôsobí ako tumor-supresorový gén. Jeho tumor-supresorová aktivita je daná schopnosť regulovať RAS proto-onkogény (Hras, Kras a Nras) urýchľovaním inaktivácie proto-onkogénu p21ras. Inaktivácia neurofibromínu je alternatívnym mechanizmom na hyperaktiváciu MAP-kinázovej dráhy. Okrem toho NF1 inhibuje signalizačnú kaskádu NGF receptoru a strata NF1 umožňuje prežívanie embryonálnych periférnych neurónov nezávisle od NGF [30].
Gén NF1 obsahuje 57 kódujúcich exónov a je jedným z najdlhších v ľudskom genóme. Mutačnú analýzu komplikuje aj prítomnosť 36 pseudogénov. Objasnenie vzťahu genotyp-fenotyp u pacientov s NF1, navyše sťažuje nízka prevalencia FEO, ktorá sa odhaduje na 0,1 – 5,7 % [31,32]. Mutačná analýza tak predstavuje pomerne veľký technický a logistický problém. V štúdii 37 pacientov s FEO asociovanými s NF1 napriek rôznym stupňom poškodenia génu NF1 Bausch et al nepozorovali výraznejšie rozdiely vo fenotype [31,32]. Stupeň fenotypových zmien pravdepodobne modulujú rôzne delécie génov v okolí NF1 [33]. Hoci ozrejmené mutácie postihujú celú kódujúcu sekvenciu génu bez špecifických horúcich mutačných miest, najviac mutácií sa nachádza v oblasti domény bohatej na cysteín a serín, tzv. CSR domény (CSD). Výsledky štúdie naznačujú, že CSD je predominantnou mutačnou oblasťou minimálne v prípade FEO asociovaných s NF1, ak nie u všetkých vrodených a sporadických FEO [32]. Pomerne novým zistením týkajúcim sa klinickej manifestácie NF1 je asociácia s gastrointestinálnymi stromálnymi nádormi (GIST). V priebehu poslednej dekády bolo publikovaných niekoľko prípadov GIST u pacientov s NF1, či už s alebo bez FEO [34]. Dôkaz somatickej inaktivácie génu NF1 v tkanivách nádorov, pri absencii mutácii génov KIT alebo PDGFRA, jednoznačne priraďujú GIST do spektra klinických symptómov NF1 [35].
Syndrómy s paragangliómami
O genetickej predispozícii na paragangliómy (PGL), najmä hlavy a krku, sa vie minimálne 20 rokov [4], avšak iba nedávno boli identifikované gény, ktoré sú v kauzálnom vzťahu s výskytom paragangliómov. Ich podkladom sú (až na výnimky) mutácie podjednotiek B, C a D sukcinyldehydrogenázy (SDH). Gény kódujúce podjednotky SDH súhrnne zastrešuje skratka SDHx. SDH je enzýmový komplex mitochondriálneho dýchacieho reťazca, nevyhnutný v citrátovom cykle. V nádoroch deficitných pre SDH inhibuje akumulácia sukcinátu funkciu dioxygenáz, čo vedie k hyperaktivite HIF. Mutácia ktorejkoľvek z podjednotiek SDH alebo strata SDH pri poškodení chromozómu vedie k poruche Krebsovho cyklu a vyúsťuje do pseudohypoxie. Preto pacienti s predispozíciou na FEO alebo PGL s mutáciami génov VHL, SDHA, SDHB, SDHC alebo SDHD zdieľajú poruchu systému monitorujúceho hladinu kyslíka v bunkách.
Syndrómy sa aktuálne rozčleňujú na PGL 1 (s mutáciami génu podjednotky SDHD), PGL 2 (mutácie génu SDH5), PGL 3 (mutácie podjednotky SDHC) a PGL 4 (mutácie podjednotky SDHB). Ako zriedkavá príčina PGL bola potvrdená aj mutácia génu pre podjednotku SDHA [4,36]. Charakteristika fenotypov jednotlivých syndrómov je uvedená v tab. 1 a 2.
Paragangliomatóza 1. typu (PGL 1)
Podkladom ochorenia sú germinatívne mutácie génu SDHD podjednotky D pre SDH, keď menší podiel doteraz tvoria jedinečné mutácie (14 %) oproti rekurentným mutáciám (86 %) [4]. Riziko vývoja viacpočetných primárnych nádorov a FEO u nositeľov mutácií génu SDHD zvyšujú mutácie vedúce ku skráteniu bielkoviny a život vo vysokej nadmorskej výške. Veľmi nízka nadmorská výška v Holandsku (nízka nadmorská výška zmierňuje hypoxickú stimuláciu paraganglií), v spojitosti s missense mutáciami opakovane dedenými tzv. efektom zakladateľa sú preto možným vysvetlením pre nezvykle vysoké zastúpenie germinatívnych mutácií medzi zdanlivo sporadickými prípadmi HNPGL a celkovo nízke riziko FEO u holandských nositeľov mutácií SDHD génu. Výhradný paternálny prenos ochorenia vysvetľuje selektívna somatická strata celého maternálneho chromozómu 11 postihujúca jednak divú alelu SDHD a maternálne exprimovaný (zatiaľ neznámy) tumor-supresorový gén umiestnený v blízkosti SDHD, ktorý pôsobí cis - alebo aj trans-regulačne. Možným vysvetlením fenotypovej variability syndrómu potom môže byť práve veľkosť a lokalizácia somatických delécií normálnej alely [37].
Paragangliomatóza 2. typu (PGL 2)
Podkladom syndrómu je germinatívna mutácia génu SDH5, kódujúceho enzým SDHAF2 (SDH complex assembley factor 2). Tento je potrebný na flavináciu podjednotky SDHA, a tým aj na aktivitu SDH [38]. U 33 členov holandskej rodiny s PGL, nositeľov mutácie G78R, sa ochorenie manifestovalo, naopak u siedmych, ktorí zdedili mutáciu od matky, ochorenie nevzniklo (medián veku 74 rokov), čo svedčí pre podobný typ dedičnosti ako pri syndróme PGL 1 [38].
Paragangliomatóza 3. typu (PGL 3)
Podkladom ochorenia sú germinatívne mutácie génu SDHC pre podjednotku C pre SDH. U doteraz identifikovaných pacientov, ktorých počet je zatiaľ malý, dominujú jedinečné mutácie [4].
Paragangliomatóza 4. typu (PGL 4)
Podkladom ochorenia sú germinatívne mutácie génu SDHB pre podjednotku B pre SDH. Podobne ako syndróme PGL 1 dominujú rekurentné mutácie (73 %), svedčiace pre efekt zakladateľa [4].
Aktuálne pribúdajú dôkazy o tom, že predispozíciou na nádory môžu byť aj mutácie iných génov zúčastňujúcich sa HIF signalizačnej kaskády. Pri normoxických podmienkach sú podjednotky HIF-α-hydroxylované dioxygenázami. PHD proteíny sú dioxygenázy, ktoré hydroxylujú kľúčové prolínové rezíduá HIF-α, čím umožňujú naviazanie HIF - α na proteín VHL. Spomedzi viacerých PHD proteínov je kritickým senzorom kyslíka označujúcim HIF-1α na deštrukciu dioxygenáza PHD2. V roku 2008 bol publikovaný prípad pacienta s erytrocytózou a recidivujúcim PGL, ktorý bol nositeľom germinatívnej mutácie génu PHD2. Mutovaná alela (bodová mutácia c.1121A→G, p.H374R) sa spolu so stratou heterozygozity pre PHD2 dokázala aj v tkanive nádoru, z čoho vyplýva, že PHD2 môže mať funkciu tumor-supresorového génu [39].
Posledným potvrdeným podkladom PGL syndrómu je mutácia génu SDHA, kódujúceho flavoproteínovú podjednotku A komplexu SDH. Germinatívna mutácia R589W, potvrdená u francúzskej pacientky s extraadrenálnym abdominálnym PGL, viedla pri funkčných in vivo a in vitro štúdiách k rovnakým zmenám (t.j. pseudohypoxii a angiogenéze), aké spôsobujú mutácie ďalších SDHx génov [36]. Poruchy ďalších metabolických génov citrátového cyklu (napr. izocitrátdehydrogenáz IDH1, IDH2), ktoré by mohli podmieňovať pseudohypoxický fenotyp a vznik FEO/ PGL, sú vyslovene zriedkavé [40,41].
Analýza klinických dát a genetických nálezov doteraz identifikovaných pacientov so syndrómom PGL viedla k rozpoznaniu vzťahu genotyp-fenotyp. Nositelia mutácií SDHD a SDHC majú pozitívnu rodinnú anamnézu častejšie (60 %) ako pacienti s mutáciami SDHB (30 %). Medián veku v čase diagnózy prvého nádoru pri SDHB a SDHD je 32, resp. 33 rokov oproti 38 rokom pri SDHC. Primárne nádory pri SDHD sú často viacpočetné (79 %). Naopak, 67 % pacientov s SDHB, resp. 73 % pacientov so SDHC má solitárne nádory. Malígny potenciál PGL/ FEO pri syndrómoch PGL 1 a PGL 4 stúpa v rade HNPGL < FEO < PGL [4]. Zaujímavá je spojitosť germinatívnych mutácií SDH s inými nádormi. Najznámejšou, iba nedávnou ozrejmenou, je dyáda PGL a GIST – Carney-Stratakisov syndróm (synonymum Carney-Stratakisova dyáda) [42,43], resp. Carneyho triáda, pri ktorej sú u pacientov navyše prítomné benígne pľúcne chondrómy [44]. Na rozdiel od sporadických GIST, je pri GIST manifestovaných v rámci Carneyho triády častá generalizácia lymfatickými cievami (asi 29 % vs ≤ 2 %) [45]. Vyšetrenie expresie D2-40 (podoplanínu) v nádorových bunkách GIST možno využiť na identifikáciu syndromologických GIST, pozitivita D2-40 v tkanive GIST môže navyše naviesť k aktívnemu pátraniu po PGL [45].
Vzhľadom na finančne nákladnú molekulárnu diagnostiku sú vypracované štatistické modely priority vyšetrovania kandidátnych génov v závislosti od klinického fenotypu. Prediktormi prítomnosti germinatívnych mutácií sú vek <45 rokov, mnohopočetný FEO, extraadrenálna lokalizácia a anamnéza paragangliómov hlavy a krku (head and neck paraganglioma – HNPGL).
Pri splnení minimálne 1 z týchto kritérií je indikované genetické vyšetrenie kandidátnych génov SDHx, RET a VHL podľa algoritmu uvedeného na obr. 1 [8,46]. Pri bilaterálnych a familiárnych adrenálnych FEO sa mutácie génov SDHx vyskytujú menej často ako mutácie génov RET a VHL. Naopak, prevalencia SDHx mutácií je vyššia pri sporadických extraadrenálnych nádoroch, malígnych nádoroch a u nádorov manifestovaných v detskom veku. Vo všetkých týchto prípadoch je vysoká pravdepodobnosť nálezu mutácií SDHB génu.
![Algoritmus genetického vyšetrenia pri zdanlivo nesyndrómovom FEO/PGL – podľa [8].](https://www.prelekara.sk/media/cache/resolve/media_object_image_large/media/image/c7f3133e52fe57a357849347268988bb.jpg)
Iné nádory vychádzajúce z drene nadobličiek
Z nervového tkaniva môžu okrem feochromocytómu a/ alebo paragangliómu vzniknúť aj neuroblastóm, ganglioneuroblastóm, ganglioneuróm, resp. zmiešané nádory. Základ zmiešaných nádorov tvorí feochromocytóm, ktorý dopĺňa neuroblastómová, ganglioneuroblastómová alebo ganglioneurómová zložka. Adrenálne ganglioneurómy sú vzácne, väčšinou ide o sporadické prípady, boli však publikované aj popisy familiárnych nádorov [7,47]. V rámci syndrómu MEN 2 boli popísané zmiešané nádory [48,49], ale aj čisté ganglioneurómy [50]. Preto je možné, že za určitých okolností môžu u pacientov s MEN 2 aktivačné mutácie RET-protoonkogénu postihnúť okrem chromafinných aj iné bunky odvodené od neurálnej lišty [50]. Adrenálne neuroblastómy predstavujú nádory vzniknuté z hniezd neurálnych buniek v dreni nadobličky. Vrodená predispozícia podmieňuje menej ako 5 % prípadov neuroblastómov a je heterogénna. Výskyt neuroblastómov v spojitosti s kongenitálnym centrálnym hypoventilačným syndrómom a s Hirschprungovou chorobou nasvedčuje tomu, že by mohlo ísť o poruchu génov angažovaných vo vývoji buniek odvodených od neurálnej lišty. Prvým identifikovaným génom predisponujúcim na vrodené neuroblastické nádory je gén PHOX2B (16p12 – 13) [51 – 56]. Mutácie génu PHOX2B narušujú homeodoménu PHOX2B proteínu. V nádoroch sa nedokázala alelická strata lokusu PHOX2B, onkogénny efekt týchto mutácií preto podmieňuje haploinsuficiencia, aktivačná mutácia alebo dominantne negatívny efekt. Podarilo sa lokalizovať ďalšie 2 lokusy predisponujúce na familiárny výskyt neroblastómov [57], konkrétne gény však zatiaľ identifikované nie sú.
prim. MUDr. Peter Vaňuga, PhD.
www.nedu.sk
e-mail: peter.vanuga@nedu.sk
Doručeno do redakce: 30. 9. 2010
Sources
1. Fränkel F. Ein Fall von doppelseitigem, völlig latent verlaufenen Nebennierentumor und gleichzeitiger Nephritis mit Veränderungen am Circulationsapparat und Retinitis. Arch Pathol Anat Physiol Klin Med 1886; 103 : 244 – 263.
2. Neumann HP, Vortmeyer A, Schmidt D et al. Evidence of MEN ‑ 2 in the original description of classic pheochromocytoma. N Engl J Med 2007; 357 : 1311 – 1315.
3. Karagiannis A, Mikhailidis DP, Athyros VG et al. Pheochromocytoma: an update on genetics and management. Endocr Relat Cancer 2007; 14 : 935 – 956.
4. Pasini B, Stratakis CA. SDH mutations in tumorigenesis and inherited endocrine tumours: lesson from the phaeochromocytoma ‑ paraganglioma syndromes. J Intern Med 2009; 266 : 19 – 42.
5. Erlic Z, Neumann HP. Familial pheochromocytoma. Hormones 2009; 8 : 29 – 38.
6. Koch CA, Pacak K, Chrousos GP. The molecular pathogenesis of hereditary and sporadic adrenocortical and adrenomedullary tumors. J Clin Endocrinol Metab 2002; 87 : 5367 – 5384.
7. Neumann HP, Bausch B, McWhinney SR et al. Germ‑line mutations in nonsyndromic pheochromocytoma. N Engl J Med 2002; 346 : 1459 – 1466.
8. Erlic Z, Rybicki L, Peczkowska M et al. Clinical predictors and algorithm for the genetic diagnosis of pheochromocytoma patients. Clin Cancer Res 2009; 15 : 6378 – 6385.
9. Dahia PL, Hao K, Rogus J et al. Novel pheochromocytoma susceptibility loci identified by integrative genomics. Cancer Res 2005; 65 : 9651 – 9658.
10. Opocher G, Schiavi F, Iacobone M et al. Familial nonsyndromic pheochromocytoma. Ann NY Acad Sci 2006; 1073 : 149 – 155.
11. Woodward ER, Maher ER. Von Hippel ‑ Lindau disease and endocrine tumour susceptibility. Endocr Relat Cancer 2006; 13 : 415 – 425.
12. Brauch H, Kishida T, Glavac D et al. Von Hippel ‑ Lindau (VHL) disease with pheochromocytoma in the Black Forest region of Germany: evidence for a founder effect. Hum Genet 1995; 95 : 551 – 556.
13. Eisenhofer G, Huynh TT, Pacak K et al. Distinct gene expression profiles in norepinephrine ‑ and epinephrine producing hereditary and sporadic pheochromocytomas: activation of hypoxia‑driven angiogenic pathways in von Hippel ‑ Lindau syndrome. Endocr Relat Cancer 2004; 11 : 897 – 911.
14. Langer P, Cupisti K, Bartsch DK et al. Adrenal involvement in multiple endocrine neoplasia type 1. World J Surg 2002; 26 : 891 – 896.
15. Woodward ER, Eng C, McMahon R et al. Genetic predisposition to phaeochromocytoma: analysis of candidate genes GDNF, RET and VHL. Hum Mol Genet 1997; 6 : 1051 – 1056.
16. Vanhorne JB, Andrew SD, Harrison KJ et al. A model for GFRα4 function and a potential modifying role in multiple endocrine neoplasia 2. Oncogene 2005; 24 : 1091 – 1097.
17. Plaza ‑ Menacho I, Burzynski GM, de Groot JW et al. Current concepts in RET‑related genetics, signalling and therapeutics. Trends Genet 2006; 22 : 627 – 636.
18. Lesueur F, Cebrian A, Robledo M et al. Polymorphisms in RET and its coreceptors and ligands as genetic modifiers of multiple endocrine neoplasia type 2A. Cancer Res 2006; 66 : 1177 – 1180.
19. Cerrato A, De Falco V, Santoro M. Molecular genetics of medullary thyroid carcinoma: the quest for novel therapeutic targets. J Mol Endocrinol 2009; 43 : 143 – 155.
20. Toledo RA, Wagner SM, Coutinho FL et al. High penetrance of pheochromocytoma associated with the novel C634Y/ Y791F double germline mutation in the RET protooncogene. J Clin Endocrinol Metab 2010; 95 : 1318 – 1327.
21. Agarwal SK, Mateo CM, Marx SJ. Rare germline mutations in cyclin‑dependent kinase inhibitor genes in multiple endocrine neoplasia type 1 and related states. J Clin Endocrinol Metab 2009; 94 : 1826 – 1834.
22. Pellegata NS, Quintanilla ‑ Martinez L, Siggelkow H et al. Germ‑line mutations in p27Kip1 cause a multiple endocrine neoplasia syndrome in rats and humans. Proc Natl Acad Sci USA 2006; 103 : 15558 – 15563.
23. Georgitsi M, Raitila A, Karhu A et al. Germline CDKN1B/ p27Kip1 mutation in multiple endocrine neoplasia. J Clin Endocrinol Metab 2007; 92 : 3321 – 3325.
24. Owens M, Stals K, Ellard S et al. Germline mutations in the CDKN1B gene encoding p27 Kip1 are a rare cause of multiple endocrine neoplasia type 1. Clin Endocrinol (Oxf) 2009; 70 : 499 – 500.
25. Igreja S, Chahal HS, Akker SA et al. Assessment of p27 (cyclin‑dependent kinase inhibitor 1B) and aryl hydrocarbon receptor ‑ interacting protein (AIP) genes in multiple endocrine neoplasia (MEN1) syndrome patients without any detectable MEN1 gene mutations. Clin Endocrinol (Oxf) 2009; 70 : 259 – 264.
26. Fritz A, Walch A, Piotrowska K et al. Recessive transmission of a multiple endocrine neoplasia syndrome in the rat. Cancer Res 2002; 62 : 3048 – 3051.
27. Franklin DS, Godfrey VL, O’Brien DA et al. Functional collaboration between different cyclin‑dependent kinase inhibitors suppresses tumor growth with distinct tissue specificity. Mol Cell Biol 2000; 20 : 6147 – 6158.
28. Molatore S, Kiermaier E, Jung CB et al. Characterization of a naturally ‑ occurring p27 mutation predisposing to multiple endocrine tumors. Mol Cancer 2010; 9 : 116.
29. Toledo RA, Mendonca BB, Fragoso MC et al. Isolated familial somatotropinoma: 11q13 - LOH and gene/ protein expression analysis suggests a possible involvement of AIP also in non‑pituitary tumorigenesis. Clinics 2010; 65 : 407 – 415.
30. Vogel KS, Brannan CI, Jenkins NA et al. Loss of neurofibromin results in neurotrophin‑independent survival of embryonic sensory and sympathetic neurons. Cell 1995; 82 : 733 – 742.
31. Bausch B, Koschker AC, Fassnacht M et al. Comprehensive mutation scanning of NF1 in apparently sporadic cases of pheochromocytoma. J Clin Endocrinol Metab 2006; 91 : 3478 – 3481.
32. Bausch B, Borozdin W, Mautner VF et al. Germline NF1 mutational spectra and loss ‑ of ‑ heterozygosity analyses in patients with pheochromocytoma and neurofibromatosis type 1. J Clin Endocrinol Metab 2007; 92 : 2784 – 2792.
33. Jenne DE, Tinschert S, Reimann H et al. Molecular characterization and gene content of breakpoint boundaries in patients with neurofibromatosis type 1 with 17q11.2 microdeletions. Am J Hum Genet 2001; 69 : 516 – 527.
34. Bouhanick B, Berry M, Hascouet S et al. Intestinal obstruction and pheochromocytoma in a patient suffering from von Recklinghausen’s disease. Clin Med Insights Endocrinol Diab 2009; 2 : 35 – 41.
35. Maertens O, Prenen H, Debiec ‑ Rychter M et al. Molecular pathogenesis of multiple gastrointestinal stromal tumors in NF1 patients. Hum Mol Genet 2006; 15 : 1015 – 1023.
36. Burnichon N, Brière JJ, Libé R et al. SDHA is a tumor suppressor gene causing paraganglioma. Hum Mol Genet 2010; 19 : 3011 – 3020.
37. Hensen EF, Jordanova ES, van Minderhout IJ et al. Somatic loss of maternal chromosome 11 causes parent ‑ of ‑ origin‑dependent inheritance in SDHD‑linked paraganglioma and phaeochromocytoma families. Oncogene 2004; 23 : 4076 – 4083.
38. Hao HX, Khalimonchuk O, Schraders M et al. SDH5, a gene required for flavination of succinate dehydrogenase, is mutated in paraganglioma. Science 2009; 325 : 1139 – 1142.
39. Ladroue CL, Carcenac R, Leporrier M et al. PHD2 mutation and congenital erythrocytosis with paraganglioma. N Engl J Med 2008; 359 : 2685 – 2692.
40. Gaal J, Burnichon N, Korpershoek E et al. Isocitrate dehydrogenase mutations are rare in pheochromocytomas and paragangliomas. J Clin Endocrinol Metab 2010; 95 : 1274 – 1278.
41. Yao L, Barontini M, Niederle B et al. Mutations of the metabolic genes IDH1, IDH2, and SDHAF2 are not major determinants of the pseudohypoxic phenotype of sporadic pheochromocytomas and paragangliomas. J Clin Endocrinol Metab 2010; 95 : 1469 – 1472.
42. McWhinney SR, Pasini B, Stratakis CA. International Carney Triad and Carney ‑ Stratakis Syndrome Consortium. Familial gastrointestinal stromal tumors and germ‑line mutations. N Engl J Med 2007; 357 : 1054 – 1056.
43. Pasini B, McWhinney SR, Bei T et al. Clinical and molecular genetics of patients with the Carney ‑ Stratakis syndrome and germline mutations of the genes coding for the succinate dehydrogenase subunits SDHB, SDHC, and SDHD. Eur J Hum Genet 2008; 16 : 79 – 88.
44. Matyakhina L, Bei TA, McWhinney SR et al. Genetics of carney triad: recurrent losses at chromosome 1 but lack of germline mutations in genes associated with paragangliomas and gastrointestinal stromal tumors. J Clin Endocrinol Metab 2007; 92 : 2938 – 2943.
45. Agaimy A, Carney JA. Lymphatics and D2 – 40/ podoplanin expression in gastrointestinal stromal tumours of the stomach with and without lymph node metastasis: an immunohistochemical study with special reference to the Carney triad. J Clin Pathol 2010; 63 : 229 – 234.
46. Neumann HP, Erlic Z, Boedeker CC et al. Clinical predictors for germline mutations in head and neck paraganglioma patients: cost reduction strategy in genetic diagnostic process as fall‑out. Cancer Res 2009; 69 : 3650 – 3656.
47. Koch CA, Vortmeyer AO, Zhuang Z et al. New insights into the genetics of familial chromaffin cell tumors. Ann NY Acad Sci 2002; 970 : 11 – 28.
48. Brady S, Lechan RM, Schwaitzberg SD et al. Composite pheochromocytoma/ ganglioneuroma of the adrenal gland associated with multiple endocrine neoplasia 2A: case report with immunohistochemical analysis. Am J Surg Pathol 1997; 21 : 102 – 108.
49. Matias ‑ Guiu X, Garrastazu MT. Composite phaeochromocytoma ‑ ganglioneuro-blastoma in a patient with multiple endocrine neoplasia type IIA. Histopathology 1998; 32 : 281 – 282.
50. Lora MS, Waguespack SG, Moley JF et al. Adrenal ganglioneuromas in children with multiple endocrine neoplasia type 2: a report of two cases. J Clin Endocrinol Metab 2005; 90 : 4383 – 4387.
51. Trochet D, Bourdeaut F, Janoueix ‑ Lerosey I et al. Germline mutations of the paired‑like homeobox 2B (PHOX2B) gene in neuroblastoma. Am J Hum Genet 2004; 74 : 761 – 764.
52. Mosse YP, Laudenslager M, Khazi D et al. Germline PHOX2B mutation in hereditary neuroblastoma. Am J Hum Genet 2004; 75 : 727 – 730.
53. van Limpt V, Schramm A, van Lakeman Aet al. The Phox2B homeobox gene is mutated in sporadic neuroblastomas. Oncogene 2004; 23 : 9280 – 9288.
54. Bourdeaut F, Trochet D, Janoueix ‑ Lerosey I et al. Germline mutations of the paired‑like homeobox 2B (PHOX2B) gene in neuroblastoma. Cancer Lett 2005; 228 : 51 – 58.
55. Perri P, Bachetti T, Longo L et al. PHOX2B mutations and genetic predisposition to neuroblastoma. Oncogene 2005; 24 : 3050 – 3053.
56. Serra A, Häberle B, König IR et al. Rare occurrence of PHOX2b mutations in sporadic neuroblastomas. J Pediatr Hematol Oncol 2008; 30 : 728 – 732.
57. Longo L, Panza E, Schena F. Genetic predisposition to familial neuroblastoma: identification of two novel genomic regions at 2p and 12p. Hum Hered 2007; 63 : 205 – 211.
Labels
Diabetology Endocrinology Internal medicineArticle was published in
Internal Medicine

2010 Issue 12
Most read in this issue
- Extrémně nízké hladiny SHBG jako důsledek polymorfizmu Pro156Leu v genu pro SHBG – kazuistiky dvou žen se syndromem polycystických ovarií
- Plicní forma histiocytózy z Langerhansových buněk – hodnocení aktivity nemoci a léčebné odpovědi pomocí PET‑CT (indexu SUVmax Pulmo/ SUVmax Hepar). Popis vlastních zkušeností a přehled literatury
- Aktivita osi hypotalamus- hypofýza- nadoblička u pacientov s reumatoidnou artritídou
- HIV lipodystrofie