
Imunoendokrinní vztahy u nadledvin
Immunoendocrine associations in adrenal glands
Immune and endocrine systems are basic regulatory mechanisms of organism and, including the nervous system, maintain the organism’s homeostasis. The main immune system representatives are mononuclear cells, T ‑ and B ‑ cells and their products, in the endocrine system the main representatives are cells of the glands with inner secretion and their products. One of the most important glands for maintaining homeostasis are adrenal glands. It has been proven that either cells of the immune system, either endocrine cells can, although in trace amounts, produce mutually mediators of both systems (hormones, cytokines). Disorders in one system can lead to pathological symptoms in the other system. Also here represent adrenals an important model.
Key words:
adrenal glands – Addison’s disease – lymphocytes – cytokines – glucocorticoids – autoantibodies – autoantigens
Authors:
I. Šterzl; P. Hrdá
Authors‘ workplace:
Endokrinologický ústav Praha, ředitel doc. MUDr. Vojtěch Hainer, CSc.
Published in:
Vnitř Lék 2010; 56(12): 1286-1291
Category:
Celebration
Overview
Imunitní a endokrinní systém jsou základními regulačními mechanizmy organizmu a spolu s nervovým systémem udržují homeostázu organizmu. Hlavními reprezentanty imunitního systému jsou mononukleární buňky, lymfocyty a jejich produkty, v endokrinním systému jsou to buňky žláz s vnitřní sekrecí a jejich produkty. Jednou z nejdůležitějších endokrinních žláz v udržení homeostázy jsou nadledviny. Prokázalo se, že jak buňky imunitního systému, tak endokrinní buňky dokážou, byť i ve stopových množství, produkovat vzájemně mediátory obou systémů (hormony, cytokiny). Poruchy funkce jednoho sytému mohou vést k patologickým projevům v systému druhém. I zde nadledviny sehrávají významný model.
Klíčová slova:
nadledviny – Addisonova choroba – lymfocyty – cytokiny – glukokortikoidy – autoprotilátky – autoantigeny
Obecný úvod
V 50. letech 19. století se jako jeden z prvních v moderní medicíně zabýval chorobami nadledvin dr. Thomas Addison [1]. Jeho pacienti se známkami selhání nadledvin měli nejčastěji tuberkulózu (TBC) a nádory nadledvin, ale ve svém souboru nalezl i některé, u kterých docházelo k atrofii nadledvin a které nazýval „idiopatické“. Dá se tedy říct, že je to poprvé, kdy bylo popsáno onemocnění nadledvin jako autoimunitní adrenalitida. Později v roce 1926 Schmidt pospal pacienty s lymfocytární infiltrací jak štítné žlázy, tak nadledvinové kůry [2]. Toto zjištění bylo mezi 30. a 50. lety minulého století následováno řadou publikací, které popisovaly lymfocytární infiltraci řady dalších orgánů, jako je štítná žláza, pankreas, hypofýza a játra. V roce 1964 Carpenter pozoroval, že na inzulinu závislý diabetes je také vázán na sníženou funkci nadledvin a štítné žlázy, tedy na již známý Schmithův syndrom [3]. V roce1926 byl tedy poprvé popsán autoimunitní polyglandulární syndrom (APS), který byl v roce 1980 Neufeldem a Blizzardem rozdělen na 2 typy, APS I. a II. typu [4]. V té době po ústupu tuberkulózy, která byla dříve hlavní příčinou Addisonovy choroby, se autoimunita stává hlavní příčinou poškození nadledvin.
Vliv imunitního systému na nadledviny
V 90. letech minulého století se prokázalo, že na funkci nadledvin a hladiny kortikoidů nepůsobí pouze poškození nadledvin, ale že nadledviny a produkce glukokortikoidů mohou být ovlivněny přímo imunitními regulačními mechanizmy, které cestou cytokinů, např. ve stresových situacích nebo při infekci, ovlivňují hypotalamo-hypofyzárně nadledvinovou osu. K porozumění vzájemného vlivu imunitního a endokrinního systému je nutno připomenout, že nejen endokrinní a neuroendokrinní systém produkuje hormony, neuropeptidy a cytokiny, které mají své receptory i na lymfoidních buňkách imunitního systému a jsou schopny ovlivnit jejich reakce na vnější stimuly, ale také že řada hormonů je produkována buňkami imunitního systému. Jde především o pro opio-melanokortikotrofní hormon (POMC), adrenokortikotropní hormon (ACTH), růstový hormon (GH), prolaktin, tyreostimulační hormon (TSH), luteinizační hormon (LH), folikuly stimulační hormon (FSH). Tato produkce hormonů imunokompetentními buňkami je oproti endokrinním buňkám pouze stopová, ale nabývá významu v cílových tkáních, kde se lymfocyty, např. při zánětu, hromadí a hladina hormonu se signifikantně zvýší [5].
Vliv glukokortikoidů (GK) na imunitní systém
Vliv GK na imunitní systém je znám již dříve než z poloviny minulého století. Steroidy účinkují v cílových buňkách především mechanizmem stimulace nebo inhibice genové exprese, a to od samého počátku, tedy od transkripce příslušného genu do mRNA. Tento účinek je zprostředkován nitrobuněčnými receptory, jejichž přítomnost je znakem cílových buněk pro tyto hormony. Mezi proteiny, jejichž exprese je pod kontrolou glukokortikoidů, patří enzymy, hormony a další regulační molekuly (faktory) a jejich receptory. Produkty exprese spolu mohou interagovat a jejich působení je navzájem propojeno. Trojrozměrná struktura receptorů pro steroidy (a rovněž pro tyreoidální hormony, patřící do téže „rodiny“ spolu s receptory pro látky skupiny vitaminu D a s deriváty kyseliny retinové), byla popsána v 2. polovině 80. let minulého století, včetně úplné charakteristiky příslušných genů. V roce 1924 Jaffe popsal hypertrofii thymu na zvířecím modelu po adrenalektomii. V 70. letech minulého století Basedovsky et al předložili představu, že imunosupresivní efekt GK patří do fyziologické regulace imunitního systému a ochraňuje ho před jeho nepřiměřenou reakcí vedoucí např. k autoimunitám. Tato teorie se postupně potvrzovala a ukázalo se, že GK mají také významný protizánětlivý efekt a nedostatek GK u infikovaných zvířat vedl ke zvýšení prozánětlivých cytokinů TNF α, INF γ a IL-1. Potvrdilo se tedy, že fyziologické hladiny GK ochraňují jedince před těžkým zánětem a jeho imunitní odpovědí [6].
Též bylo prokázáno, že imunosupresivní aktivita může být pozměněna např. exogenně přidávanými cytokiny, a v takovém případě GK působily synergicky. Příkladem je synergický efekt GK a IL-1 a IL-6 na lidské B lymfocyty, kde tato kombinace vedla k aktivaci produkce IgM a IgG. GK s IL-4 dokonce stimulovaly produkci IgE protilátek. Tento synergický efekt je velice důležitý např. v terapeutickém podávání GK pacientům s 1. typem imunopatologické reakce [6]. Obdobný synergizmus u GK jsme prokázali při vzájemném podání metabolitu DHEA při stimulaci dynamiky tvorby protilátek IgG a IgM v lymfocytární kultuře [7].
GK mají velký význam při aktivaci subpopulací TH1 a TH2 lymfocytů, kdy TH1 populace produkuje především IL-2, INF γ a TNF β, a tato buněčná linie se významně uplatňuje v buňkami zprostředkované imunitní odpovědi a oddáleného typu přecitlivělosti. TH2 subpopulace produkuje především IL-4, IL-5, IL-6, IL-10, IL-13, které aktivují proliferaci B lymfocytů a uplatňují se při humorální odpovědi a alergických reakcích. GK převážně aktivují TH1 odpověď a inhibují produkci TH2 cytokinů. GK směřují diferenciaci T-lymfocytů do TH2 subpopulace, inhibují produkci IL-2 a zvyšují produkci IL-4. GK inhibují produkci IL-12 a zdá se, že tato inhibice může být hlavním mechanizmem, jak GK vstupují do balance TH1 a TH2 odpovědi. Dlouhodobé působení může vést k indukci produkce IL-4 a následně i produkci i IgE periferními mononukleárními buňkami. To by mohlo vysvětlit i postupnou rezistenci alergických pacientů na terapii GK [6].
Proto v poslední době není pohled na působení GK zdaleka tak jasný, jak se zdálo v 70. letech 20. století a ukazuje se, že protizánětlivá a imunosupresivní aktivita GK je především závislá na dávce a délce jejich podávání. Na příkladu typu oddálené přecitlivělosti lze demonstrovat, že malé dávky GK ji stimulují, zatímco vysoké dávky ji suprimují. Obdobně je tomu při stresovém modelu, kdy krátký stres oddálený typ přecitlivělosti stimuluje, zatímco chronický stres ji inhibuje. Dá se tedy říci, že prozatím nevíme, jak endogenní GK modulují imunitní systém. Pravděpodobně působí mnohem složitějším způsobem nežli pouhou cestou suprese imunitního systému GK, kde by tato převážně inhibiční forma jejich působení mohla jedince spíše poškodit [6].
Primární nadledvinová nedostatečnost jako autoimunitní onemocnění
Primární nadledvinová nedostatečnost, Addisonova choroba, je vzácné onemocnění, ale současné epidemiologické studie ukazují narůstající prevalenci v rozvinutých zemích. Ta je odhadovaná mezi 110 a 140 případů/ 1 milion obyvatel. Incidence choroby je opisována kolem 4,7 – 6,2 případů/ 1 milion obyvatel ročně [8]. Až 99 % případů primární nadledvinové nedostatečnosti je v současnosti autoimunitního původu, zprostředkované poškozením buněk nadledvin imunocytoxickými lymfocyty. Detekce autoimunitního procesu nadledvinové nedostatečnosti je postaveno na přítomnosti cirkulujících protilátek proti kůře nadledvin a na průkazu normální nebo atrofické kůry nadledvin. K identifikaci autoimunitní podstaty choroby přispívají i další klinické (věk nástupu, další autoimunitní asociované choroby, hypogonadizmus, kandidóza) a genetické nálezy (mutace AIRE genu, HLA DR, CTLA 4) [9].
Autoimunitní Addisonova choroba se může vyskytovat v rámci 4 rozdílných klinických forem: izolovaná Addisonova choroba, autoimunitní polyglandulární syndrom (APS) I. typu, APS II. typu, APS IV. typu (tab. 1).
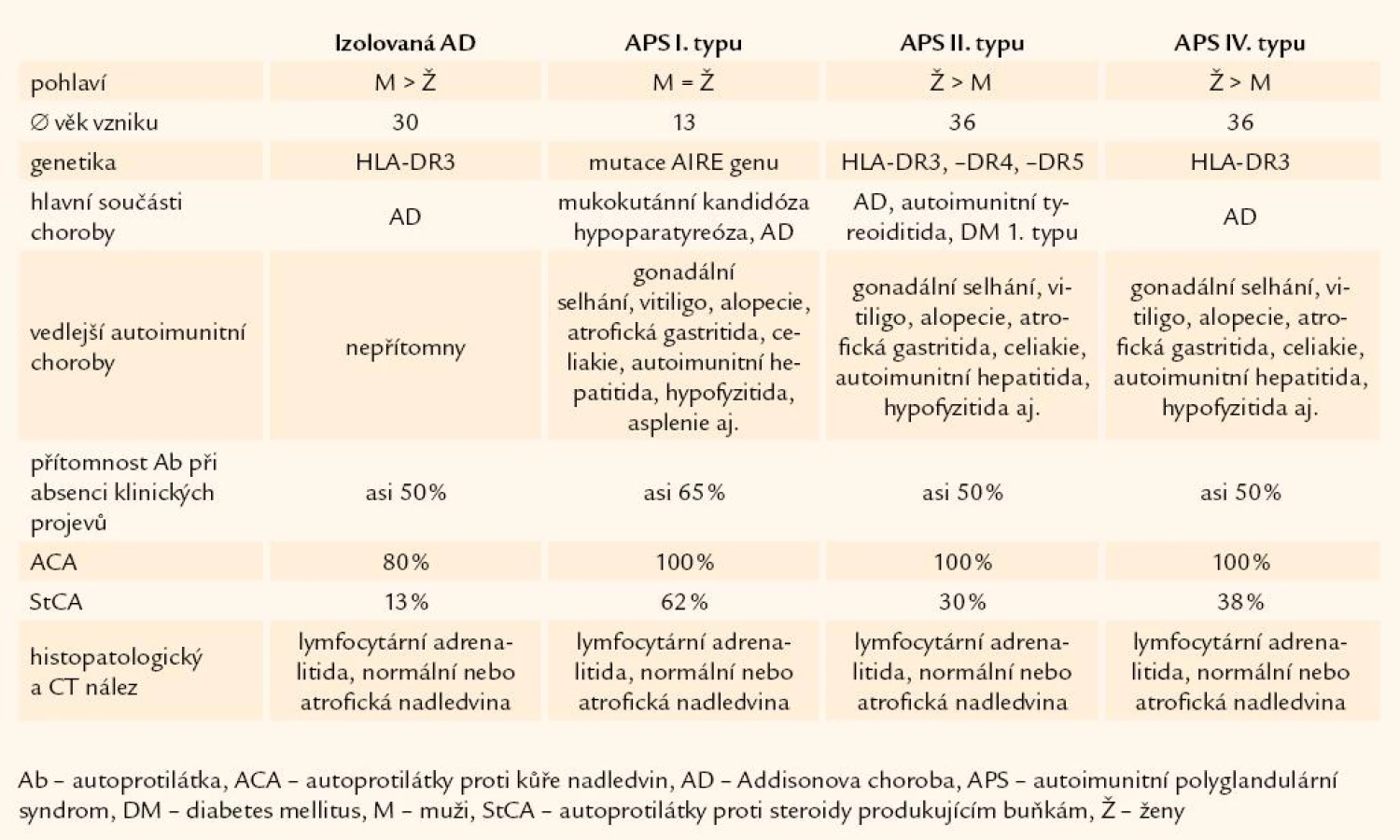
Etiopatogeneze autoimunitní nadledvinové nedostatečnosti
Autoimunitní onemocnění nadledvin má jako ostatní autoimunitní choroby multifaktoriální etiologii, s účastí faktorů vnitřních a zevních. Jak jsme již dříve informovali, při primárním poškození byla nalezena infiltrace lymfocytů, mononukleárních buněk, plazmatických buněk a makrofágů v kůře nadledvin a bylo prokázáno, že tyto buňky sehrávají významnou roli při poškození nadledvin. Jedná se o primární buněčné poškození buněk nadledvin cytotoxickými T-lymfocyty, k jejichž aktivaci je nezbytná aktivace především TH1 lymfocytů. Sled patogenních událostí, zapojených v autoimunitní destrukci kůry nadledvin, ale není stále zcela jasný. Autoprotilátky nalezené při této destrukci mají především diagnostický význam.
Příkladem eitopatogeneze Addisonovy choroby je APS I. typu, jehož je součástí. Toto onemocnění je monogenní, a proto mnohem lépe charakterizovatelné než polygenní onemocnění. Je způsobené mutací AIE genu. Genetická predispozice spojená s homozygotní mutací AIRE genu může být v případě APS I. typu podstatný faktor, který navozuje ztrátu tolerance k vlastním antigenům [10].
V případě autoimunitního postižení nadledvin, které není součástí APS I. typu, je genetická predispozice vázána především na HLA antigeny II. třídy, a to HLA-DR3, - DR4, - DR5. Tato predispozice je nezbytná, ale ne dostačující. K rozvoji choroby je potřeba i vliv zevních faktorům jako např. infekce, léky, vliv stravy nebo stres [11]. Genetické riziko pro autoimunitní postižení nadledvin je modulováno i polymorfizmem cytotoxického lymfocytárního antigenu 4 (CTLA4), i když výsledky některých prací nejsou ve shodě [12].
Autoprotilátky proti nadledvinovým antigenům ve většině případy nemají patogenní význam, slouží jako diagnostický a prognostický markér nemoci a jejich vymizení neznamená vyloučení pokračování nemoci. Autoprotilátky jsou většinou podtřídy IgG1 a způsobují antigen specifický na TH lymfocytech dependentní switching (přepnutí) tříd. Aktivované cytotoxické T-lymfocyty (TC lymfocyty) a aktivované makrofágy jsou rozhodujícími mediátory, zodpovědnými na destrukci nadledvinové tkáně. Tento proces je antigen specifický, řízení TH lymfocyty. T-lymfocyty pravděpodobně rozpoznávají vlastní antigeny kůry nadledvin ve vazbě s antigeny HLA II. třídy [13]. V periferní krvi pacientů s Addisonovou chorobou byla prokázána přítomnost 21 - OH specifických autoreaktivních T-lymfocytů [14]. Aktivované TH lymfocyty vedou k produkci řady cytokinů, především IL-2, které indukují aktivaci TC lymfocytů a B lymfocytů, schopných vytvářet specifické protilátky proti nadledvinám. Na rozdíl od tyreoidální autoimunity je sled událostí v případě nadledvinové autoimunity pouze hypotetický a nebyl doposud studován experimentálně [13].
Význam autoprotilátek proti nadledvinám
Přestože Addisonova choroba je zapříčiněna destrukcí tkáně cytotoxickými lymfocyty, je detekce toho cytotoxického na specifický autoantigen vázaného klonu velmi obtížná, v běžné laboratorní diagnostice téměř nemožná. Z toho důvodu je nutno využít detekci autoprotilátek, které samostatně i ve vzájemné kombinaci mohou diagnózu Addisonovy choroby přiblížit.
Autoprotilátky nalezené u autoimunitního postižení nadledvin jsou:
- autoprotilátky proti kůře nadledvin(ACA) nebo proti 21 - hydroxyláze (21 - OH),
- autoprotilátky proti steroidy produkujícím buňkám (StCA).
Cílovými autoantigeny jsou 3 enzymy z rodiny P450 cytochromů, 21 - OH, 17α-hydroxyláza (17αOH), cytochrom P450 scc (P450 scc), které jsou lokalizované v endoplazmatickém retikulu nebo mitochondriích. 21 - OH je nadledvinově specifický enzym, 17αOH je exprimována v nadledvinách a gonádách a P450 scc je enzym přítomný nadledvinách, gonádách a placentě. V kůře nadledvin jsou všechny 3 enzymy ubiquitní, nicméně 21 - OH a P450 scc jsou lokalizovány zejména v zona glomerulosa, fasciculata a reticularis, zatímco 17αOH v zona fasciculata a reticularis. Tyto enzymy jsou zapojeny do syntézy 4 hlavních steroidních hormonů, kortisolu, aldosteronu, androsteronu a dehydroepiandrosteronu. Patogenní role autoprotilátek na enzymatickou aktivitu těchto enzymů nebyla in vivo prokázána [13].
Autoprotilátky proti kůře nadledvin (ACA) proti 21 - OH
- U pacientů s klinicky vyjádřenou Addisonovou chorobou ACA reagují se všemi 3 vrstvami kůry nadledvin a v současnosti je nejužívanější metodou k jejich detekci nepřímá imunofluorescence (NIF) s užitím kryostatických řezů zvířecích nadledvin. V roce 1992 byla ve 3 nezávislých studiích identifikována jako hlavní nadledvinový autoantigen u pacientů s Addisonovou chorobou 21 - OH, s výjimkou APS I [15 – 17]. Reaktivita proti jiným enzymům steroideogeneze, jako je 11α-hydroxyláza, aromatáza a adrenodoxin, nebyla u pacientů s autoimunitní Addisonovou chorobou nalezena [18,19]. Stanovení ACA u pacientů s klinicky manifestovanou Addisonovou chorobou má význam k ozřejmění autoimunitního původu choroby. Pozitivita ACA u pacientů s autoimunitní Addisonovou chorobu má sklon přetrvávat i delší dobu od začátku choroby např. v porovnání s pozitivitou autoprotilátek proti ostrůvkovým antigenům u pacientů s diabetes mellitus (DM) 1. typu [13].
- U pacientů bez klinických projevů Addisonovy choroby výskyt ACA nevylučuje možné počínající subklinické poškození nadledvin. Případně mohou reprezentovat „bystanders“ efekt, který je využíván např. u izohormonální terapie. Nejvyšší prevalence je popisována u pacientů s idiopatickou hypoparatyreózou (48 %) a předčasným ovariálním vyhasnutím (9 %). ACA se mohou vyskytovat i u 4 % nejbližších příbuzných pacientů s Addisonovou chorobou. ACA jsou popisovány i u 4 % hospitalizovaných pacientů a u 0 – 0,6 % zdravé populace [20]. Význam pozitivity ACA u pacientů bez Addisonovy choroby není zatím zcela jasný. Část těchto ACA pozitivních pacientů má porušenou adrenokortikální rezervu během ACTH testu, nebo u nich k tomu později dojde [21,22]. V několika pracích se ukázalo, že někteří ACA pozitivní pacienti se subklinickým hypokortikalizmem se mohou stát ACA negativními s plnou obnovou funkce nadledvin po imunosupresivní léčbě pro aktivní endokrinní orbitopatii [23,24]. Autoprotilátky proti 21-OH jsou dobrým markerem možného vývoje Addisonovy choroby. Riziko progrese do nadledvinové nedostatečnosti závisí na titru autoprotilátek (vysoké titry), věku pacienta (mladší věk), genetickém pozadí (především HLA-B8 a HLA-DR3) a přítomnosti jiného orgánově specifického autoimunitního onemocnění, zejména přítomnost hypoparatyreózy nebo autoimunitní tyreoiditidy nebo DM 1. typu [13].
Na základě dlouhodobých sledování pacientů s autoimunitními endokrinopatiemi v Endokrinologickém ústavu jsme v minulosti prokázali, že současný výskyt protilátek proti dalším endokrinním orgánům – polyglandulární aktivace autoimunity (PAA) není jen náhodný laboratorní výskyt (laboratorní autoimunitní syndrom), ale že charakterizuje probíhající autoimunitní proces. Poškození adrenálních buněk vedoucí k subklinické formě projevu může být i u klinicky vyjádřené autoimunitní tyreoiditidy. Přítomnost obou autoprotilátek měla vliv na funkční odpověď nadledvin, a to na poměr bazální hladiny ACTH/ kortisol. Tato skupina pacientů měla statisticky významnou vazbu na chronickou únavu. Významným nálezem, který odlišoval PAA a APS II, se ukázala přítomnost autoprotilátek proti 21-OH, která je vázána s rozvojem Addisonovy choroby a je významným laboratorním prognostickým ukazatelem klinického rozvoje APS II. typu. U našich pacientů s PAA jsme pomocí imunblotu prokázali, že přítomné nadledvinové autoprotilátky jsou proti jiným antigenům, než je 21 - OH, jedná se o antigeny z oblasti 40 – 60 kDa [25].
Autoprotilátky proti steroidy produkujícím buňkám (StCA)
- U pacientů s Addisonovou chorobou a hyperandrogenním hypogonadizmem. Někteří pacienti mají autoprotilátky, které reagují s ostatními steroidy produkujícími buňkami (StCA), jako jsou Leydigovy buňky testes, thékální buňky ovarií, syncytiotrofoblast placenty. ACA i StCA reagují s kůrou nadledvin a vykazují identický imunofluorescenčí obraz, k rozlišení může sloužit užití tkáně nadledvin a pak gonád. ACA se mohou vyskytovat bez přítomnosti StCA, ale StCA jsou vždy asociovány s ACA [13]. Řada studií potvrdila vztah mezi StCA a předčasným ovariálním vyhasnutím u žen [26 – 28]. U mužů je výskyt StCA výjimečný, jsou li však přítomny, mohou být považovány za markér primární gonadální nedostatečnosti [6]. U pacientů s APS I. typu je výskyt StCA 60 – 80 %, u APS II. typu 25 – 40 % a u pacientů s izolovanou AD kolem 18 % [29]. Pomocí imunblotových technik se ukázalo, že StCA jsou namířeny zejména proti dvěma antigenům, a to 17αOH a P450 scc [13].
- U pacientů s Addisonovou chorobou bez klinicky vyjádřeného hyperandrogenního hypogonadizmu. StCA byly popsány asi u 10 – 43 % pacientů s autoimunitní Addisonovou chorobou bez gonadálního selhání. I v těchto případech je výskyt StCA asociován s 17αOH a P450 scc. Sledování StCA pozitivních pacientů s autoimunitní Addisonovou chorobou bez gonadálního selhání ukazuje vysoké riziko vzniku selhání gonadálních funkcí u žen, ale ne u mužů [13].
- U pacientů s klinicky vyjádřeným hyperandrogenním hypogonadizmem bez Addisonovy choroby. Část pacientek s předčasným ovariálním vyhasnutím bez Addisonovy choroby má jiné autoimunitní onemocnění, převážně v subklinické formě. Nejčastější je tyreoidální autoimunita, následuje gastroparietální autoimunita, DM 1. typu a myasthenia gravis. U těchto pacientek se StCA nacházejí s nízkou frekvencí okolo 7 % a zároveň se u nich vyskytují ACA. Riziko vzniku Addisonovy choroby je v těchto případech vysoké [30].
Předčasné ovariální vyhasnutí na podkladě lymfocytární ooforitidy je úzce spjato s výskytem ACA a StCA a k poškození ovarií dochází pomocí cytotoxických mechanizmů spojených s T-lymfocyty. Patogeneze ovariálního selhání u pacientek s předčasným ovariálním vyhasnutím bez přítomnosti StCA a chromozomálních abnormalit zůstává nejasná [31].
Závěr
Nadledviny sehrávají významnou roli v udržení homeostázy v lidském organizmu. Jedním z nejvýznamnějších systémů podílejících se na udržení homeostázy je imunitní systém, a proto není divu, že jejich funkce se navzájem tolik prolínají. To nejen na fyziologické úrovni, kdy imunitní systém může ovlivnit funkci nadledvin za řady fyziologických stavů, jako je stres, stárnutí, vývoj, tak i za patologických stavů, kde především v poslední době po ústupu těžkých infekcí typu TBC, se stává nejvýznamnější etiopatogenetickou příčinou poškození nadledvin. Produkty nadledvin, zvláště GK, na druhé straně významně ovlivňují imunitní reakce i průběh imunopatologických stavů. GK prodělaly již několik vln oslav a zatracení, přesto je to jeden z nejvýznamnějších léků imunopatologických onemocnění (alergií). Jeho schéma působení v rámci řady synergií (IL-12), antagonizmů (DHEA) a jeho role stále ještě velkou výzvou.
Autoimunitní poškození nadledvin je zprostředkováno buněčnou imunopatologií, ale pro detekci onemocnění laboratorně jsou pro nás nenahraditelné autoprotilátky. Detekce autoprotilátek napomáhá v běžné praxi odhalit etiologii nadledvinové nedostatečnosti, identifikuje pacienty se subklinickým postižením nadledvin a umožňuje časnou léčbu a prevenci.
Poděkování
Chceme velice poděkovat prof. Stárkovi, že vedl a vybudoval problematiku steroidních hormonů a nadledvin a zapsal náš ústav nesmazatelným písmem do vědy v endokrinologii a vědy obecně. Jsme rádi, že jsme se i této oblasti za jeho vedení přiblížili a tuto část endokrinologie stále považujeme za nesmírně zajímavou a perspektivní.
doc. MUDr. Ivan Šterzl, CSc.
www.endo.cz
e-mail: isterzl@endo.cz
Doručeno do redakce: 4. 10. 2010
Sources
1. Addison T. On the constitutional and local effects of disease of the suprarenal capsules. In a collection of the published writings of the late Thomas Addison, MD, physician to Guy’s Hospital, London. London: New Sydenham Society 1986 (reprinted in Medical Classics 1937, 2, 244 – 293).
2. Schmidt MB. Eine biglandulate Eokankung (Nebennieren ind Schildruse) bei Morbus Addisonii. Verh Dtsch Ges Pathol 1926; 21 : 212 – 221.
3. Carpenter C, Solomon N, Silverberg S et al. Schmidt’s syndrome (thyroid and adrenal insufficiency). A review of the literature and a report of 15 new cases including 10 instances of co ‑ existend diabetes mellitus. Medicien (Baltimore) 1964; 43 : 153 – 180.
4. Neufeld M, Blizzard RM. Polyglandular autoimmune disease. In: Pinchera A, Doniach D, Fenzi GF et al (eds). Symposium on Autoimmune Aspects of Endocrine Disordes. New York: Acadenic Press 1980 : 357 – 365.
5. Šterzl I. Přehledná imunoendokrinologie. Praha: Maxdorf 2006 : 39 – 41.
6. Šterzl I. Přehledná imunoendokrinologie. Praha: Maxdorf 2006 : 48 – 50.
7. Šterzl I, Hampl R, Sterzl J et al. 7Beta‑OH ‑ DHEA counteracts dexamethasone induced suppression of primary immune response in murine spleenocytes. J Steroid Biochem Mol Biol 1999; 71 : 133 – 137.
8. Coco G, Dal Pra C, Presotto F et al. Estimated risk for developing autoimmune Addison’s disease in patients with adrenal cortex autoantibodies. J Clin Endocrinol Metab 2006; 91 : 1637 – 1645.
9. Betterle C, Dal Pra C, Mantero F et al. Autoimmune adrenal insufficiency and autoimmune polyendocrine syndromes: autoantibodies, autoantigens, and their applicability in diagnosis and disease prediction. Endocr Rev 2002; 23 : 327 – 364.
10. Kahaly GJ. Polyglandular autoimmune syndromes. Eur J Endocrinol 2009; 161 : 11 – 20.
12. Erichsen MM, Løvås K, Skinningsrud B et al. Clinical, immunological, and genetic features of autoimmune primary adrenal insufficiency: observations from a Norwegian registry. J Clin Endocrinol Metab 2009; 94 : 4882 – 4890.
12. Brozzetti A, Marzotti S, Tortoioli C et al. Cytotoxic T-lymphocyte antigen ‑ 4 Ala17 polymorphism is a genetic marker of autoimmune adrenal insufficiency: Italian association study and meta‑analysis of European studies. Eur J Endocrinol 2010; 162 : 361 – 369.
13. Betterle C, Dal Pra C, Mantero F et al. Autoimmune adrenal insufficiency and autoimmune polyendocrine syndromes: autoantibodies, autoantigens, and their applicability in diagnosis and disease prediction. Endocr Rev 2002; 23 : 327 – 364.
14. Bratland E, Skinningsrud B, Undlien DE et al. T cell responses to steroid cytochrome P450 21 ‑ hydroxylase in patients with autoimmune primary adrenal insufficiency. J Clin Endocrinol Metab 2009; 94 : 5117 – 5124.
15. Winqvist O, Karlsson FA, Kämpe O. 21 ‑ Hydroxylase, a major autoantigen in idiopathic Addison’s disease. Lancet 1992; 339 : 1559 – 1562.
16. Bednarek J, Furmaniak J, Wedlock N et al. Steroid 21 ‑ hydroxylase is a major autoantigen involved in adult onset autoimmune Addison’s disease. FEBS Lett 1992; 309 : 51 – 55.
17. Baumann‑Antczak A, Wedlock N, Bednarek J et al. Autoimmune Addison’s disease and 21 ‑ hydroxylase. Lancet 1992; 340 : 429 – 430.
18. Song YH, Connor EL, Muir A et al. Autoantibody epitope mapping of the 21 ‑ hydroxylase antigen in autoimmune Addison’s disease. J Clin Endocrinol Metab 1994; 78 : 1108 – 1112.
19. Peterson P, Uibo R, Peränen J et al. Immunoprecipitation of steroidogenic enzyme autoantigens with autoimmune polyglandular syndrome type I (APS I) sera; further evidence for independent humoral immunity to P450c17 and P450c21. Clin Exp Immunol 1997; 107 : 335 – 340.
20. Betterle C, Coco G, Zanchetta R. Adrenal cortex autoantibodies in subjects with normal adrenal function. Best Pract Res Clin Endocrinol Metab 2005; 19 : 85 – 99.
21. Scherbaum WA, Berg PA. Development of adrenocortical failure in non‑Addisonian patients with antibodies to adrenal cortex. A clinical follow‑up study. Clin Endocrinol (Oxf) 1982; 16 : 345 – 352.
22. Ketchum CH, Riley WJ, Maclaren NK. Adrenal dysfunction in asymptomatic patients with adrenocortical autoantibodies. J Clin Endocrinol Metab 1984; 58 : 1166 – 1170.
23. Laureti S, De Bellis A, Muccitelli VI et al. Levels of adrenocortical autoantibodies correlate with the degree of adrenal dysfunction in subjects with preclinical Addison’s disease. J Clin Endocrinol Metab 1998; 83 : 3507 – 3511.
24. De Bellis AA, Falorni A, Laureti S et al. Time course of 21 ‑ hydroxylase antibodies and long‑term remission of subclinical autoimmune adrenalitis after corticosteroid therapy: case report. J Clin Endocrinol Metab 2001; 86 : 675 – 678.
25. Šterzl I, Hrdá P, Matucha P et al. Polyglandulární aktivace autoimunity jako projev subklinických endokrinopatií. Čas Lék Čes 2007; 146 : 256 – 261.
26. Weetman AP. Autoimmunity to steroid ‑ producing cells and familial polyendocrine autoimmunity. Baillieres Clin Endocrinol Metab 1995; 9 : 157 – 174.
27. Chen S, Sawicka J, Betterle C et al. Autoantibodies to steroidogenic enzymes in autoimmune polyglandular syndrome, Addison’s disease, and premature ovarian failure. J Clin Endocrinol Metab 1996; 81 : 1871 – 1876.
28. Betterle C, Volpato M, Greggio AN et al. Type 2 polyglandular autoimmune disease (Schmidt’s syndrome). J Pediatr Endocrinol Metab 1996; 9 (Suppl 1): 113 – 123.
29. Bottazo GF, Mirakian R, Drexhage HA. Adrenalitis, oophoritis and autoimmune polyglandular disease. In: Rich RR, Fleisher TA, Schwarz DB et al (eds). Clinical immunology, principles and practice. St Louis: Mosby 1996 : 1523 – 1536.
30. Dal Pra C, Chen S, Furmaniak J et al. Autoantibodies to steroidogenic enzymes in patients with premature ovarian failure with and without Addison’s disease. Eur J Endocrinol 2003; 148 : 565 – 570.
31. Hoek A, Schoemaker J, Drexhage HA. Premature ovarian failure and ovarian autoimmunity. Endocr Rev 1997; 18 : 107 – 134.
Labels
Diabetology Endocrinology Internal medicineArticle was published in
Internal Medicine

2010 Issue 12
Most read in this issue
- Extrémně nízké hladiny SHBG jako důsledek polymorfizmu Pro156Leu v genu pro SHBG – kazuistiky dvou žen se syndromem polycystických ovarií
- Plicní forma histiocytózy z Langerhansových buněk – hodnocení aktivity nemoci a léčebné odpovědi pomocí PET‑CT (indexu SUVmax Pulmo/ SUVmax Hepar). Popis vlastních zkušeností a přehled literatury
- Aktivita osi hypotalamus- hypofýza- nadoblička u pacientov s reumatoidnou artritídou
- HIV lipodystrofie