
Odlišné průběhy recidivující anebo multisystémové formy histiocytózy z Langerhansových buněk u dospělých osob – popis 22 případů z jednoho pracoviště
Different courses of recurrent or multisystem Langerhans cells histiocytosis in adults – description of 22 cases from one centre
Since its establishment in 1990, a total of 22 patients with confirmed Langerhans cell histiocytosis (LCH) have been monitored and treated at the Clinic of Internal Medicine Haemato-Oncology in Brno. In 5 patients, the disease was diagnosed in childhood and 2 of these 5 patients had late neurodegenerative changes in the CNS with a typical picture on MR and a typical PET-CT imaging fluorodeoxyglucose hypometabolism in the cerebellar area. In 5 patients from the cohort of 22, the disease had unifocal form, dominant in the area of skeleton with no recurrence after the treatment. However, in 12 patients, the disease affected a number of organs simultaneously (multifocal form of LCH). The aim of the description below is to characterise the monitored cohort of 22 patients and describe the very different courses of multifocal forms of LCH in 12 patients.
Key words:
Langerhans cell histiocytosis – diabetes insipidus – panhypopituitarizm – 2-chlorodeoxyadenosine – high-dose chemotherapy
:
Z. Adam 1; M. Krejčí 1; L. Pour 1; P. Szturz 1; J. Neubauer 2; T. Nebeský 2; Z. Řehák 3; R. Koukalová 3; J. Mayer 1; J. Vorlíček 1
:
Interní hematoonkologická klinika Lékařské fakulty MU a FN Brno, pracoviště Bohunice, přednosta prof. MU Dr. Jiří Vorlíček, CSc. 2Radiologická klinika Lékařské fakulty MU a FN Brno, pracoviště Bohunice, přednosta prof. MU Dr. Vlastimil A. Válek, CSc. 3Odd
1
:
Vnitř Lék 2010; 56(6): 542-556
:
65th Birthday - Petr Svacina, MD
Od založení Interní hematoonkologické kliniky v Brně v roce 1990 bylo na tomto pracovišti sledováno a léčeno celkem 22 pacientů s prokázanou histiocytózou z Langerhansových buněk (LCH). Nemoc byla u 5 z nich diagnostikována již v dětském věku, přičemž u 2 z těchto 5 pacientů se objevily pozdní neurodegenerativní změny v CNS s typickým obrazem na MR a typickým hypometabolizmem fluorodeoxyglukózy v oblasti mozečku při PET‑CT vyšetření. U 5 dospělých z 22členého kolektivu měla nemoc unifokální formu postižení, dominantně v oblasti skeletu, bez další recidivy po léčbě. U 12 osob však tato nemoc postihovala více orgánů současně (multifokální forma LCH). Cílem následujícího popisu je charakterizovat sledovaný soubor 22 pacientů a popsat velmi odlišné průběhy multifokálních forem LCH u 12 dospělých osob.
Klíčová slova:
histiocytóza z Langerhansových buněk – diabetes insipidus – panhypopituitarizmus – 2‑chlordeoxyadenosin – vysokodávkovaná chemoterapie
Úvod
Histiocytóza z Langerhansových buněk (LCH) je nozologická jednotka o velmi širokém spektru forem a závažnosti klinických projevů. Některé formy spontánně regredují, jiné opakovaně recidivují a některé formy jsou léčbou velmi špatně ovlivnitelné a vedou ke smrti nemocného. Projevy LCH u dospělých počínají náhodným RTG nálezem jednoho osteolytického ložiska, ale mohou mít podobu i generalizované systémové choroby. Údaje o incidenci u dospělých v ČR nemáme, němečtí autoři uvádějí 1 – 2 případy/ 1 milion obyvatel [1,2]. V České republice není incidence této nemoci známa, nicméně o existenci této choroby svědčí četné popisy jednotlivých případů [1 – 9].
Dle rozšíření nemoci se popisuje:
- lokalizované postižení – u pacienta je nalezeno pouze jedno solitární ložisko, např. jedno izolované kostní ložisko
- multifokální postižení – v rámci jednoho orgánu či tkáně je více ložisek této nemoci, např. více kostních ložisek
- multisystémové postižení – nemoc postihuje více orgánů či tkání (např. kosti, kůži a plíce současně)
Rozsah postižení je velmi úzce spojen s prognózou. V rámci klinických studií bylo zjištěno, že existují některé „rizikové orgány“, při jejichž postižení je prognóza velmi nepříznivá a často dochází k úmrtí. To vedlo k vytvoření 3 prognostických skupin pro multifokální či multisystémové formy této nemoci.
- Do skupiny s vysokým rizikem spadají pacienti s multisystémovým postižením, u nichž nemoc postihuje také jeden či více z následujících rizikových orgánů: kostní dřeň, slezinu, játra a plíce.
- Do skupiny se středním rizikem spadají pacienti s multisystémovými projevy nemoci, ale bez postižení takzvaných „rizikových orgánů“.
- Do třetí skupiny spadají pacienti s multifokálním postižením skeletu. Tato skupina má nejpříznivější prognózu.
Dále byly popsány kostní lokalizace, při jejichž postižení se zvyšuje riziko následujícího postižení CNS touto nemocí. Jedná se o postižení očnice, mastoidea, klínové kosti, jařmového oblouku, horní čelisti, paranazálních dutin a také postižení přední či střední jámy lební s intrakraniální propagací nádorových mas.
Z hlediska četnosti postižení orgánů jsou v literatuře divergentní údaje, pro ilustraci uvádíme pouze výsledky analýzy provedené Baumgartnerem [2]: v 80 % bývají postiženy kosti, v 60 % kůže, ve 33 % játra, slezina a uzliny, ve 30 % kostní dřeň, ve 25 % plíce, ve 25 % orbita, ve 20 % zevní zvukovod a přiléhající okolí a také orodentální oblast, kde nemoc může zapříčinit uvolňování zubů. Ale také může být infiltrována stopka hypofýzy, což se projeví jako diabetes insipidus.
U dospělých pacientů má onemocnění velmi různorodý průběh. U některých postihne jenom jedno ložisko a po léčbě se již neobjeví, u jiných má recidivující charakter [9 – 13] a případně infaustní průběh.
Cílem následujícího článku je velmi stručně informovat o souboru pacientů sledovaných na našem pracovišti a v diskuzi pak rozvést možné projevy této nemoci. Nemoc patří do skupiny krevních chorob dle WHO klasifikace z roku 2010, projevy nemoci jsou však natolik různorodé, že spadají téměř do všech specializací medicíny, proto je důležitý multioborový přístup k těmto nemocným.
Popisy případů
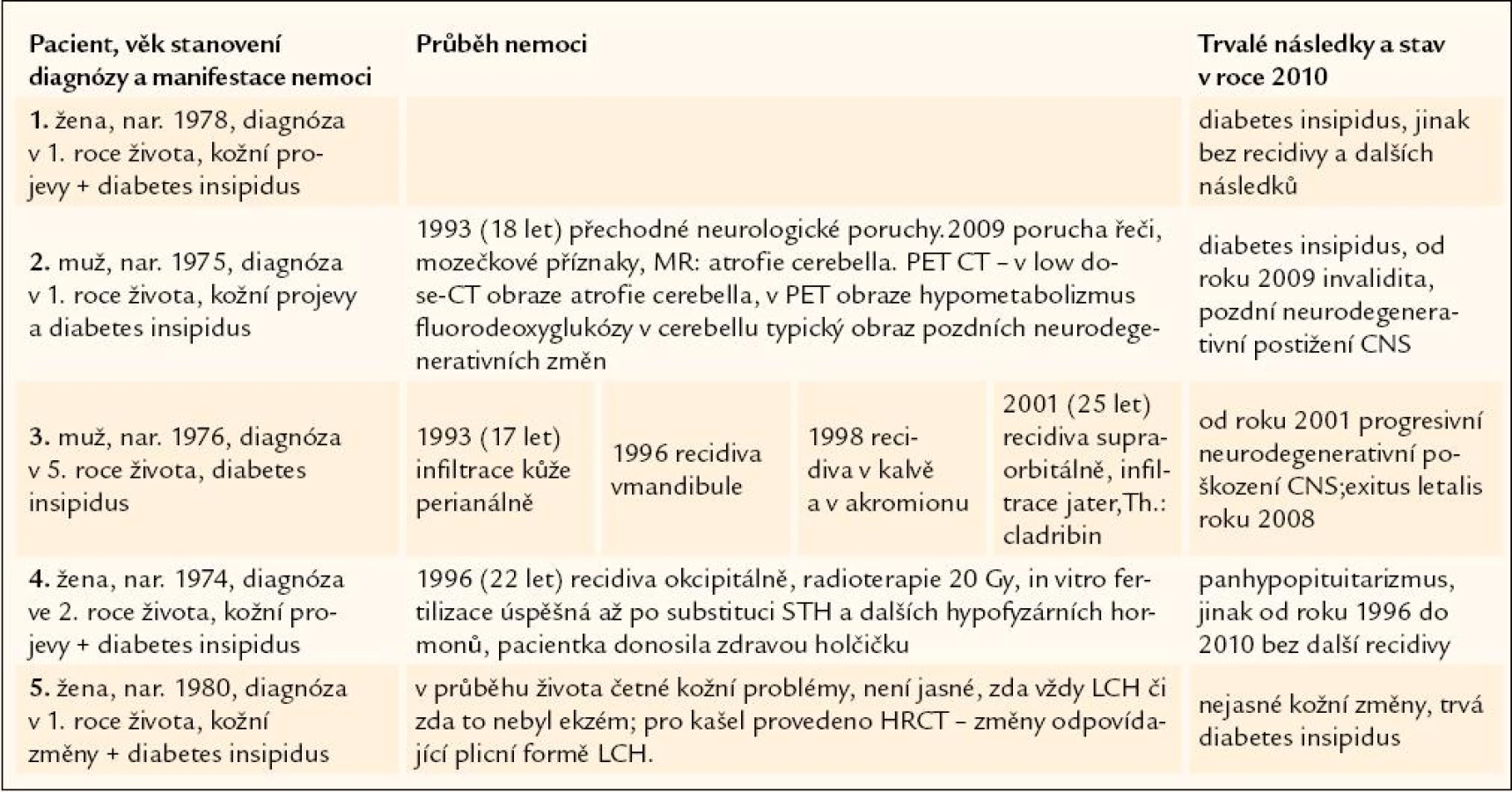
Dospělé osoby s LCH diagnostikovanou v dětském věku
Na našem pracovišti jsme sledovali za uvedené období celkem 5 pacientů s diagnózou stanovenou v dětském věku. Pouze jedna pacientka z nich je zcela bez známky recidivy této nemoci. Další pacientka měla jenom jednu izolovanou recidivu v kalvě, ale jinak z mládí přetrvává diabetes insipidus. Tato nemocná se pokoušela několikrát marně o in vitro fertilizaci. Ta byla úspěšná až po endokrinologickém přešetření, které prokázalo kompletní panhypopituitarizmus, a tedy i nutnost substituce somatotropního hormonu pro úspěšné provedení in vitro fertilizace. Třetí nemocná s LCH diagnózou stanovenou v dětství má zřejmě kožní recidivu a je podezření i na plicní recidivu.
U 2 pacientů s diagnózou stanovenou v dětství jsme pozorovali v dospělosti pozdní následky nemoci – atrofii cerebella s dysartrií a poruchou hybnosti. Prvními příznaky této vzácné komplikace jsou porucha řeči a hybnosti. U jednoho z nich byla tato porucha diagnostikována v roce 2002, od té doby se progresivně zhoršovaly funkce CNS, pacient byl upoután na lůžko a v roce 2008 roce zemřel. U druhého byla tato pozdní neurologická komplikace diagnostikována v roce 2009 a zatím vedla pouze k tomu, že pacient, dříve práce schopný, se stal trvale práce neschopným. Pro tuto pozdní formu je typický jak obraz při MR zobrazení, tak obraz při PET‑CT zobrazení. PET‑CT umí v low dose znázornit atrofické tkáně a v PET obraze je zde typický hypometabolizmus fluorodeoxyglukózy.
Osudy 5 nemocných s diagnózou LCH stanovenou v dětském věku shrnuje tab. 1.
Jednoložisková forma LCH s první manifestací v dospělém věku, bez dalších recidiv
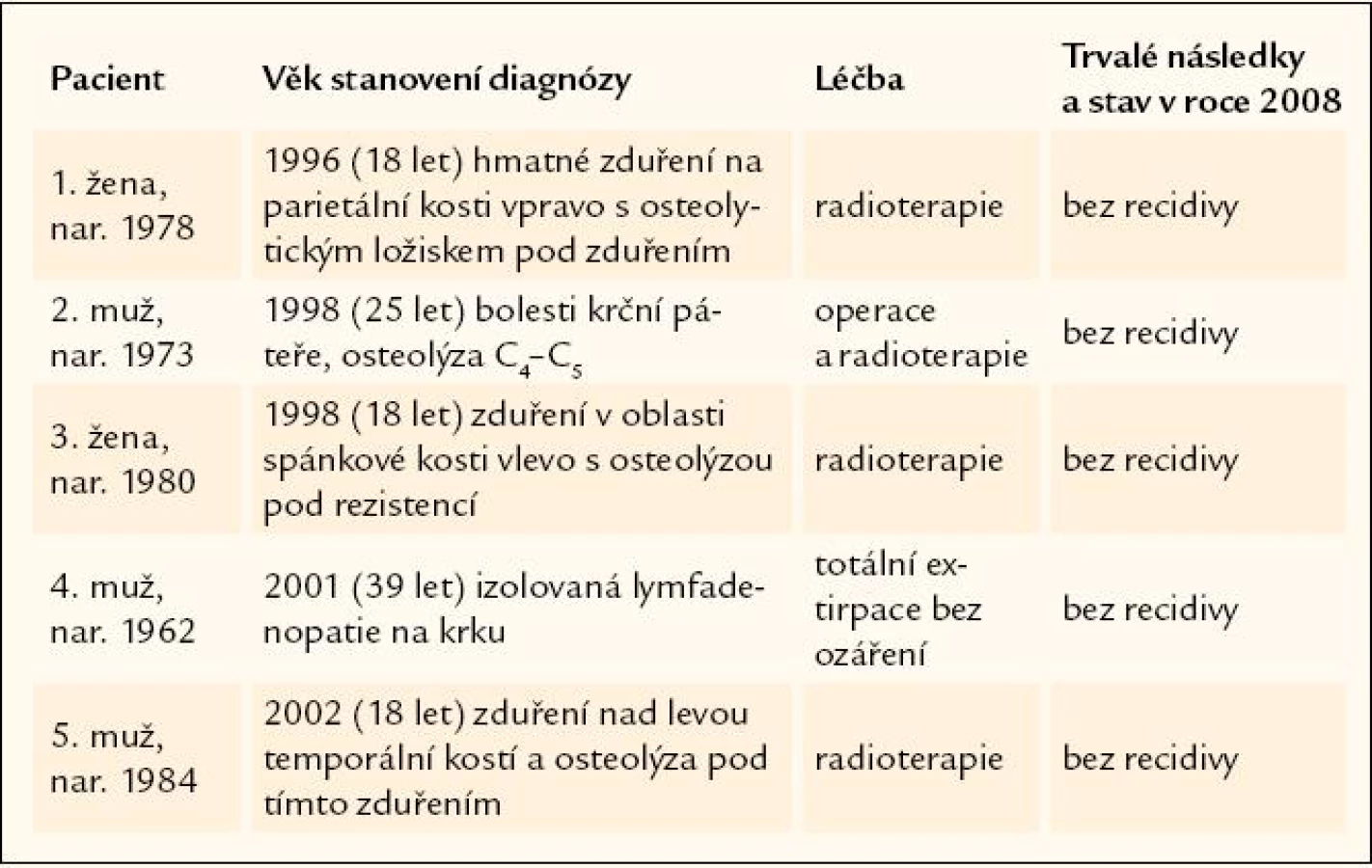
Celkem 5 pacientů je u nás sledováno se jednoložiskovou formou nemoci, která vícekrát nerecidivovala.
Z těchto 5 osob se jednalo v prvním případně o izolovaný proces, poškozující krční páteř, v druhém případě o izolované postižení jedné uzliny na krku, bez dalších ložisek nemoci. Ve 3 případech šlo o postižení jednoho ložiska v oblasti kalvy, které bylo léčeno zářením, a nemoc již nikdy více nerecidivovala. Ve stručnosti je průběhu shrnut v tab. 2.
Nemocní s první manifestací LCH v dospělém věku a s opakovanými recidivami nemoci anebo s multisystémovým postižením
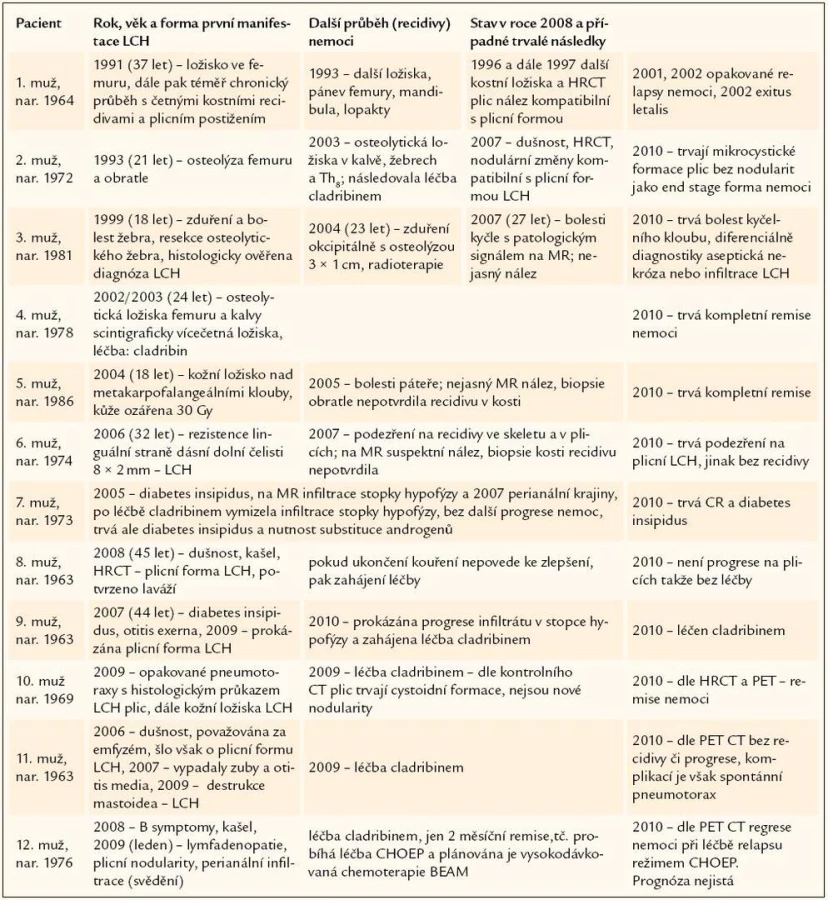
Celkem 12 pacientů mělo vícesystémové postižení LCH anebo formu nemoci, která postupně recidivovala v dalších oblastech. Pro přehlednost jsme jejich osudy znázornili v tab. 3. Průběh onemocnění u každého je zcela osobitý, liší se formou postižení organizmu i mírou agresivity nemoci. Proto považujeme za edukačně přínosné uvést podrobněji průběh nemoci u jednotlivých osob.
Pacient 1 – recidivující multifokální postižení kostí a plic
Velmi komplikovaný průběh měla LCH u muže narozeného v roce 1964. Tento mladý muž neměl do svých 37 let závažnější onemocnění. V roce 1991 (ve 37 letech věku) bylo nalezeno osteolytické ložisko ve femuru – řešeno ortopedicky, histologicky ověřena LCH. Po operaci následovala radioterapie. Za 2 roky, v roce 1993, byla RTG zjištěna další ložiska v oblasti pánve a obou femurů. Scintigrafie kostí popsala ještě ložiska v mandibule a pravé lopatce. Ložiska v pánvi a v L5 byla ověřena CT zobrazením.
U této víceložiskové formy jsme se rozhodli pro podání 7 cyklů chemoterapie CHOP (adriamycin, vinkristin, cyklofosfamid a prednison). První remise trvala 3 roky.
V březnu roku 1996 byla zjištěna nová ložiska v oblasti žeber a Th páteře – řešeno zářením a podáváním vinblastinu 1krát týdně po dobu 6 týdnů.
V červenci roku 1997 byla zjištěna opět nová ložiska na paži v hlavici humeru a v.s. i další ložiska v páteři. Zároveň nově se objevil neproduktivní dráždivý kašel, námahová dušnost, bolesti na hrudníku. Proto bylo provedeno vyšetření plic metodou HRCT, výsledek byl kompatibilní s plicní formou histiocytózy, ale nebyl dále ověřen. Následovala chemoterapie (etoposid a prednison) a radioterapie na oblast humeru. Vzhledem k častým recidivám a neuspokojivému výsledku běžné chemoterapie byl v září roku 1997 proveden sběr kmenových krvetvorných buněk a následně podána první vysokodávkovaná chemoterapie (melfalan a etoposid) s podporou transplantace autologních krvetvorných buněk. Remise po této léčbě trvala jenom 2,5 roku, do června roku 2001, kdy byla scintigraficky zjištěna nová ložiska. Následovaly tedy opět 3 cykly chemoterapie (etoposid + dexametason) a ozáření těchto ložisek. Po krátkém klidovém období se v červenci roku 2002 objevila ložiska na žebrech a páteři, zřetelná nejen na scintigrafii, ale i na RTG snímku. Pro tento relaps byla opět provedena stimulace s opakovaným sběrem kmenových krvetvorných buněk z periferní krve a následně podána druhá vysokodávkovaná chemoterapie.
Po této 2. vysokodávkované chemoterapii měla remise jen krátké trvání, několik měsíců, a opět se objevovala další kostní ložiska, takže dále pokračovala jen paliativní perorální chemoterapie melfalanem. V únoru roku 2002 byl pacient s aktivním onemocněním naposledy na naší ambulanci a v roce 2002 zemřel.
Pacient 2 – recidivující víceložiskové postižení kostí a později i plic
V roce 1993 (21 let) vznikla u pacienta narozeného roku 1972 první ataka LCH, která se projevila bolestí v pravé stehenní kosti a dále v obratli Th12. Osteolytická ložiska byla operována, čímž byla zjištěna diagnóza a léčena operačně alogeními štěpy. Pak byl pacient pouze sledován bez další léčby.
V roce 2003, po 10 letech, se nově objevila bolest žeber, páteře a kalvy. Na RTG snímcích bylo zjištěno osteolytické ložisko v kalvě, které bylo potvrzeno CT zobrazením, průměr ložiska byl 21 mm. Na pravé straně hrudníku bylo velké osteolytické ložisko v 7. žebru a velmi suspektní nález byl také na 6., 5. a 4. žebru. Na páteři byla nápadná nerovná dolní krycí ploténka Th8 a byla zřetelná přestavba krčku pravého femuru. V těle L4 CT prokázalo 3 osteolytická ložiska, největší 15 × 14 × 9 mm a další ložiska byla i v těle obratle L5. Zobrazení páteře pomocí MR potvrdilo ložiska také v obratlích Th11, L2, L4, L5. Vzhledem k desetiletému klidovému intervalu bylo resekováno 7. žebro. Histologické vyšetření prokázalo recidivu LCH.
Protože zobrazovací metody (RTG, scintigrafie skeletu, CT a MR) prokázaly mnohočetné osteolytické postižení skeletu, přistoupili jsme k systémové chemoterapii. Pacient dostal 5 cyklů chemoterapie (2‑chlordeoxyadenosin 5 mg/ m2 i.v. 1. – 5. den). Léčba byla ukončena v listopadu roku 2003.
Kontrolní PET vyšetření bylo v prosinci roku 2005 negativní a MR páteře neprokázalo žádnou progresi, jsou však stále zřetelná původní ložiska, remise trvá déle než 2 roky. V roce 2007 při kontrole nespecifické potíže, dušnost a bolesti v oblasti hrudního koše. Zobrazovací vyšetření nepopsala progresi ve skeletu. Pro dušnost bylo provedeno HRCT vyšetření, na němž byly popsány cystické okrsky různé velikosti, místy splývající, dále četné nodularity i fibrózní proužky. Tento nález je kompatibilní s nálezem plicní formy histiocytózy z Langerhansových buněk. Mladý muž podstoupil také bronchoskopii s bronchoalveolární laváží, v níž převažovaly makrofágy. Nebylo však provedeno cytochemické vyšetření přítomnosti buněk exprimujících CD1 antigen a protein S100, bez nichž se nemůže hodnotící vyjádřit k přítomnosti či nepřítomnosti plicní formy LCH.
Mladému muži jsme doporučili nekouřit, protože plicní forma LCH vzniká velmi často v souvislosti s kouřením. V červenci roku 2009 provedeno HCTC plic vyšetření se závěrem: diskrétní nodularity a trvání mnohočetných plicních cyst beze změn. Funkční plicní vyšetření bylo také v červnu roku 2009 beze změny. V lednu roku 2010 proběhla kontrolní PET‑CT. V low‑dose CT obraze jsou zřetelné mnohočetné tenkostěnné cysty, ale nejsou přítomna nodulární ložiska v plicním parenchymu. V PET obraze nebylo zřetelné žádné ložisko zvýšeného vychytávání fluorodexyglukózy.
Dle těchto vyšetření soudíme, že počátkem roku 2010 není LCH aktivní ani v oblasti plic, ani jinde v těle. V březnu roku 2010 se pacient objevil s novým problémem – svědění perianální oblasti, a tak v současnosti řešíme otázku, zda je to způsobené infiltrací kůže LCH, či ne.
Pacient 3 – recidivující postižení kostí
V případě muže narozeného roku 1981 se první příznak objevil v 18. roce života (1999): bolest a rezistence v oblasti 8. žebra. Žebro bylo totálně resekováno a histologicky stanovena diagnóza unifokální LCH. Další ložisko nebylo v té době patrné. Po 5 letech, v roce 2004, přišel s bolestmi v oblasti temene, kde si nahmatal rezistenci. Na RTG snímku kalvy měl neostré mapovité projasnění o velikosti 3 × 1 cm, odpovídající kostní formě nemoci. Následovala radioterapie, cílená na kostní ložisko. Při kontrole v dubnu roku 2007 měl mladý nejasný nález na MR zobrazení pravého kyčelního kloubu – obraz patologického signálu při okraji hlavice femuru vpravo s malým výpotkem v kloubu. Při zobrazení metodou PET byla jen lehce zvýšená aktivita v oblasti pravého kyčelního kloubu.
Kontrolní MR kyčelních kloubů bylo provedeno 18. 9. 2008 s nálezem již pokročilého vývoje aseptické nekrózy hlavice femuru vpravo.
Zda proces v oblasti hlavice byl opravdu jenom aseptickou nekrózou nebo souvisel s LCH, přesně nevíme, protože nebyla dělána histologie tohoto procesu. Nicméně bolesti v pravém kyčelním kloubu trvají stále i počátkem roku 2010. Zřejmě bez odběru histologie nebude možné přesně určit, zda se jedná opravdu o aseptickou nekrózu nebo zda je to projev LCH. Pro přetrvávající bolesti v oblasti pravého kyčelního kloubu budou další kontroly metodou RTG, MR a PET‑CT.
Pacient 4 – mnohočetné postižení kostí s intrakraniální expanzí z kostního ložiska
Stejně tak mladý muž, narozený roku 1978, neměl v předchozím životě žádné vážné onemocnění, až v letech 2002/ 2003 (24 let) bylo nalezeno osteolytické postižení femuru vlevo. V lednu roku 2003 byla provedena v nemocnici Na Bulovce v Praze exkochleace a zpevnění hřebem. Histologie ložiska: eozinofilní granulom. Již asi od roku 2002 si povšiml rezistencí (bulek) na hlavě. Největší byla v okcipitální krajině a zvětšovala se. Způsobovala bolest hlavy a posléze i poruchy zraku. Z chirurgicky odstraněných hmot opět histologicky vyšel eozinofilní granulom (leden roku 2003). Rozsah kostního postižení: na RTG snímku lebky osteolytické ložisko okcipitálně o průměru 2 cm. V levé lopatě kyčelní bylo ložisko 8 × 4 cm. Celotělová MIBI scintigrafie: vyšší aktivita v oblasti pánve, další ložisko v oblasti levé orbity, pokračující do os temporale.
Scintigrafie skeletu: ložiska v kalvě, frontoparietálně a okcipitálně, další ložisko v kosti spánkové vpravo, velké ložisko v lopatě kosti kyčelní vlevo. Dále ložiska v pravém femuru. Vyšší aktivita také v oblasti kolenního a talokrurálního kloubu vpravo.
CT kalvy a mozku: defekty v okcipitální oblasti oboustranně.
MR mozku: extraaxiální expanzivní proces oboustranně parasagitálně okcipitálně, šířící se defektem kalvy epidurálně až do oblasti zadní jámy lební, bez infiltrace dury. Suspektní další kostní ložiska frontoparietálně vpravo a parietálně vlevo.
Vzhledem k mnohočetným kostním ložiskům, která však u tohoto nemocného měla již i mimokostní šíření, byl proveden sběr periferních kmenových buněk po předchozí stimulační chemoterapii (etoposid a cyklofosfamid). Pak následovaly 4 cykly chemoterapie – 2‑chlordeoxyadenosin 5 mg/ m2 i.v. inf. 1. – 5. den v měsíčních intervalech. Léčba byla ukončena v srpnu roku 2003.
Po ukončení léčby při kontrolním vyšetření bylo nalezeno: MR – jen velmi drobné reziduum měkkotkáňové expanze. V rozsahu původního defektu kalvy okcipitálně nyní jen pruh středního signálu v rozsahu diploe, bez známek sycení či expanzivních projevů, mající charakter původního infiltrátu, nebo regresivních změn v místě původního infiltrátu, což nelze na MR odlišit. Kontrolní MIBI skeletu – je patrná difuzní kumulace aktivity v páteři, náznakem v pánvi. Nejsou však patrná původní ložiska v kalvě, zřetelná při prvním MIBI vyšetření. PET po léčbě – 12. 2. 2007 bez ložisek aktivity nemoci, PET vyšetření před léčbou bohužel nebylo provedeno. Remise trvá (leden roku 2010 s negativním nálezem na PET‑CT).
Pacient 5 – izolované kožní ložisko a suspektní kostní postižení
S primárně kožním projevem LCH přišel muž, narozený v roce 1986. Tento muž žádné vážnější onemocnění dříve neprodělal. V roce 2004 (18 let) se objevilo nejasné kožní ložisko v oblasti metakarpofalangeálních kloubů. Morfa byla excidována a histolog morfu popsal jako dermatofibrom s příměsí Langerhansových buněk. Elektronová mikroskopie potvrdila Birbeckova granula, takže diagnóza z tohoto ložiska byla nakonec uzavřena jako kožní forma LCH. Následovalo vyšetření s cílem potvrdit či vyloučit generalizaci nemoci (zobrazení skeletu, CT břicha). Místo původního kožního defektu bylo i s bezpečnostním lemem ozářeno dávkou 30 Gy.
V roce 2005 se objevily bolesti bederní páteře. RTG snímek neprokázal patologii, zato MR bederní páteře prokázalo difuzní změny kostní dřeně, které jsou jednoznačně abnormálním nálezem. Pro podezření na nepoznanou kostní formu histiocytózy provedli ortopedové cílenou biopsii kosti s odběrem materiálu na histologii. Histologie však byla nekonkluzivní, LCH nebyla potvrzena. PET byl před biopsií negativní. Vzhledem k mladému věku nemocného je pravděpodobné, že změny v oblasti páteře jsou následkem předchozí ataky kostní formy této nemoci a nekonkluzivostí histologického odběru lze mimo jiné vysvětlit tím, že se jednalo o staré ložisko již ve formě jizvení, kdy převládá zánětlivá celulizace starých LCH ložisek v páteři. Další sledování ukáže, zda opravdu šlo o jedno kožní ložisko, nebo zda časem vzniknou další nová kostní ložiska.
Pacient 6 – izolovaný infiltrát v oblasti dásní a podezření a plicní postižení
Muž, narozený v roce 1974, byl dříve bez vážnějších nemocí, v roce 2006 se mu objevila rezistence v oblasti dásní, na linguální straně ( – 7) velikosti asi 8 × 2 mm. Provedena resekce a zjištěna histiocytóza z Langerhansových buněk. Exstirpace provedena do zdravé tkáně, takže dále nebylo pokračováno v léčbě. V roce 2007 přichází s nespecifickými problémy. Jednak měl zduření a bolest pod sternomanubriálním skloubením. Scintigraficky zde byla aktivita a MR zobrazení prokázalo patologický signál, stejně tak patologický signál byl z několika dalších obratlů. Biopsie sterna a její histologické hodnocení nepřinesly objasnění, stejně jako HRCT plic nepřineslo nic patologického, pouze funkční testy prokázaly zvýšenou bronchokonstrikci. Předpokládáme, že v tomto případě teprve další průběh nemoci ukáže, zda se jednalo jednoložiskové postižení, či zda bude docházet k recidivám, nebo zda první dechové potíže signalizovaly riziko plicní formy LCH.
Pacient 7 – postižení stopky hypofýzy a kůže v perianální krajině
Primární postižení CNS v dospělosti je velmi výjimečné. Setkali jsme se s ním u mladého muže narozeného roku 1972, který si poprvé v létě roku 2005 všiml, že více močí, v listopadu roku 2007 byla internisty uzavřena diagnóza diabetes insipidus. První CT vyšetření v roce 2005 nepopsalo nic patologického, až MR mozku (2006) prokázalo rozšíření stopky hypofýzy. S tímto nálezem byl sledován bez dalšího zákroku. Etiologie infiltrátu v oblasti stopky hypofýzy nebyla jasná a bez poškození nemocného nebyla ověřitelná. V době vzniku diabetes insipidus začaly také bolestivé morfy na hýždích, přesněji řešeno v perianální oblasti. Byly hodnoceny jako anální fisury. Z původních fisur však vyrostl květákový útvar velikosti 4 cm zaujímající celé anální okolí. Makroskopicky to bylo hodnoceno jako akuminátní kondylomy. Excize z této morfy byla provedena v březnu roku 2007 – histiocytóza z Langerhansových buněk – a s touto diagnózou byl poslán k nám.
Byla provedena vyšetření s cílem zjistit přesně rozsah nemoci – vyjma popsaného nebyla jiná postižení prokázána. Mimo jiné provedeno FDG - PET 14. 4.2007, které v oblasti CNS nezachytilo aktivitu odpovídající maligní tkáni, zatímco v oblasti perianální bylo jasné solitární ložisko zvýšeného vychytávání fluorodexyglukózy. (Pozn.: Fluorodeoxyglukóza ve stavu nalačno proniká fyziologicky do mozku, zatímco do ostatních fyziologických tkání neproniká, proniká však to těch patologických tkání, které se vymykají regulačním dějům organizmu, tedy do zánětlivých ložisek a od nádorových ložisek, proto je PET‑CT optimální metodou k detekci nádorových či zánětlivých ložisek).
Pacient dostal 4 cykly chemoterapie 2‑chlordeoxyadenosinem, po níž se morfa v perianální krajině zmenšila na 50 %. Léčbu dokončila radioterapie 20 Gy. Při kontrolním vyšetření v květnu roku 2008 je anální krajina bez patologického nálezu, mladý muž může opět vychutnávat jízdu na kole. Kontrolní MR vyšetření zobrazilo již normální stopku hypofýzy. Ta byla původně rozšířena na 4,5 mm a nyní jsou její rozměry v mezích normy – 1,5 mm. Uvedené 4 cykly 2‑chlordeoxyadenosinu vedly k vymizení infiltrátu v oblasti stopky hypofýzy, ale sekrece antidiuretického hormonu se neobnovila. Poškození této oblasti je zřejmě nevratné.
Při zahájení léčby si nemocný stěžoval na viklavost zubů, stav byl stomatologem hodnocen jako náhle vzniklá a výrazná parodontóza v levém dolním kvadrantu, hodnoceno jako paradontitis chronica. Tělo mandibuly bylo bez patologie. Nebyla odebrána žádná histologie. Takže není jasné, zda to byla koincidence nebo zda to byly projevy nemoci v oblasti čelisti, jak jsou popisovány v literatuře a jak je uvádíme v diskuzi.
Poslední kontrolní PET‑CT vyšetření bylo provedeno v březnu roku 2010 – bez patologického obrazu těla v CT obraze a bez patologického vychytávání fluorodeoxyglukózy.
Pacient 8 – izolované postižení plicního parenchymu
Pacient, narozený v roce 1963, byl rela-tivně zdráv až do března roku 2008.Pro bronchitidu byl přeléčen antibio-tiky a pro přetrvávající kašel odeslán naplicní oddělení, kde po RTG snímku plic provedli následně HRCT a diagnos-tikovali mikronodulární rozsev do 5 mmbilaterálně. Následně byla provedena u tohoto nemocného bronchoalveo-lární laváž. Z cytologického vyšetření získané tekutiny bylo konstatováno, že jsou přítomny s velkou převahou makrofágy a téměř všechny elementy jsou protein S 100 pozitivní, CD1 pozitivní, což svědčí pro Langerhansovy buňky.
Tímto vyšetření byla potvrzena dia-gnóza. Pacient, kuřák, přestal po vysvětlení kouřit a po několika měsících by měl mít kontrolní vyšetření, po němž se dle vývoje rozhodne, zda nemoc vyžaduje či nevyžaduje medikamentózní léčbu. V červenci roku 2008 provedeno PET‑CT, kde nebylo ani na CT, ani na PET nic patologického popsáno a podobný nález byl v únoru roku 2010 – bez jakéhokoliv nového suspektního ložiska a na plicích byly patrné pouze drobné cysty, nikoliv však nodularity. Domníváme se, že u tohoto nemocného aktivita plicní formy nemoci vyhasla díky tomu, že pacient sám přestal kouřit a pečlivě se vyhýbá místnostem, které jsou zamořeny toxickým cigaretovým kouřem.
Pacient 9 – postižení stopky hypofýzy, plic a velmi pravděpodobně také zevních zvukovodů
Pacient, narozený roku 1974, kuřák, byl zdráv, až do prosince roku 2007, kdy u něj byla prokázán diabetes insipidus. Při vyšetření na endokrinologii byl zjištěn deficit dalších hormonů, dominantně androgenů. Při vyšetření mozku byla zjištěna rozšířená stopka hypofýzy s patologickým infiltrátem dle MR zobrazení. Při opakovaném probírání anamnézy jsme zjistili, že od roku 2007 se také datuje chronický zánět v oblasti obou zvukovodů. Byl vyšetřen na ORL pracovišti, kde sice neprokázali diagnózu LCH, ale také mu neudělali histologické ani cytologické vyšetření, byl vyšetřen jen klinicky. Dalším problémem bylo svědění kůže v oblasti perianální.
V rámci diferenciální diagnostiky patologické infiltrace v oblasti hypofýzy byl odeslán v únoru roku 2009 na naše pracoviště. Protože odběr vzorku z oblasti stopky hypofýzy pacientům spíše ublíží než prospívá, tedy velmi často nepřináší diagnózu a zákrok sám o sobě způsobuje komplikace, rozhodli jsme se hledat extrakraniální projevy té nemoci, která způsobila infiltraci stopky hypofýzy. Pacient byl vyšetřen metodou PET‑CT vyšetření (březen roku 2009) s následujícím výsledkem: v oblasti plicního parenchymu byly prokázány četné drobné cystické útvary různého tvaru, velikosti 0,8 – 0,9 cm, některé s poměrně dobře vyznačenou stěnou. Současně ojedinělé menší acinární opacity, nodularity a okrsky zesíleného intersticia. Pleurální prostor bez výpotku. V mediastinu nebyly zvětšené uzliny. V PET obraze byla jedině zvýšená kumulace radioaktivní glukózy v oblasti nazofaryngu, ale jen hraniční. Výsledkem PET‑CT vyšetření bylo tedy jednoznačný průkaz patologických změn plicního parenchymu.
To bylo potvrzeno HCRT vyšetřením plicního parenchymu, které přineslo obraz mnohočetných kavitovaných uzlů, respektive silnostěnných dutin s centrilobulární distribucí v relativně typické lokalitě horních laloků a proximálních segmentů laloků dolních. Obraz byl typický pro plicní formu LCH.
U pacienta byla proto v dubnu roku 2009 provedena bronchoalveolární laváž s průkazem 10 % CD1 pozitivních elementů s obsahujících protein S 100. Tento nález odpovídal plicnímu postižení LCH. Funkční plicní z 29. 10. 2009 bez patologických změn.
V prosinci roku 2009 bylo provedeno kontrolní MR mozku. Při srovnání s předchozími MR z března roku 2008 a prosince roku 2008 je patrné, že stále chybí typický hyperintenzivní signál neurohypofýzy. Adenohypofýza má normální velikost bez zřetelného ložiska. Bobulovitě rozšířené infundibulum se mírně oproti předchozí kontrole zvětšilo o 1 mm z 4,5 na 5,5 mm. Výraznější je i sycení ložiska postkontrastně a jeho demarkace vůči proximální části stopky. Jako vedlejší nález popsáno drobné necharakteristické ložisko 3 mm pareventrikulárně vlevo, které dříve nebylo.
Při stanovení diagnózy na jaře roku 2009 jsme vzhledem k přání pacienta založit rodinu odložili zahájení léčbu až do známek progrese nemoci. Po zjištění dalšího progrese infiltrátu v CNS jsme v březnu roku 2010 zahájili léčbu 2‑chlordeoxyadenosinem.
Pacient 10 – pneumotorax jako první příznak nemoci
Pacient, narozený roku 1969, neměl v anamnéze žádné vážnější nemoci. První pneumotorax jej překvapil v roce 2008 v březnu a od té doby byl sledován na plicní klinice. Pneumotorax s nutností léčebné intervence se celkem 3krát opakoval. Teprve po 3. spontánním pneumotoraxu bylo rozhodnuto o provedení biopsie plic. Ta na základně histologického hodnocení prokázala LCH.
V červenci roku 2008 byla na dermatologickém pracovišti provedena kožní excise ze kštice pro makulopapulozní vyrážku. Překvapením byl průkaz kožní formy LCH. Vstupní HRCT plic z května roku 2009 popisuje oboustranně v plicních křídlech četné splývané dutiny se zesílenou stěnou 1 – 2 mm a septy. Maximum je v horních lalocích pod ventrální stěnou hrudníku a dále difuzně v dolních lalocích. Buly dosahují velikosti až 2 – 3 cm v periferii a pod interlobiem mají splývající charakter a vytváření obraz hnízd. Největší dutiny dosahují velikosti 5 cm. Mediastinum není rozšířené a uzliny nejsou zvětšené.
20. května 2009 bylo provedeno PET‑CT s nálezem mnohočetným bul s maximem v horních a středních polích. Radioaktivní glukóza se kumulovala mírně jedině v oblasti infrahyoidních svalů.
Funkční plicní vyšetření: středně těžká restrikční ventilační porucha (inspirační vitální kapacita 2,51 = 47 % náležití hodnoty a TLC – total lung capacity 5,15 = 68 % náležité hodnoty).
V tomto případně jsme měli před sebou pacienta s postižením 2 orgánů, kůže a plic.
Léčba byla proto zahájena v dubnu roku 2009 2 - chloradenosin (5 mg/ m2) v kombinaci s cyklofosfamidem (300 mg celková dávka) a dexametasonem 20 mg i.v. Celkem podáno VI cyklů léčby, poslední v září roku 2009. V průběhu léčby ustoupily kožní projevy této nemoci. Dle kontrolního PET‑CT provedeného v březnu roku 2010 je bez nových ložisek nemoci. V plicním parenchymu přetrvávají mnohočetné cystoidní formace, při srovnání s předchozími obrazy je patrné splývaní drobnějších bul ve větší formace, nebyl však popsán výsev nových nodularit.
Pacient 11 – postižení plic, dásní, zevního zvukovodu a později mastoidea
Prvním problémem nemocného, narozeného roku 1963, byla dušnost, a to taková dušnosti, že vedla k hospitalizaci na interním oddělení v roce 2006. Nějakou dobu před tím mu začaly vypadávat zuby, příčina nebyla rozpoznána.
Výsledek diagnostického vyšetření byl závěr, který neodpovídal pravdě – rozedma plic. A z toho plynuly jedině kontroly u místních plicních specialistů. Asi od počátku roku 2008 se přidalo mokvání a výtok z ucha. Byl opakovaně ošetřován na ORL ambulanci. Následně byl předán na ORL kliniku, kde mu udělali CT vyšetření, nalezli rozsáhlou destrukci pyramid vlevo.
Vlevo bylo zastření zevního zvukovodu s destrukcí jeho dorzální stěny v rozsahu kolem 8 mm v.s. s rozpadovou dutinkou v procesus mastoideus velikosti 2 cm. Porušeno bylo kostní ohraničení kraniálního části svislého úseku canalis nervi facialis. Kostní ohraničení struktur vnitřního ucha včetně polokulovitých kanálků se zdálo být zachovalé. Rozrušena byla i dorzální stěna středouší, ventrální kortika procesus mastoideus a v krátkém úseku i přední hrana pyramidy v sousedství Eustachovy trubice. Stručně řečeno rozsáhlý destruktivní proces levé pyramidy.
ORL lékaři tak stáli před otázkou: Je to zánět, nebo tumor? Odpověď na tuto otázkou přineslo histologické vyšetření resekátu patologických tkání, které prokázalo LCH. S touto diagnózou byl poslán na naše pracoviště.
V rámci pátrání po rozsahu nemoci provedena na jaře roku 2009 následující vyšetření:
HRCT plic – difuzní postižení plicního parenchymu – mnohočetné silnostěnné formace oboustranně velikosti 3 – 80 mm, maximální změny dorzobazálně, kde cysty splývají a je zřetelná voštinovitá struktura plic s podílem fibrózních změn. Dále jsou patrné drobné nodularity v plicním parenchymu a subpleurálně a zřetelné zesílení nástěnné plesury dorzobazálně bilaterálně. Na scintigrafii skeletu byla aktivita jedině v oblasti levého procesus mastoideus.
PET‑CT vyšetření potvrdilo patologické změny plicního parenchymu, hraniční konzumaci glukózy v oblasti levého středního ucha – v defektu po antromastoidektomii.
Kontrolní PET‑CT vyšetření bylo provedeno v srpnu roku 2009 a již na něm nebyla aktivita v oblasti levého ucha a ubylo nodularit v plicním parenchymu.
Spirometrické vyšetření: restrikční porucha mírného stupně, inspirační vitální kapacita byla 3,66 = 70 % náležité hodnoty, TLC – total lung capacity 4 – 65 % náležití hodnoty.
Léčba: Vzhledem k postižení dvou orgánů – plic a ORL – oblasti použita radioterapie na oblasti levého ucha v dávce 20 Gy a celková chemoterapie 2‑chlordeoxyadenosinem v monoterapii 5 mg/ m2 5 dní po sobě celkem 6 cyklů.
Kontrolní PET‑CT bylo provedeno v březnu roku 2010. Low dose CT vyšetření popsalo stacionární obraz sledovaných kavit v obou plicních křídlech, ale nyní bez ložiskových nodularit v plicích, které byly zřetelné na předchozích snímcích. Při PET zobrazení bez patologické kumulace radiofarmaka. I přes nepřítomnost zřetelné aktivity nemoci však došlo v březnu roku 2010 k pneumotoraxu, který vyžadoval hospitalizaci na chirurgii.
Pacient č. 12 – generalizovaná lymfadenopatie, postižení plic a později i kostní postižení
Mladý muž, narozený roku 1973, byl bez vážnějších chorob až do října roku 2008, kdy si poprvé všiml zvětšených uzlin v oblasti pravého třísla a postupně zjišťoval, že zvětšené uzliny má nejen v třísle, ale také na krku. Měl bolesti v oblasti pravého třísla a bederní krajiny a od listopadu roku 2008 se začaly objevovat undulující teploty dosahují až 38 °C. První uzlina (z nadklíčku) byla extirpována v listopadu roku 2008 a histologický závěr zněl: histiocytóza z Langerhansových buněk. První návštěva pacienta na našem pracovišti byla v únoru roku 2009. V té době měl generalizované zvětšení uzlin na krku, axilách a v tříslech, velikosti kolem 2 cm.
Dle vstupního PET‑CT bylo v CT obraze zřetelné patologické zvětšení uzlin na krku, v axilách, dále v oblasti dutiny břišní, retroperitoneálně a v malé pánvi, kde byla naměřena největší velikost uzlin – 3,5 cm v průměru. Tyto uzliny byly aktivní i při PET zobrazení, aktivita se pohybovala mezi 5,0 SUV v axilách a 12,3 SUV v oblasti pravých ilických uzlin. Kostní postižení nebylo v té době detekováno. Dle low dose CT při PET CT vyšetření byly v plicích popsány miliární stínky, tedy zřejmě již zárodek plicní formy LCH. Klinický obraz zcela jednoznačně svým průběhem odpovídal malignímu lymfomu či lymfogranulomu s B symptomy. S generalizovaným postižením uzlin bez výrazného postižení dalších tkání jsme se zatím u pacientů s LCH nesetkali. Proto jsme diagnózu ověřili excizní další uzliny z třísla a dále byla provedena biopsie perianální kůže. Jak v tříselné uzlině, tak v perianální kůži byla nalezena infiltrace LCH.
Vzhledem k agresivní formě nemoci jsme se rozhodli pro provedení sběru periferních kmenových buněk po stimulačním režimu obsahujícím etoposid a cyklofosfamid, protože další léčba 2‑chlordeoxyadenosinem by výrazně snížila vyhlídky na úspěšný sběr. Ihned po podání této chemoterapie ustaly teploty a došlo k výraznému zmenšení uzlin. Pak následovala chemoterapie 2‑chlordeoxyadenosin 5 mg/ m2 5 dní po sobě, cyklofosfamid 300 mg 5 dní po sobě a solumedrol 250 mg, celkem 6 cyklů, chemoterapie byla ukončena dne 26. 8. 2009. Pro vyhodnocení účinnosti léčby bylo po 4 cyklech provedeno v pořadí 2. PET‑CT vyšetření. Bylo konstatováno, že ubylo miliárních stínů na plicích a dále lymfatické uzliny již byly normální velikosti. V PET obraze došlo k normalizaci původně zvýšené kumulace glukózy. Další kontrolní PET‑CT měl být s odstupem za delší dobu až po ukončení léčby.
Jenže 5. 11. 2009 byl pacient přijat pro akutní zhoršení stavu (dušnost, teploty) k diferenciální diagnostice, zda se jedná o infekční či neoplastický původ těchto potíží. V pořadí třetí PET‑CT prokázalo v low dose CT obraze stále nezvětšené lymfatické uzliny, novinkou však byla osteolytická ložiska v kostech kyčelních, dříve nedekovaná. PET vyšetření však již odpovídalo relapsu nemoci v doposud nezvětšených uzlinách a nově byl zřetelný na PET i CT obraze relaps v kyčelních kostech. Současně provedeno také HRCT vyšetření, kde jsou popsány četné nodularity, místy s tvorbou drobných cystických formací v horních lalocích. Patologická infiltrace pánve potvrzena i na MR obraze.
Pro velmi časnou recidivu nemoci jsme tedy zahájili chemoterapii CHOEP.
Pro časné vyhodnocení léčebné účinnosti této chemoterapie bylo provedeno po 2 cyklech kontrolní PET‑CT vyšetření (26. 1. 2010). Low dose CT vyšetření zobrazilo přetrvávající mnohočetné nodularity v oblasti plic, do 8 mm, které nabývají cystoidní vzhled. Na rozdíl od předchozího vyšetření však na PET zobrazení již nebyl patrný hypermatabolizmus. Toto vyšetření potvrdilo tedy účinnost etoposid obsahující chemoterapie. V plánu je ukončení chemoterapie vysokodávkovanou chemoterapií dle protokolu BEAM.
Diskuze
Záměrem tohoto textu bylo podat obecný přehled rozmanitost projevů LCH a velmi stručně zmínit zkušenosti s touto nemocí u souboru 22 pacientů sledovaných na Interní hematoonkologické klinice v Brně. Léčebné postupy jsme uvedli jinde [15].
Kostní projevy
Histiocytóza z Langerhansových buněk v dospělosti postihuje dominantně skelet, vytváří osteolytická ložiska, podobná osteolýze při mnohočetném myelomu. Nejčastěji je uváděno postižení kalvy, následuje pak osteolytické postižení žeber. S menší frekvencí bývají postiženy další části skeletu [16 – 18].
U některých pacientů jsou kostní ložiska různého stáří, hojící se místa mají sklerotický lem. Ne všechna musí bolet. Zduření tkání, přiléhajících ke kosti, signalizuje, že choroba prorůstá do okolí a mnohdy zduření nad kostí upozorní na ložisko. Klasickou metodou průkazu ložisek je RTG snímek [19].
Kostní ložiska často způsobují zvýšený kostní obrat, a proto jsou znázornitelná metodou scintigrafie skeletu pomocí technecium pyrofosfátu [20,21]. Novou metodou, která detekuje ložiska LCH v kostech i jinde, je FDG - PET [22]. Tato metoda vypovídá o aktivitě či neaktivitě ložiska a může pomoci při plánování radioterapie a vyhodnocování léčebné odpovědi [23,24].
Velmi citlivou metodou zobrazení patologického děje v kosti je však magnetická rezonance (MR) [25,26], která je optimální pro plánování rozsahu radioterapie [26].
Za zvláště riziková kostní ložiska jsou považovány kostní defekty v oblasti orbitální se supraorbitálními infiltráty, dále ložiska v kosti temporální. Uvedené postižení je spojené s vyšší pravděpodobností pozdějšího postižení CNS, hlavně cerebella [13].
V našem souboru 22 nemocných se postižení kalvy (primární ložisko či místo recidivy) vyskytlo celkem u 7 (32 %) nemocných. U jednoho pacienta se objevilo ložisko supraorbitálně několik měsíců před první manifestací poškození CNS. Potvrdila se tedy v literatuře popisovaná zkušenost, že supraorbitální infiltrace často předchází pozdějšímu postižení CNS.
Z podskupiny nemocných s LCH dia-gnostikovanou v dospělém věku [17] byly přítomny kostní projevy u 10 (59 %) nemocných ze 17, přičemž 7 ze 17 (41 %) mělo v době stanovení diagnózy přítomno jen jedno kostní ložisko a 3 ze 17 (18 %) měli v době stanovení diagnózy vícečetné kostní postižení. Kostní forma LCH vzniklá v dospělosti recidivovala u 3 (17 %) ze 17 nemocných.
Z podskupinky nemocných s LCH diagnostikovanou v dětském věku byly pozdní kostní recidivy v dospělém věku diagnostikovány u 2 z 5 (40 %).
Kožní projevy
Infiltrace kůže buňkami Langerhansovy histiocytózy dominuje u dospělých v oblasti mediální roviny a často postihuje intertriginózní oblasti (perianální krajinu, třísla, pupek, vulvu) [28 – 34].
Kožní manifestace jsou u Langerhansovy histiocytózy velmi časté a mohou být vůbec prvními zachytitelnými projevy nemoci. Infiltrace kůže buňkami Langerhansovy histiocytózy převládá v oblasti mediální roviny a často postihuje intertriginózní oblasti (perianální oblast, vulva, třísla, pupek). Typickou morfou je hnědorůžová papula velikosti 1 – 5 mm, při tendenci ke splývání – zvláště v oblasti kůže kštice – se objevuje i šupení. Popisovány jsou i vezikuly a pustuly. Papulózní projevy jsou často hodnoceny jako nespecifické či ekzémové. Šupící plošky jsou zaměňovány se seboroickou dermatitidou, zvláště u kojenců a malých dětí při postižení vlasaté části hlavy. Vezikulózní projevy mohou napodobovat varicelu i ekzematizovaný scabies. U nejbenignější formy choroby, Hashimoto - Pritzkerovy nemoci, se již krátce po narození objevují mnohočetné nebo solitární červenohnědé uzlíky. Kožní projevy se mohou sdružovat s kostním či viscerálním postižením, v případě závažnějšího průběhu splývají, exulcerují a stav se může komplikovat bakteriální či mykotickou superinfekcí [35,36].
Kožní projevy se mohou sdružovat s kostním či viscerálním postižením, může však jít také o izolovanou morfu, která často spontánně regreduje [37 – 40]. Mimo kůže vznikají někdy změny i na nehtech [41,42].
V našem souboru jsme se kožními projevy setkali celkem u 5 (18 %). U 2 byly kožní projevy ve formě svědící ploché infiltrace perianální kůže, makroskopicky se podobající ekzému. Jeden pacient měl v perianální krajině verukózní výrůstky, které taktéž byly formou LCH. Jeden pacient měl izolovanou infiltraci kůže na rukou a jednoho nemocného jsme histologicky prokázali infiltraci kůže ve vlasaté části hlavy a v intertriginózních oblastech.
Plicní projevy
Respirační cesty jsou postiženy častěji u dospělých než u dětí, 60 – 100 % pacientů s plicní formou jsou kuřáci. Incidence plicního postižení mezi všemi pacienty s LCH se udává kolem 20 %. Pacienti přicházejí s anamnestickým údajem dušnosti, ale i bolesti na hrudníku, neproduktivním kašlem, někdy udávají teploty a úbytek hmotnosti.
Infiltrace vyvolává restrikční změny, které předcházejí RTG změnám. Radiografický nález je tvořen cystami a intersticiálními nodulárními opacitami, obvykle blíže k hilům. Optimálním prostředkem pro diagnostiku plicní formy histiocytózy je HRCT, s jehož pomocí je možno rozpoznat tenkostěnné cysty, mikronoduly, opacity a zesílení intersticiálních prostorů. Cysty jsou více koncentrované v horních lalocích a středních plicních polích a často vynechávají kostodiafragmatický úhel. Mikronoduly miliární velikosti 0,5 – 4,0 mm, s typickou distribucí, umožňující dle některých autorů rozlišit pulmonální formu LCH od jiných nodulárních chorob. Zesílení intersticia je při HRCT vyšetření zřetelné hlavně bazálně [43,44]. Prasknutí cyst a jejich komunikace s pleurální dutinou způsobí spontánní pneumotorax [45,46]. Postižení plic histocytózou často komplikuje nasedající oportunní infekce. Odlišit ji může být problém, neboť teplota a váhový úbytek mohou být jak prvními projevy plicní histiocytózy, tak mohou mít i jiné (infekční) příčiny [48 – 58].
V diferenciální diagnostice pomůže buď bronchoalveolární laváž, nebo torakoskopie s excizí materiálu na histologické vyšetření.
Bronchoalveolární laváž je přínosná jedině v tom případě, pokud se provedou odpovídající imunohistochemické vyšetřovací metody, samotná bronchoskopie s biopsií bronchiální sliznice je pro průkaz LCH zcela nepřínosná. Langerhansovy buňky lze identifikovat flowcytometricky anebo v sedimentu z laváže imunohistochemicky průkazem makrofágů s pozitivitou znaku CD1a proteinu S 100 [59,60]. Tato metoda hodnocení je však dostupná jen na ojedinělých pracovištích v ČR. Udává se, že v případě plicní formy histiocytózy bývá v laváži více než 5 % CD1a+ buněk, zatímco u zdravých pacientů je počet CD1a+ menší než 1 %. Dále bývá zvýšen počet buněk v aspirované tekutině nad 106/ ml, s prevalencí alveolárních makrofágů. Makrofágová alveolitis je totiž přítomna u kuřáků s LCH a chybí u vzácných případů plicní LCH nekuřáků [60].
Dle nálezů z konce 90. let minulého století se plicní Langerhansova histiocytóza dospělých liší od ostatních forem tím, že proliferující histiocyty jsou polyklonální, zatímco u ostatních forem jsou histiocyty spíše monoklonální. Plicní forma Langerhansovy histiocytózy se považuje za reaktivní proces na kouření či jiné stimuly. U této formy bylo také potvrzeno, že v případech pacientů se silnou vůlí, kteří dokázali přestat kouřit, dle RTG nálezů došlo k spontánní regresi nemoci. Plicní forma se považuje za relativně příznivou, pokud jde o izolovanou formu u kuřáka. Jestliže však jde o plicní formu navazující na generalizované postižení skeletu a dalších orgánů nemocí, tak průběh je relativně nepříznivý [61 – 63]. V plicním parenchymu však může vzniknout i Erdheimova ‑ Chesterova choroba [64].
Pro sledování vývoje plicní formy LCH se používají jak funkční testy, tak HRCT [64].
V našem souboru nemocných byla plicní forma identifikována pomocí HRCT u 7 (32 %) z 22 pacientů. Cytologicky byla plicní forma LCH prokázána ve 2 případech (průkaz CD1a pozitivních elementů v bronchoalveolární laváži, jednou byla prokázána histologicky ve vzorku odebraném torakoskopicky). U zbývajících 5 pacientů s plicním postižením byla diagnóza LCH ověřena histologickým vyšetřením jiné, snáze dostupné tkáně. K závěru, že jde o plicní formu LCH, se došlo na základně histologického průkazu přítomnosti LCH v jiném orgánu či tkáni a typickém nálezu nodularit a cyst na HRCT plic.
Postižení endokrinního systému
Diabetes insipidus je typickým projevem dětské formy LCH, výjimečně může vzniknout i v dospělosti, v literatuře se popisuje při multifokálním kostním poškození. Zvláště v dětském věku však nepostihuje pouze sekreci adiuretinu, ale občas vzniká v důsledku nemoci deficit i ostatních hormonů, včetně poškození tvorby somatotropinu [65,66]. Tyto případy uvádí literatura formou popisů případů [69 – 73].
V našem souboru byl diabetes insipidus přítomen u 3 ze 5 nemocných s diagnózou zjištěnou v dětském věku. U pacientů s diagnózou této nemoci v dětském věku je nutné vždy pátrat po deficitu ostatních hypofyzárních hormonů. Ale také u dospělých může být nově vzniklý diabetes insipidus prvním projevem této nemoci [67,68].
V dospělosti vzniklý diabetes insipidus, čili postižení stopky hypofýzy byl prvním příznakem nemoci u 2 pacientů (tedy u 12 % z 17 pacientů s LCH diagnostikovanou v dospělém věku).
Oba pacienti měli rozšíření stopky hypofýzy.
Biopsie z infiltrátu v hypofýze by byla spojena s komplikacemi. Diagnóza LCH byla u 1. pacienta ověřena histologickým vyšetřením kožních morf v oblasti anální krajiny a u 2. bronchoalveolární laváží, která prokázala přítomnost plicní formy nemoci.
Zajímavé je, že ani jeden z mužů s diabetes insipidus na podkladě v dospělosti vzniklé nemoci neměl kostní postižení. Velmi příznivé je, že po léčbě cladribinen se šíře stopky hypofýzy upravila na normu, což signalizuje, že infiltrace v CNS příznivě reagovala na léčbu cladribinem, i když diabetes insipidus stále trvá. V literatuře je popsán případ, kdy diabetes insipidus vymizel po léčbě cladribinem [74]. V našem případě se tak nestalo a můžeme spekulovat, že je to způsobeno zahájením léčby až po 2 letech trvání diabetes insipidus, kdy poškození hypotalamických struktur se stalo nevratným.
Mozek
Velmi vzácnou manifestací LCH v dospělosti je postižení CNS. Podezření na tuto formu je nutno mít u všech nemocných s LCH, u nichž se objeví jakékoliv neurologické příznaky. Infiltráty lze znázornit pomocí MR a CT s aplikací kontrastní látky. Expanzivní ložiska jsou prokazatelná těmito metodami u 50 % pacientů s poruchou funkce hypofýzy. Disperzní infiltraci prokáže jedině autopsie. Není jasné, proč Langerhansovy buňky mají takovou afinitu k hypotalamu a jeho stopce.
Poškození CNS však může být jak přechodné [75], tak velmi vzácně i trvalé [76]. Vhledem k nemožnosti zkoumat průběh těchto komplikací opakovanými biopsiemi musí poznání vycházet ze zobrazovacích metod, nemnohých výsledků biopsií a autoptických studií a případně analýz cerebrospinálního moku [77,78]. Dle posledních informací je možné diferencovat 3 typy postižení:
- ohraničené granulomy v oblasti mozkové pojivové tkáně, zřetelné na MR zobrazení, odpovídající tumorózním ložiskům v meningách nebo v chorioideálním plexu; jejich histologie odpovídá Langerhansovým granulomům v pojivové tkáni,
- granulomy v oblasti pojivové mozkové tkáně s částečnou infiltrací šedé hmoty mozkové jak patologickými (CD1a+), tak i reaktivními histiocyty; tyto infiltráty byly provázené výraznou T buněčnou infiltrací a neurodegenerací, ztrátou neuronů, axonů a gliózou,
- neurodegenerativní ložiska postrádající CD1a+ buňky, nejčastěji postihující cerebellum, nucleus dentatus, cerebellární bílou hmotu a mozkový kmen, s výraznou zánětlivou infiltrací, obsahující CD8 lymfocyty; tento proces vede k degeneraci a glióze nervové tkáně. Neurodegenerativní proces se objevuje na základě T buněčného zánětlivého procesu. Je provázen destrukcí neuronů a axonů se sekundární demyelinizací, připomínající paraneoplastickou encefalitidu. Tento typ postižení se objevuje až za dlouhou dobu od stanovení diagnózy, je progredující a nevratný. Projevuje se hyporeflexií, ataxií, závratěmi, dysartrií, nystagmem, tremorem, diploopií, psychomotorickou retardací a neuropsychologickými defekty [78 – 84].
V našem souboru 5 nemocných, u nichž LCH vzniká v dětském věku, se pozdní neurodegenerativní poškození vyskytlo u 2 (40 %) osob.
V jednom případě (pacient narozen roku 1975) byla v anamnéze ataka neurologických potíží typu ataxie netypických pro mladého muže. Tyto potíže výrazně ustoupily, byť ne zcela ad integrum. Kontrolní CT mozku bylo dle lékařských zpráv negativní, nicméně vzhledem k obtížnosti interpretací změn na CT při neurodegenerativních změnách v důsledku LCH se domníváme, že pravděpodobně šlo o nerozpoznanou mozkovou ataku LCH, podobně jako to popisuje Imashuku [85]. V roce 2009 však došlo k další progresi mozkového poškození, které již bylo jasně potvrzeno nálezem atrofie cerebella na MR zobrazení a hypometabolizmem v oblasti cerebella na PET‑CT vyšetření mozku.
V případě muže narozeného roku 1976 je evidentní, jak původně dětská forma LCH přešla v dospělosti do závažného chronického onemocnění. Zpočátku jsme se domnívali, že recidivy budou omezené pouze na skelet. Výjimečné bylo, že mnohočetné kostní recidivy byly prokázány vždy pouze v oblasti kalvy. Po poslední recidivě v oblasti oka (rok 2001) následovalo za několik měsíců těžké zhoršení stavu s diplopií, skandovanou řečí a příznaky z postižení cerebella a progresivním zhoršováním funkce CNS tak, že nemocný nebyl schopen samostatné chůze, neovládal svěrače a komunikace byla velmi omezená. Tento nemocný zemřel v dubnu roku 2008. MR obraz i další průběh odpovídal neurodegenerativnímu poškození CNS, pro nějž v době stanovení diagnózy neexistovala žádná účinná léčba [85].
Lymfadenopatie
Histiocytóza obvykle nepůsobí výraznou lymfadenopatii, pokud ano, jde spíše o ložiskové než generalizované postižení [86].
V našem případně jsme se setkali s jedním případem izolované lymfadenopatie, která byla, jak se zdá, definitivně vyřešena chirurgickým odstraněním uzliny. V druhém případě šlo o průběh nemoci připomínající maligní lymfom provázený B symptomy (subfebrilie, velký úbytek hmotnosti) s generalizovanou lymfadenopatií. Postižení bylo histologicky ověřené jak z uzliny extirpované z krku, tak z uzliny extirpované z třísla. Tato forma nemoci provázená B symptomy měla velmi agresivní průběh.
Uši
Poruchy sluchu mohou nastat jak postižením zevního sluchového kanálu, tak poruchou středního či vnitřního ucha propagací choroby z processus mastoideus. Infiltrace je nebolestivá a postupně vede k hluchotě. Časté jsou sekundární infekce, které jsou příčinou záměny za chronickou otitidu [87,88]. Problém bývá s diagnózou, protože bez odebrání histologie a bez cíleného vyšetření odebraného histologického vzorku není možno přesné pojmenování této nemoci.
V našem souboru jsme se setkali s jedním pacientem, u něhož nebylo postižení zevního zvukovodu včas rozpoznáno, ač pilně navštěvoval ORL lékaře. K rozpoznání LCH došlo, až proces postihl celý procesus mastoideus a vyžádal si operaci. V našem souboru ještě jeden pacient je dlouhodobě sledován na ORL s diagnózu otitis externa, ale zatím u něj nebyla LCH v zevním zvukovodu histologicky prokázána (nebyla dělána histologie).
Oči
Intraokulární postižení je vzácné, zatímco infiltrace orbitálního prostoru je relativně častá. Dětští lékaři se s ní setkávají u 20 – 30 % nemocných Langerhansovou histiocytózou. Projevuje se ptózou víčka, edémem papily a poruchou funkce VII. nervu. V některých případech může být poškozen optický nerv, což si někdy kromě systémové léčby vynutí i akutní léčbu nitroložiskovou aplikací kortikosteroidů a radioterapii [89 – 92]. U dospělých jsme se s těmito projevy nesetkali.
Játra a slezina
Játra i slezina mohou být touto chorobou postižena, což se projeví jejich zvětšením. Velmi silná infiltrace jater pak může vyvolat příznaky jaterního selhání (pokles albuminu, snížení aktivity koagulačních faktorů, žloutenku bez výrazného zvýšení jaterních enzymů). V chronických formách může vzniknout periportální fibrotizace s příznaky shodnými se sklerotizující cholangitidou a obstrukční biliární žloutenkou, kterou je nutno na základě biopsie odlišit od primární sklerotizující cholangitidy a adekvátně léčit [93 – 99].
V našem případně jsme na postižení jater bez histologického průkazu usuzovali u muže narozeného roku 1976, u něhož v roce 2001 prudce vzestoupily hodnoty aktivity jaterních enzymů, aniž by se našlo jiné vysvětlení než infiltrace jater LCH.
Proto tento pacient byl léčen 2‑chlordeoxyadenosinem, po němž se hodnoty jaterních enzymů upravily.
Dutina ústní a trávicí trakt
Počínající infiltrace se v této oblasti projevuje zduřením dásní a sliznice patra. Může dojít k postižení kostí a uvolňování zubů či hypertrofii dásní [100]. Progrese infiltrátů pak vytváří ulcerace v ústech [101,102]. U dětí je diagnosticky významná předčasná druhá dentice.
V našem souboru je jeden nemocný (narozen roku 1974), u něhož byla diagnostikována infiltrace lingvální části dásně a kompletně excidována. U dalších dvou pacientů jsme na tuto nemoc měli podezření, protože u nich došlo k náhlému uvolňování zubů. U jednoho pacienta proces pracovně nazvaný parodontóza zastavil a nešlo ke ztrátě zubů, u druhého nemocného došlo ke ztrátě asi poloviny zubů. Vzhledem k časové koincidenci se vznikem jiných projevů LCH a problémy s uvolňováním zubů se obou pacientů nabízí příčinná souvislost, byť nebyla stomatology potvrzena. Žádný vzorek z dutiny ústní však nebyl k histologickému vyšetření odeslán. Pokud stomatolog nevěděl o formě LCH, postihující dásně a čelist, vedoucí k uvolňování zubů, nemohl ji ani diagnostikovat. Potvrdit či nepotvrdit by ji mohla jedině histologie z postižených dásní a panoramatický snímek čelisti.
Sliznice střevního traktu je postižena jen zřídka. Prvními příznaky je celkové neprospívání, hubnutí. Klasické projevy malabsorpce se objevují až při rozsáhlejším postižení trávicího traktu. Anální kanál a perianální oblast jsou infiltrovány často, a tvoří tak součást kožního postižení [102 – 105].
Sledování nemocných
Zatím nemáme v praxi dostupný žádný laboratorní parametr, takže nezbývá nic jiného než používat odpovídající zobrazovací metody a klinická vyšetření. Zatím se popsané použití sérové hladiny proteinu S ‑ 100 nedostalo do rutinní praxe [106].
Závěry pro praxi
Histiocytóza z Langerhansových buněk je výjimečná nemoc svou nízkou incidencí, ale také výjimečná mnoha podobami, které tato nemoc může nabývat. V textu jsou uvedeny ty formy, s nimiž jsme se na našem pracovišti setkali za 20 let. V diskuzi jsme podali přehled manifestací u dospělých, jak je uvádí literatura, a srovnali je s naším pozorováním.
- Každá bolest páteře či kostí, které trvá déle než 1 měsíc, by měla být vyšetřena základními zobrazovacími metodami (RTG skeletu, scintigrafie skeletu pomocí Tc ‑ pyrofosfátu). Nejasný nález pak upřesněn pomocí MR, CT, případně PET‑CT. Pokud se prokáže osteolýza, musí následovat odběr vzorku na histologické vyšetření patologické tkáně. Další léčba se pak odvíjí od výsledku histologie. Osteolytické ložisko může být mimo jiné projevem krevní nemoci (LCH nebo mnohočetného myelomu nebo lymfomu nebo angiomatózy).
- U každého diabetes insipidus vzniklého v dospělosti je vhodné pomýšlet na LCH. Optimální je provést PET‑CT vyšetření, které může odhalit extrakraniální projevy nemoci se snazším přístupem pro histologickou verifikaci.
- U pacienta s chronickým zánětem zevního zvukovodu je vhodné jej histologicky vyšetřit, průkaz LCH pak upřesní formu léčby.
- U mladého pacienta s ložiskovým onemocněním dásní a s vypadáváním zubů, či dokonce s destrukcí čelisti je vhodné odebrat histologii a nechat vyšetřit znaky LCH.
- U mladého pacienta s dušností je vhodné provést HRCT. Nález nodularit a cyst na HRCT by měl vést k ověření diagnózy LCH biopsií a bronchoalveolární laváží, a ne jej pouze sledovat jako emfyzém.
- Pokud vznikne spontánní pneumotorax u mladého člověka, je vhodné vyšetřit plíce metodou HRCT, a pokud obraz připouští možnost LCH, provést histologické vyšetření.
- Každá nejasná kožní morfa, zvláště v anogenitální, ale i intertriginózní oblasti by měla být histologicky komplexně vyšetřena, včetně průkazu LCH.
Histiocytóza z Langerhansových buněk je vzácná nemoc, kterou lze diagnostikovat pouze tehdy, pokud lékař indikuje histologické vyšetření tkáně a doporučí patologovi, aby provedl mimo jiné ta vyšetření, která prokazují LCH.
Tato publikace byla připravena v rámci projeku MUNI/ A/ 1012/ 2009 s názvem „Optimalizace diagnostiky a terapie maligních chorob a komplikací, které tyto maligní nemoci provázejí, s využitím nových molekulárně biologických metod“ a také je součástí aktivit v rámci grantů IGA MZ: NR9225, NS10387 a NS10406.
prof. MU Dr. Zdeněk Adam, CSc.
www.fnbrno
e‑mail: z.adam@fnbrno.cz
Doručeno do redakce: 5. 5. 2010
Sources
1. Nicholson HS, Egeler RM, Nesbit ME. The epidemiology of Langerhans cell histiocytosis. Hematol Oncol Clin North Amer 1998; 12 : 379 – 348.
2. Baumgartner I, von Hochstetter A, Baumert B et al. Langerhan’s cell histiocytosis in adults. Med Pediatric Oncol 1997; 28 : 9 – 14.
3. Bubanska E, Stančokova T, Dluholucky S. Histiocytóza z Langerhansových buněk. Čes Slov Pediat 1998; 53 : 18 – 19.
4. Mottl H, Koutecký J, Ganevová M. Strategie léčby histiocytózy z Langerhansových buněk u dětí. Čes Slov Pediat 1994; 49 : 81.
5. Mottl H, Mracek J, Kabelka Z et al. Histiocytóza z Langerhansových buněk u dětí. Čs Pediat 1992; 47 : 530 – 533.
6. Mottl H, Rob L, Stary J et al. Langerhans cell histiocytosis of vulva in adolescent. Int J Gynecol Cancer 2007; 17 : 520 – 524.
7. Šímová B, Mališ J, Neuwirt J. Klinické projevy histiocytózy z Langerhansových buněk. Zdrav Nov ČR Lék Listy 2003; 49 : 18.
8. Tichy M Jr, Tichy M, Krč I et al. Multicentrická retikulohistiocytóza. Česk Dermatol 1999; 74 : 168 – 171.
9. Toušovská K, Slavík Z. Histiocytóza z Langerhansových buněk v dětském věku. Lék Zprav Lék Fak Univ Karlovy Hr Králové 1997; 42 : 127 – 132.
10. Pacovska V, Bortlova A, Homolka J et al. Granulomatóza z Langerhansových buněk. Trendy Med 2002; 4 : 59 – 61.
11. Götz G, Fichter J. Langerhans’ ‑ cell histiocytosis in 58 adults. Eur J Med Res 2004; 9 : 510 – 514.
12. Malpas JS. Langerhans cell histiocytosis in adults. Hematol Oncol Clin North Am 1998; 12 : 259 – 268.
13. Stockschlaeder M, Sucker C. Adult Langerhans cell histiocytosis. Eur J Haematol 2006; 76 : 363 – 368.
14. Imashuku S, Ishida S, Koike K et al. Cerebellar ataxia in pediatric patients with Langerhans cell histiocytosis. J Pediatr Hematol Oncol 2004; 26 : 735 – 739.
15. Adam Z, Krejčí M, Vorlícek J. Maligní krevní nemoci. Praha: Grada publishing 2008.
16. Cheung N, Selva D, McNab AA. Orbital Langerhans cell histiocytosis in adults. Ophthalmology 2007; 114 : 1569 – 1573.
17. Smilek P, Krejčova B, Čada K et al. Histiocytóza z Langerhansových buněk, případ postižení spánkové kosti. Otorinolaryng Foniat 1994; 43 : 263 – 265.
18. Srikulmontree T, Massey HD, Roberts WN. Treatment of skeletal Erdheim ‑ Chester disease with zoledronic acid: case report and proposed mechanisms of action. Rheumatol Int 2007; 27 : 303 – 307.
19. Hoover KB, Rosenthal DI, Mankin H. Langerhans cell histiocytosis. Skeletal Radiol 2007; 36 : 95 – 104.
20. Dogan AS, Conway JJ, Miller JH et al. Detection of bone lesions in Langerhans cell histiocytosis: complementary roles of scintigraphy and conventional radiography. J Pediatr Hematol Oncol 1996; 18 : 51 – 58.
21. Howarth DM, Mullan BP, Wiseman GA et al. Bone scintigraphy evaluated in diagnosing and staging Langerhans’s cell histiocytosis and related disorders. J Nucl Med 1996; 37 : 1456 – 1460.
22. Blum R, Seymour JF, Hicks RJ. Role of FDG ‑ positron emission tomography scannin in the management of histiocytosis. Leukem Lymphoma 2002; 43 : 2155 – 2157.
23. Kaste SC, Rodriguez ‑ Galindo C, McCarville ME et al. PET‑CT in pediatric Langerhans cell histiocytosis. Pediatr Radiol 2007; 37 : 615 – 622.
24. Weiss SE, O’Connor L, Welsh JS. Refinement of radiation therapy based on PET data in an adult with Langerhans cell histiocytosis of soft tissues. Clin Adv Hematol Oncol 2006; 4 : 290 – 292.
25. Furmanczyk PS, Bruckner JD, Gillespy T 3rd et al. An unusual case of Erdheim ‑ Chester disease with features of Langerhans cell histiocytosis. Skeletal Radiol 2007; 36 : 885 – 889.
26. Goo HW, Yang DH, Ra YS et al. Whole ‑ body MRI of Langerhans cell histiocytosis: comparison with radiography and bone scintigraphy. Pediatr Radiol 2006; 36 : 1019 – 1031.
27. Olschewski T, Seegenschmiedt MH. Radiotherapy of Langerhans’ Cell Histiocytosis: Results and Implications of a National Patterns ‑ of ‑ Care Study. Strahlenther Onkol 2006; 182 : 629 – 634.
28. Fernandez Flores A, Mallo S. Langerhans cell histiocytosis of vulva. Dermatol Online J 2006; 12 : 15.
29. Ferreli C, Aste N, Pinna LA et al. Langerhans cell histiocytosis in an adult. J Eur Acad Dermatol Venereol 1997; 9 : 253 – 255.
30. Ferringer T, Banks PM, Metcalf JS. Langerhans cell sarcoma. Am J Dermatopathol 2006; 28 : 36 – 39.
31. Hagiuda J, Ueno M, Ashimine S et al. Langerhans cell histiocytosis on the penis: a case report. BMC Urol 2006; 6 : 28.
32. Mlyncek M, Uharcek P, Durcanský D. Vulvar Langerhans’ cell histiocytosis: a case report. Acta Obstet Gynecol Scand 2006; 85 : 753 – 755.
33. Tzung TY, Wu JC. Nonhealing perianal ulcers. Arch Dermatol 2005; 141 : 1161 – 1166.
34. Venizelos ID, Mandala E, Tatsiou ZA et al. Primary langerhans cell histiocytosis of the vulva. Int J Gynecol Pathol 2006; 25 : 48 – 51.
35. Fluri S, Nievergelt H, Kernland K et al .When atopic dermatitis doesn’t heal – an interesting case from the medical university polyclinic. Praxis 2006; 95 : 945 – 948.
36. Punia RS, Bagai M, Mohan H et al. Langerhans cell histiocytosis of skin: a clinicopathologic analysis of five cases. Indian J Dermatol Venereol Leprol 2006; 72 : 211 – 214.
37. Imafuku S, Shibata S, Tashiro A. Cutaneous Langerhans cell histiocytosis in an elderly man successfully treated with narrowband ultraviolet B. Br J Dermatol 2007; 157 : 1277 – 1279.
38. Lan Ma H, Metze D, Luger TA et al. Successful treatment of generalized eruptive histiocytoma with PUVA. J Dtsch Dermatol Ges 2007; 5 : 131 – 134.
39. Newman B, Hu W, Nigro K et al. Aggressive histiocytic disorders that can involve the skin. J Am Acad Dermatol 2007; 56 : 302 – 316.
40. Munn S, Chu AC. Langerhans cell histiocytosis of the skin. Hematol Oncol Clin North Am 1998; 12 : 269 – 286.
41. Ashena Z, Alavi S, Arzanian MT et al. Nail involvement in langerhans cell histiocytosis. Pediatr Hematol Oncol 2007; 24 : 45 – 51.
42. Mataix J, Betlloch I, Lucas ‑ Costa A et al. Nail changes in Langerhans cell histiocytosis: a possible marker of multisystem disease. Pediatr Dermatol 2008; 25 : 247 – 251.
43. Canuet M, Kessler R, Jeung MY et al. Correlation between high‑resolution computed tomography findings and lung function in pulmonary Langerhans cell histiocytosis. Respiration 2007; 74 : 640 – 646.
44. Leatherwood DL, Heitkamp DE, Emerson RE. Best cases from the AFIP: Pulmonary Langerhans cell histiocytosis. Radiographics 2007; 27 : 265 – 268.
45. Kim CK, Park CB, Jin U et al. Pulmonary Langerhans’ cell histiocytosis presented with recurrent pneumothorax. Interact Cardiovasc Thorac Surg 2006; 5 : 512 – 513.
46. Mendez JL, Nadrous HF, Vassallo R et al. Pneumothorax in pulmonary Langerhans cell histiocytosis. Chest 2004; 125 : 1028 – 1032.
47. Adams EP, Sauceda D, Oliver J et al. Isolated pulmonary Langerhans cell histiocytosis in a 17‑year ‑ old male. J Pediatr Hematol Oncol 2007; 29 : 121 – 124.
48. Aerni MR, Aubry MC, Myers JL et al. Complete remission of nodular pulmonary Langerhans cell histiocytosis lesions induced by 2‑chlorodeoxyadenosine in a non‑smoker. Respir Med 2008; 102 : 316 – 319.
49. Bernstrand C, Cederlund K, Sandstedt B et al. Pulmonary abnormalities at long term follow up of patients with Langerhans cell histiocytosis. Med Pediatr Oncol 2001; 36 : 459 – 468.
50. Braier J, Latella A, Balancini B et al. Isolated pulmonary Langerhans cell histiocytosis presenting with recurrent pneumothorax. Pediatr Blood Cancer 2007; 48 : 241 – 244.
51. Bittenglova R, Pešek M, Mukenšnabl P et al. Granulomatóza z Langerhansových buněk. Stud Pneumol Phtiseol 2002; 62 : 196 – 202.
52. Brown RE. Bisphosphonates as antialveolar macrophage therapy in pulmonary Langerhans cell histiocytosis. Med Pediatr Oncol 2001; 36 : 641 – 643.
53. Green MB, Allen JN. Cough, dyspnea, and reticulonodular opacities in a 58‑year ‑ old smoker. Chest 2007; 132 : 700 – 703.
54. Jülg BD, Weidner S, Mayr D. Pulmonary manifestation of a Langerhans cell sarcoma: case report and review of the literature. Virchows Arch 2006; 448 : 369 – 374.
55. Rožánek P, Molnar V, Rešl M. Tři případy plicní granulomatózy z Langerhansových buněk. Lék Zpr Lék Fak Univ Karlovy Hr Králové 1998; 43 : 127 – 132.
56. Sundar KM, Gosselin MV, Chung HL et al. Pulmonary Langerhans cell histiocytosis: emerging concepts in pathobiology, radiology and clinical evolution of the disease. Chest 2003; 123 : 1673 – 1683.
57. Tazi A. Adult pulmonary Langerhans’ cell histiocytosis. Eur Respir J 2006; 27 : 1272 – 1285.
58. Skacel Z, Marel M, Vraštilova P et al. Histiocytóza z Langerhansových buněk. Přehled literatury a vlastní pozorování. Stud Pneumol Phtiseol 2000; 60 : 150 – 156.
59. Zeppa P, Cozzolino I, Russo M et al. Pulmonary langerhans cell histiocytosis (histiocytosis X) on bronchoalveolar lavage: a report of 2 cases. Acta Cytol 2007; 51 : 480 – 482.
60. Tötsch M, Guzman J, Theegarten D et al. Bronchoalveolar lavage. Pathologe 2007; 28 : 346 – 353.
61. Hidalgo A, Franquet T, Giménez A. Smoking‑related interstitial lung diseases: radiologic ‑ pathologic correlation. Eur Radiol 2006; 16 : 2463 – 2470.
62. Liu YH, Fan XH, Fang K. Langerhans’ cell histiocytosis with multisystem involvement in an adult. Clin Exp Dermatol 2007; 32 : 765 – 768.
63. Marten K. Smoking‑related interstitial lung diseases. Rofo 2007; 179 : 268 – 275.
64. Negrin‑Dastis S, Butenda D, Dorzee Jet al. Complete disappearance of lung abnormalities on high‑resolution computed tomography: a case of histiocytosis X. Can Respir J 2007; 14 : 235 – 237.
65. Amato MC, Elias LL, Elias J et al. Endocrine disorders in pediatric – onset Langerhans Cell Histiocytosis. Horm Metab Res 2006; 38 : 746 – 751.
66. Donadieu J, Rolon MA, Pion I et al. Incidence of growth hormone deficiency in pediatric onset Langerhans cell histiocytosis: efficacy and safety of growth hormone treatment. J Clin Endokrinol Metabolism 2004; 89 : 604 – 609.
67. Makras P, Alexandraki KI, Chrousos GP et al. Endocrine manifestations in Langerhans cell histiocytosis. Trends Endocrinol Metab 2007; 18 : 252 – 257.
68. Neji S, Ben Slama C, Zidi B. Hypothalamic ‑ pituitary Langerhans cell histiocytosis in adults. Presse Med 2006; 35 : 1263 – 1266.
69. Halefoglu AM. Magnetic resonance imaging of thickened pituitary stalk proceeding to Langerhans cell histiocytosis in a child. Australas Radiol 2006; 50 : 175 – 178.
70. Horn EM, Coons SW, Spetzler RF et al. Isolated Langerhans cell histiocytosis of the infundibulum presenting with fulminant diabetes insipidus. Childs Nerv Syst 2006; 22 : 542 – 544.
71. Kandpal H, Subramanian S, Hari S. Langerhans cell histiocytosis of pituitary stalk. Neurol India 2007; 55 : 91 – 92.
72. Ouyang DL, Roberts BK, Gibbs IC et al. Isolated Langerhans cell histiocytosis in an adult with central diabetes insipidus: case report and review of literature. Endocr Pract 2006; 12 : 660 – 663.
73. Ottaviano F, Finlay JL. Diabetes insipidus and Langerhans cell histiocytosis: a case report of reversibility with 2‑chlorodeoxyadenosine. J Pediatr Hematol Oncol 2003; 25 : 575 – 577.
74. Prosch H, Grois N, Prayer D et al. Central diabetes insipidus as presenting symptom of Langerhans cell histiocytosis. Pediatr Blood Cancer 2004; 43 : 594 – 599.
75. Ryan P, Walterfang M, Scholes A et al. Recovery of cognitive function in neuropsychiatric Langerhan’s cell histiocytosis. Psychiatry Clin Neurosci 2006; 60 : 629 – 632.
76. Nanduri VR, Lillywhite L, Chapman C et al. Cognitive outcome of long term survivors of multisystem Langerhans cell histiocytosis: a single‑institution, cross ‑ sectional study. J Clin Oncol 2003; 21 : 2961 – 2967.
77. Prosch H, Feldges A, Grois N et al. Demonstration of CD1a positive cells in the cerebrospinal fluid – A clue to diagnosis of isolated Langerhans cell histiocytosis of the hypothalamic ‑ pituitary axis? Med Pediatr Oncol 2003; 41 : 474 – 476.
78. Prosch H, Grois N, Wnorowski M et al. Long‑term MR imaging course of neurodegenerative Langerhans cell histiocytosis. AJNR Am J Neuroradiol 2007; 28 : 1022 – 1028.
79. Bös M, Grothe C, Urbach H et al. Cerebellar syndromes in Langerhans‘ cell histiocytosis. Nervenarzt 2007; 78 : 437 – 440.
80. Dhall G, Finlay JL, Dunkel IJ et al. Analysis of outcome for patients with mass lesions of the central nervous system due to Langerhans cell histiocytosis treated with 2‑chlorodeoxyadenosine. Pediatr Blood Cancer 2008; 50 : 72 – 79.
81. Grois N, Prayer D, Prosch H et al Neuropathology of CNS disease in Langerhans cell histiocytosis. Brain 2005; 128 : 829 – 838.
82. Martin‑Duverneuil N, Idbaih A, Hoang ‑ Xuan K et al. French Langerhans Cell Histiocytosis Study Group. MRI features of neurodegenerative Langerhans cell histiocytosis. Eur Radiol 2006; 16 : 2074 – 2082.
83. Mittheisz E, Seidl R, Prayer D et al. Central nervous system‑related permanent consequences in patients with Langerhans cell histiocytosis. Pediatr Blood Cancer 2007; 48 : 50 – 56.
84. Steiner M, Prayer D, Asenbaum S et al. Modern imaging methods for the assessment of Langerhans’ cell histiocytosis‑associated neurodegenerative syndrome: case report. J Child Neurol 2005; 20 : 253 – 257.
85. Imashuku S, Okazaki NA, Nakayama M et al. Treatment of neurodegenerative CNS disease in Langerhans cell histiocytosis with a combination of intravenous immunoglobulin and chemotherapy. Pediatr Blood Cancer 2008; 50 : 308 – 311.
86. Edelweiss M, Medeiros LJ, Suster S et al. Lymph node involvement by Langerhans cell histiocytosis: a clinicopathologic and immunohistochemical study of 20 cases. Hum Pathol 2007; 38 : 1463 – 1469.
87. Baumann C, Reschke K, Jungehülsing M et al. Destruction of the vestibular organ by Langerhans’ cell histiocytosis. Eur Radiol 2006; 16 : 1177 – 1178.
88. Kürten T, Groeger M, Angerstein W. Frequency of hearing disorders in children with langerhans’ cell histiocytosis. Laryngorhinootologie 2008; 87 : 96 – 99.
89. Anton M, Holoušova M, Řehůřek J et al. Histiocytoza X a dětská očnice. Čs Ophthal 1992; 48 : 176 – 180.
90. Bermingham N, Townley D, Fenton S et al. Ocular langerhans cell histiocytosis. Eye 2007; 21 : 1127 – 1128.
91. D’Angio GJ. Langerhans cell histiocytosis affecting the eyes. Pediatr Blood Cancer 2006; 47 : 639.
92. Tsai JH, Galaydh F, Ching SS. Anterior uveitis and iris nodules that are associated with Langerhans cell histiocytosis. Am J Ophthalmol 2005; 140 : 1143 – 1145.
93. Dina I, Copaescu C, Herlea V et al. Liver involvement in Langerhans‘ cell histiocytosis. Case report. J Gastrointestin Liver Dis 2006; 15 : 57 – 59.
94. Griffiths W, Davies S, Gibbs P et al. Liver transplantation in an adult with sclerosing cholangitis due to Langerhans cell histiocytosis. J Hepatol 2006; 44 : 829 – 831.
95. Guthery SL, Heubi JE. Liver involvement in childhood histiocytic syndromes. Curr Opin Gastroenterol 2001; 17 : 474 – 478.
96. Choi SW, Bangaru BS, Wu CD et al. Gastrointestinal involvement in disseminated Langerhans cell histiocytosis (LCH) with durable complete response to 2‑chlorodeoxyadenosine and high‑dose cytarabine. J Pediatr Hematol Oncol 2003; 25 : 503 – 506.
97. Hait E, Liang M, Degar B et al. Gastrointestinal tract involvement in Langerhans cell histiocytosis: case report and literature review. Pediatrics 2006; 118: e1593 – e1599.
98. Hait E, Liang M, Degar B et al. Gastrointestinal tract involvement in Langerhans cell histiocytosis: case report and literature review. Pediatrics 2006; 118: e1593 – e1599.
99. Konno S, Hizawa N, Betsuyaku T et al. Adult Langerhans cell histiocytosis with independently relapsing lung and liver lesions that was successfully treated with etoposide. Intern Med 2007; 46 : 1231 – 1235.
100. Klein F, Krigar D, Petzoldt D et al. Periodontal manifestation of Langerhans‘ cell histiocytosis in a young man: case report with a 24 - month follow‑up. Quintessence Int 2006; 37 : 175 – 182.
101. Silvestros SS, Mamalis AA, Sklavounou AD et al. Eosinophilic granuloma masquerading as aggressive periodontitis. J Periodontol 2006; 77 : 917 – 921.
102. Querings K, Starz H, Balda BR. Clinical spectrum of cutaneous Langerhans’ cell histiocytosis mimicking various diseases. Acta Derm Venereol 2006; 86 : 39 – 43.
103. Aricò M, Egeler M. Clinical aspects of Langerhans cell histiocytosis. Hematol Oncol Clin North Am 1998; 12 : 247 – 258.
104. Chang KL, Snyder DS. Langerhans cell histiocytosis. Cancer Treat Res 2008; 142 : 383 – 398.
105. Henter JI, Tondini C, Pritchard J. Histiocyte disorders. Crit Rev Oncol Hematol 2004; 50 : 157 – 174.
106. Ugurel S, Pföhler G, Tilgen W et al. S100 - b serum protein – a new marker in the diagnosis and monitoring of Langerhans cell histiocytosis? Br J Dermatol 2000; 143 : 201 – 202.
Labels
Diabetology Endocrinology Internal medicineArticle was published in
Internal Medicine

2010 Issue 6
Most read in this issue
- Sepsis and the septic shock in oncological or other immunocompromised patients
-
Hyperlipoproteinemie a dyslipoproteinemie I.
Klasifikace, diagnostika, kardiovaskulární, kardiometabolické a reziduální riziko - Essential thrombocythaemia and other myeloproliferative disorders with thrombocythaemia treated with Thromboreductin. A report from the database of Register for the 1st quarter of 2010
- Different courses of recurrent or multisystem Langerhans cells histiocytosis in adults – description of 22 cases from one centre