
Trombohemoragický syndrom u nemocných s myeloproliferativním onemocněním s trombocytemií
Thrombohaemorrhagic syndrome in patients with a myeloproliferative disease with thrombocythemia
Thrombohaemorrhagic syndrome is a clinical syndrome manifesting with concurrent bleeding and thrombosis. It is associated with a range of pathological states, typically with myeloproliferative diseases, paraproteinaemia, liver disease as well as disseminated intravascular coagulation and similar syndromes (so called DIC-like syndrome). Thrombohaemorrhagic syndrome might be a symptom of chronic myeloproliferations, particularly if these are associated with thrombocythemia. It is most frequently linked to essential thrombocythemia. However, in this disease, it seems that the clinical symptoms of bleeding and thrombosis might not be directly determined by the number of platelets, as it would suggest itself, but that this can be consequent to other changes. These may include predisposition to thrombophilia, cardiovascular risk, leukocytosis etc. as well as, for example, platelet dysfunction. The present study focuses on platelet dysfunction in conjunction with clinical symptoms of bleeding and thrombosis.
Key words:
thrombohaemorrhagic syndrome – thrombosis – bleeding – myeloproliferation – thrombocythemia
Authors:
M. Penka 1; J. Kissová 1; A. Buliková, (ČHS) 1; J. Zavřelová 1; J. Ovesná 1; T. Pavlík 2
Authors‘ workplace:
Oddělení klinické hematologie FN Brno, pracoviště Bohunice, přednosta prof. MUDr. Miroslav Penka, CSc.
1; Institut biostatistiky a analýz Lékařské a Přírodovědecké fakulty MU Brno, vedoucí pracoviště doc. RNDr. Ladislav Dušek, Ph. D.
2
Published in:
Vnitř Lék 2011; 57(3): 306-311
Category:
60th birthday of prof. Mudr. Jiřího Vítovce, CSc, FESC
Overview
Trombohemoragický syndrom je klinický syndrom projevující se současným výskytem krvácení a trombózy. Vyskytuje se v souvislosti s řadou chorobných stavů a je příznačný pro myeloproliferativní choroby, paraproteinemie a jaterní choroby. Dále se objevuje v souvislosti s diseminovanou intravaskulární koagulací a jí podobnými syndromy (tzv. DIC-like syndrom). Trombohemoragický syndrom může být projevem chronických myeloproliferací, zejména jsou-li provázeny trombocytemií. Nejčastěji tedy provází esenciální trombocytemii. U tohoto onemocnění se však ukazuje, že se nemusí jednat o bezprostřední závislost klinické symptomatologie krvácení a trombózy na počtu krevních destiček, jak by se nabízelo, ale že může být důsledkem i jiných změn. Z nich se vedle současné přítomnosti rizikových trombofilních dispozic, kardiovaskulárního rizika, leukocytózy a dalších nabízejí poruchy funkce krevních destiček. Právě poruchám funkce krevních destiček v souvislosti s klinickou symptomatologií krvácení a trombózy je věnována přiložená studie.
Klíčová slova:
trombohemoragický syndrom – trombóza – krvácení – myeloproliferace – trombocytemie
Úvod
Trombohemoragický syndrom je klinický syndrom projevující se současným výskytem krvácení a trombózy. Většinou se jedná o souběžný výskyt zmíněné symptomatologie v kratším časovém úseku u jednoho a téhož pacienta, mnohdy v důsledku jedné choroby.
Trombohemoragický syndrom se vyskytuje v souvislosti s řadou chorobných stavů a provází poruchy krevního srážení primární či sekundární geneze. Z těch sekundárních se s ním setkáváme především u myeloproliferativních chorob [5], paraproteinemií a jaterních onemocnění, vyskytuje se též typicky v souvislosti s diseminovanou intravaskulární koagulací a jí podobnými syndromy (tzv. DIC-like syndromy: antifosfolipidový syndrom – APS, mikroangiopatické hemolytické syndromy – MAS, heparinem indukovaná trombocytopenie – HIT apod.).
Jak již bylo řečeno, typický je mimo jiné trombohemoragický syndrom pro chronická myeloproliferativní onemocnění provázená trombocytemií – tedy pro esenciální trombocytemii (ET), pravou polycytemii (polycytemia vera – PV), primární myelofibrózu (PMF), popřípadě i chronickou myelogenní leukemii (CML). U těchto chorob dochází ke vzniku trombózy a krvácení [14], přičemž údaje o jejich výskytu se liší (tab. 1 a 2) [7].
![Výskyt trombózy a krvácení u ET [1].](https://www.prelekara.sk/media/cache/resolve/media_object_image_large/media/image/0140d830563efe8d1ffadb39c3f34e5f.png)
![Výskyt 107 trombotických a krvácivých projevů v souboru 672 pacientů českého registru [13].](https://www.prelekara.sk/media/cache/resolve/media_object_image_large/media/image/973dc249adc9d75024df0f64d2f2dabb.png)
Klinické projevy nejsou, jak se ukazuje, pouze výrazem kolísání počtu trombocytů, i když s ním souvisejí a jistou závislost na něm, jak trefně znázornil Michiels na modelu „doutníku a klínu“ (obr. 1) [10], vykazují. Klinické projevy jsou však důsledkem i jiných okolností a faktorů či jejich kombinací. Ke krvácení dochází také v důsledku funkční nedostatečnosti krevních destiček, sekundárního von Willebrandova syndromu, současného výskytu koagulačních defektů apod. Ke klinické symptomatologii trombózy dochází u myeloproliferací s trombocytemií především u nemocných s mutací JAK2V617F, dále za současného výskytu kardiovaskulárních rizikových okolností či trombofilních dispozic [17], eventuálně leukocytózy [3]. Podle toho lze stratifikovat nemocné do rizikových pásem – na nízké, střední a vysoké riziko vzniku klinických příznaků (tab. 3)[15] – a dle zařazení nemocné léčit [19]. Sama Česká pracovní skupina pro myeloproliferativní choroby (CZEMP) rozlišuje jen pacienty bez rizika a pacienty s rizikem (tab. 4) [12]. V současné době jsou studovány příčiny vzniku klinické symptomatologie v detailních analýzách [4], proto je pochopitelné, že pozornost poutá, jak bylo již výše naznačeno, i studium funkce krevních destiček [6].
![Model „doutníku a klínu“ dle [10,12].](https://www.prelekara.sk/media/cache/resolve/media_object_image_large/media/image/883197c273cd6854465440263a3e6fb5.png)
![Prognostické faktory ET (navrženo Tefferim A 1999) [15].](https://www.prelekara.sk/media/cache/resolve/media_object_image_large/media/image/e0ddd2d06ee2566d0874e5c563ce5c82.png)

Cíl studie
V naší studii jsme se zaměřili na vyhodnocení destičkových funkcí dle výsledků vyšetření PFA-100 a agregace destiček indukované ADP a kolagenem v korelaci s počtem krevních destiček a s výskytem hemoragických a trombotických příhod a na srovnání zmíněných parametrů před zahájením a v průběhu léčby pacientů.
Soubor nemocných a metoda
Soubor nemocných
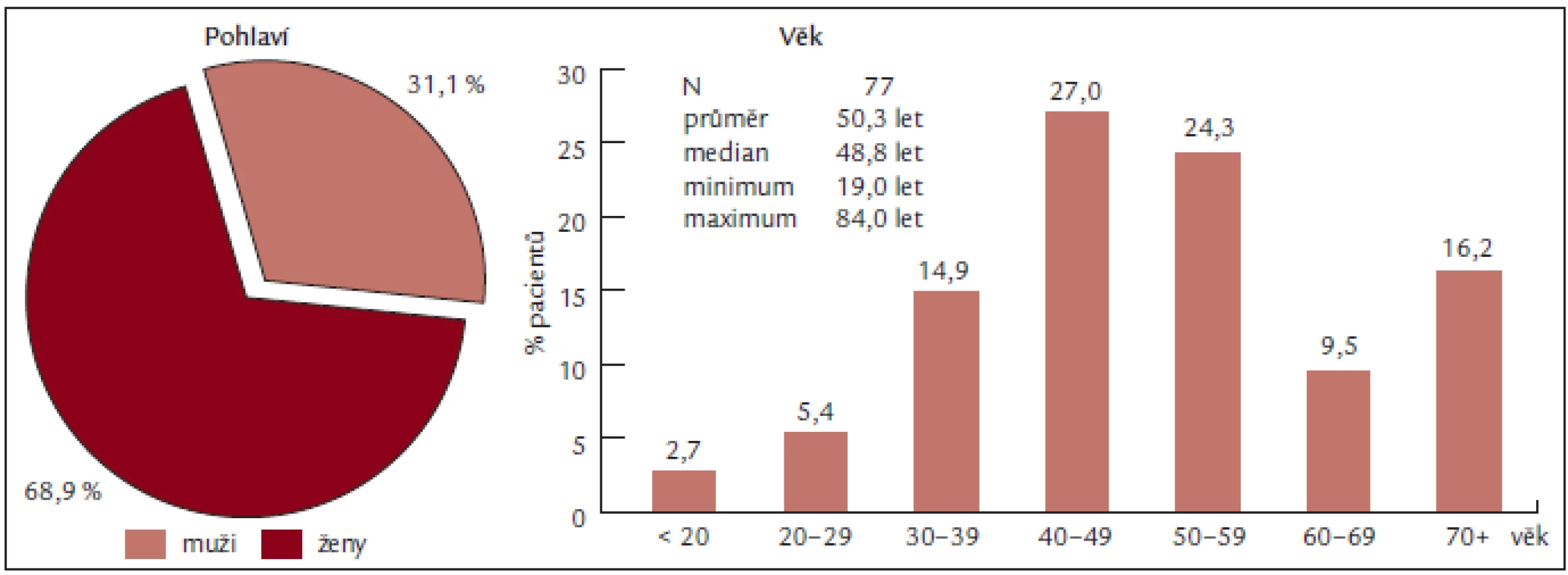
Soubor představuje 77 pacientů, z toho 53 žen a 24 mužů, s věkovým průměrem 50,3 let a mediánem věku 50 let (obr. 2), vyšetřených na Oddělení klinické hematologie FN Brno v letech 2004 až 2008. U všech nemocných byla stanovena diagnóza Ph-myeloproliferativního onemocnění (dle PVSG či WHO 2001) a u všech byla přítomna periferní trombocytemie. K léčbě byl aplikován anagrelide (Thromboreductin) a/nebo hydroxyurea (Litalir).
Panel vyšetření obsahoval:
- stanovení počtu krevních destiček (na plně automatizovaném analyzátoru krevních elementů – normální hodnota 150–350 × 109/l),
- analýzu funkce destiček (platelet function analysis 100 – PFA-100) s indukcí kolagenem/epinephrinem (normální hodnota 85–165 s),
- agregometrii dle Borna po indukci ADP 5 % a kolagenem (normální hodnoty > 60% po obou induktorech).
Vyšetření se provádělo před nasazením léčby a za 3–6 měsíců od jejího zahájení.
Statistické hodnocení
Popis souboru pacientů a sledovaných charakteristik byl proveden pomocí frekvenčních tabulek a standardních popisných statistik: průměru, mediánu, minima, maxima a kvantilů. Pro vizualizaci byly použity krabicové grafy, v případě kategoriálních dat pak kontingenční tabulky. Hodnocení vztahu dvou kategoriálních proměnných binárního charakteru bylo provedeno pomocí Fisherova exaktního testu. Pro stanovení statistické významnosti byla použita standardní hladina α = 0,05. Data byla zpracována v programu Statistica 9 [18].
Výsledky
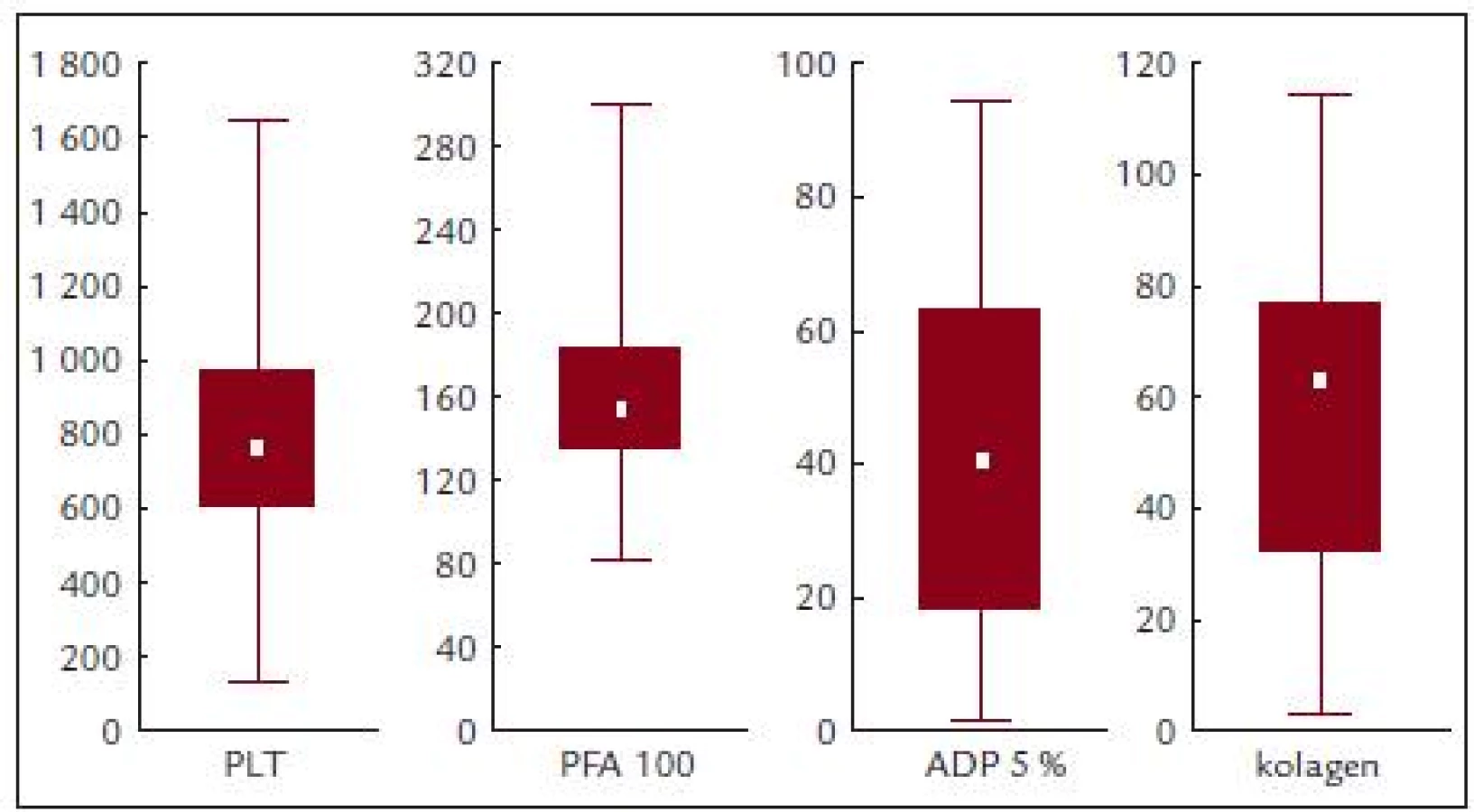
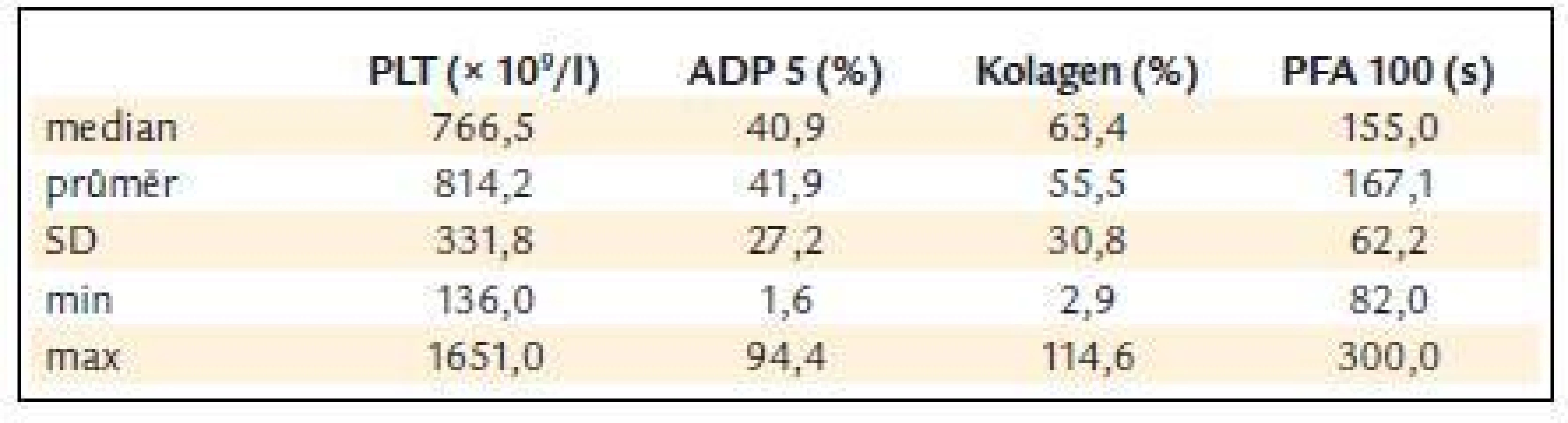
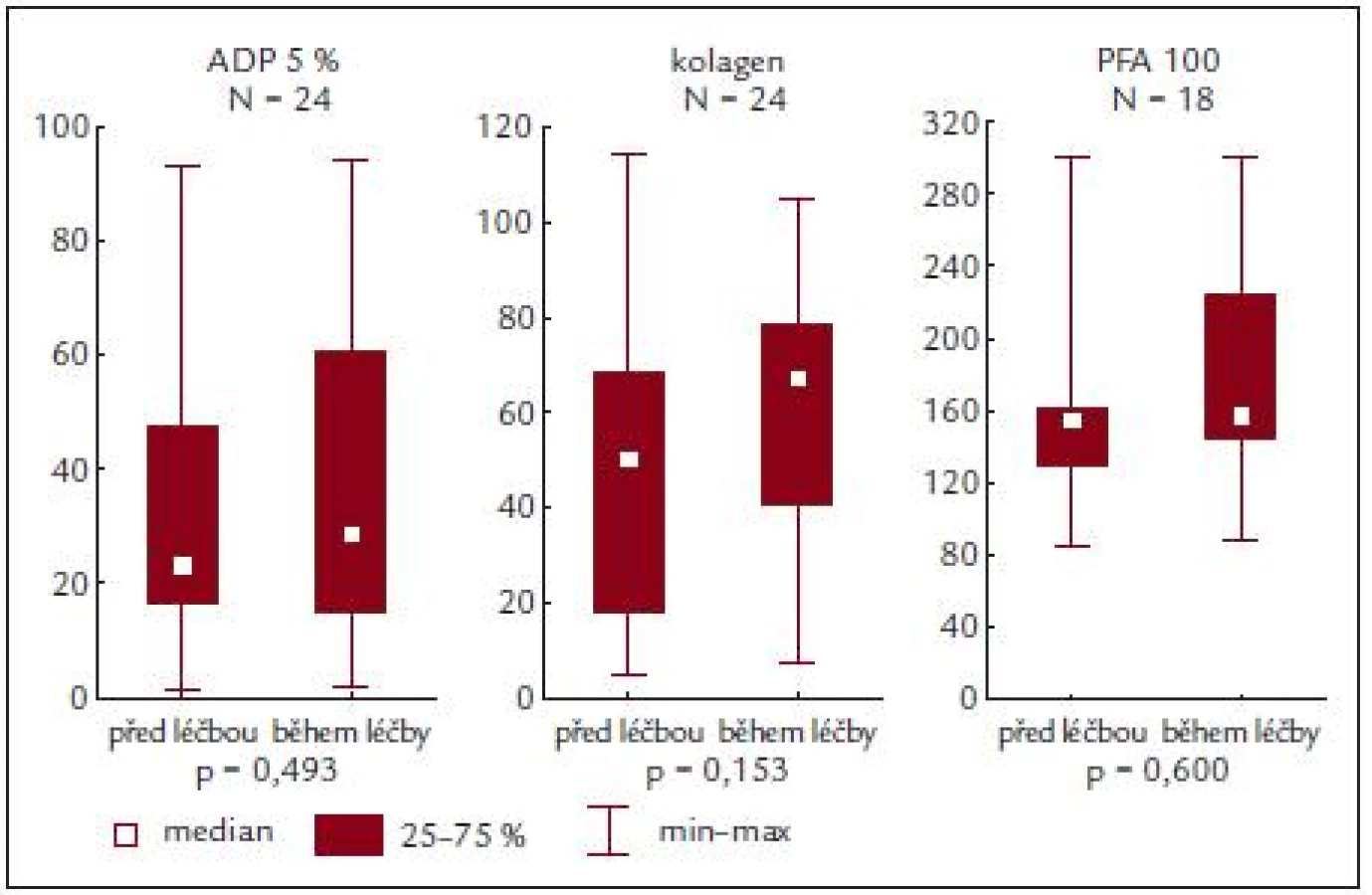
Před léčbou činila průměrná hodnota krevních destiček 814,2 ± 331,8 × 109/l, průměrná hodnota ADP indukované agregace 41,9 ± 27,2%, kolagenem indukované agregace 55,5 ± 30,8 % a průměrná hodnota PFA-100 byla 167,1 ± 62,2 s (obr. 3 a tab. 5).
U pacientů s počtem destiček nižším než 1 000 × 109/l byla ADP agregace 39,9 ± 25,9 %, kolagenem indukovaná agregace 55,2 ± 28,7 a PFA-100 172,1 ±± 63,1 s.
U pacientů s počtem destiček vyšším než 1 000 × 109/l byla ADP agregace 49,0 ± 31,3% a kolagenem indukovaná agregace 56,6 ± 37,9 a PFA-100 148,1 ±± 57,4 s (tab. 6).
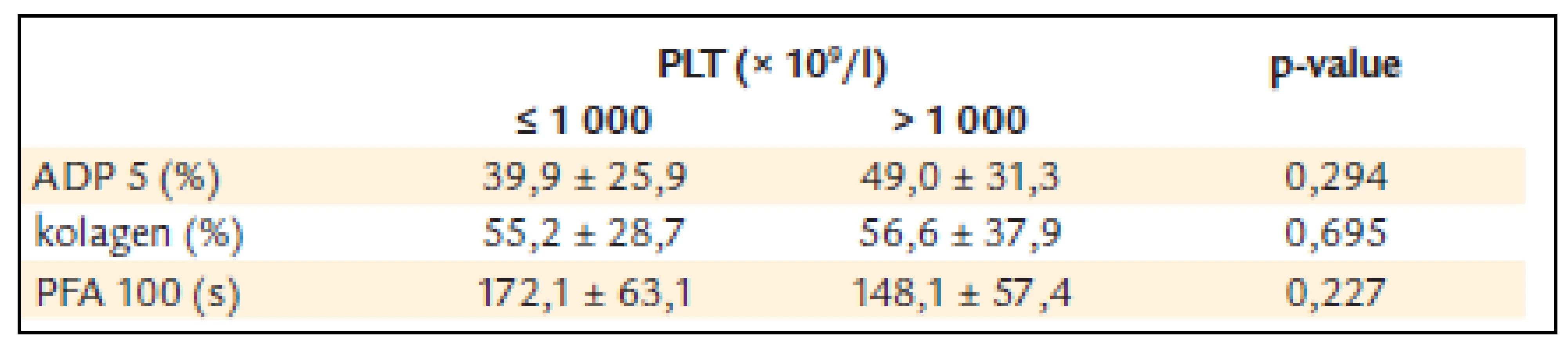
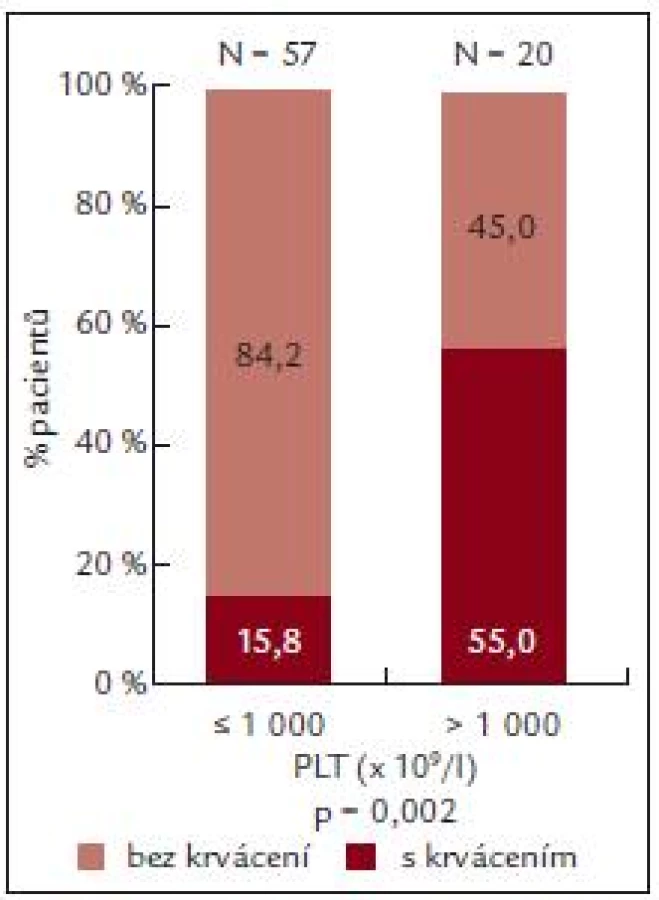
U pacientů s počtem destiček do 1 000 × 109/l byl výskyt krvácení nižší než výskyt krvácení u nemocných s výskytem destiček vyšším než 1 000 × 109/l (n = 20).
U pacientů s výskytem krvácení (n = 20) byla ADP indukovaná agregace 36,8 ± 25,05%, zatímco u pacientů bez krvácení byla ADP agregace 49,76 ± 32,21 % (tab. 7).

U pacientů s výskytem trombózy nebyla zjištěna žádná závislost na hodnotách ADP či kolagenem indukované agregace, taktéž zde nebyly zjištěny rozdíly ani ve výsledcích PFA-100.
Současný výskyt krvácení a trombózy byl pozorován u 2 nemocných bez závislosti na agregaci destiček po ADP a kolagenu či PFA-100.
Diskuze
Ze sledování vyplývá, že u našich nemocných je po stimulaci ADP a kolagenem zjišťována nižší průměrná hodnota agregace, a to jak před zahájením léčby, tak i po ní. Podobně i PFA-100 vykazuje subnormální hodnoty, ale ani zde není zjišťován rozdíl u nemocných před léčbou či v jejím průběhu.
Fenomén poruchy destičkové funkce byl však vyjádřen rozdílně u pacientů s počtem destiček vyšším než 1 000 × 109/l a u pacientů s počtem destiček nižším než 1 000 × 109/l (rovněž však statisticky nevýznamně). Byl přitom (neočekávaně) výraznější u nemocných s počtem destiček nižším než 1 000 × 109/l, u nichž se častěji setkáváme s trombózou (viz Michielsův „doutník a klín“ [10]) a kde bychom očekávali normální funkční schopnost krevních destiček.
Naopak, jak by bylo možné očekávat, u nemocných s výskytem krvácení byla zjišťována statisticky významně nižší agregace po ADP než u nemocných bez krvácivých projevů. Briere [2] se nicméně domnívá, že v souvislosti s krvácením nehraje roli porucha funkce krevních destiček, ale spíše různě vystupňovaná porucha charakteru von Wilebrandovy choroby. Z našich výsledků vyplývá, že funkční změny primární hemostázy určitou roli hrát mohou.
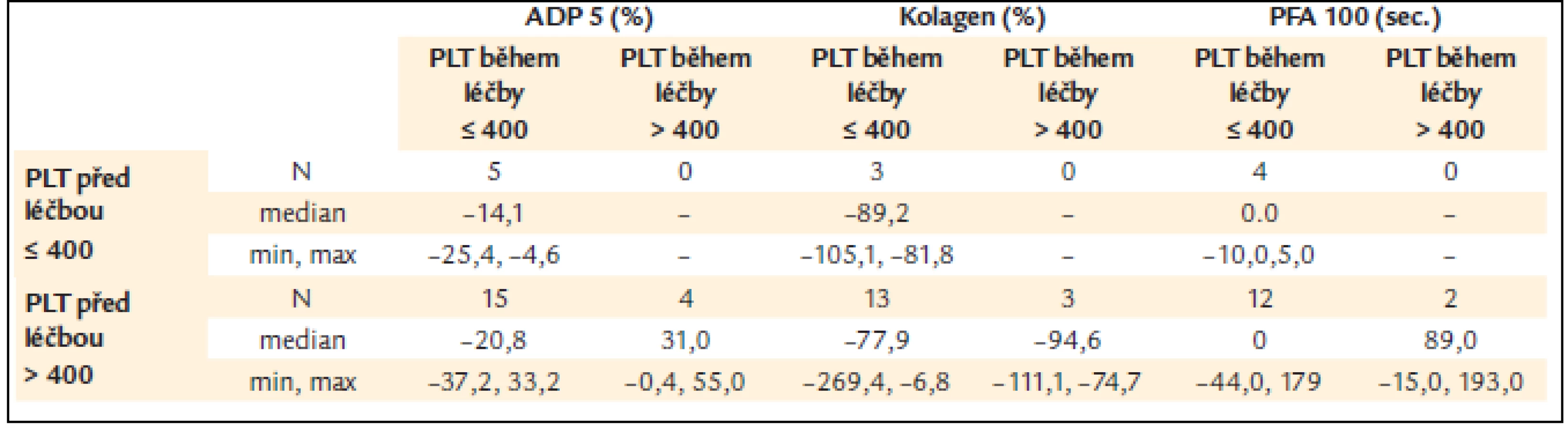
Žádné rozdíly nebyly zjištěny mezi skupinou pacientů s počtem destiček vyšším nebo nižším než 400 × 109/l (= limit kompletní léčebné odpovědi), ale počet destiček pod 400 × 109/l během léčby vykazoval nižší procento poruchy ADP indukované agregace v porovnání s pacienty s počtem destiček vyšším než 400 × 109/l (tab. 8) [13], což by odpovídalo předpokladu úpravy poruchy funkce destiček při dosažení kompletní odpovědi hodnocené pouze dle počtu krevních destiček.
Zajímavým zjištěním ale je, že se hodnoty agregace destiček či PFA-100 nemění ani v důsledku cytoredukční terapie (obr. 5) [13]. Přitom některé výsledky naznačují (viz předchozí odstavec), že se v důsledku léčby upravuje celkový profil nemoci, kdy se při adekvátní kontrole minimalizuje nebezpečí vzniku klinické symptomatologie krvácení či trombózy. Jediným signálem v tomto směru je, že se při kompletní léčebné odpovědi dané snížením počtu krevních destiček snižuje procento patologické agregace indukované ADP [20]. Poněkud odlišná však bude odezva funkce krevních destiček na specifickou antiagregační léčbu [9] a na typ této léčby. Tato okolnost však v naší studii analyzována nebyla.
Závěr
Výsledky analýzy některých destičkových funkcí ukazují možnost odhalení rizika vzniku klinické symptomatologie nezávisle na počtu krevních destiček a mohou přispět k zajištění preventivních opatření či specifické léčby [8].
Zatímco některé výsledky vyznívají v očekávaném smyslu, jiné mají nejen neočekávané, ale i protichůdné vyznění, které bude vyžadovat podrobnější studium na větším souboru a vícečetná opakovaní vyšetření při zohlednění známých okolností stavu nemoci (dosažení odpovědi, přítomnost komorbidity či rizikových faktorů [15], souběžná léčba). Zvláště souběžná léčba antiagregancii, kterou nemusejí mít pacienti paušálně podávanou, může velmi významně sledovaný profil měnit.
Destičkové funkce odrážejí tedy i terapeutické konsekvence a, i když neposkytují jednoznačné vysvětlení (nebo právě proto), budou muset být předmětem dalších studií.
Poděkování
Děkuji za spolupráci také MUDr. Jiřímu Schwarzovi, CSc., vedoucímu České pracovní skupiny pro Ph-negativní myeloproliferativní onemocnění (CZEMP) a všem jejím členům.
prof. MUDr. Miroslav Penka, CSc.
www.fnbrno.cz
e-mail: m.penka@fnbrno.cz
Doručeno do redakce: 31. 1. 2011
Sources
1. Barbui T, Barosi G, Grossi A et al. Practice guidelines for the therapy of essential thrombocythemia. A statement from the Italian Society of Hematology, the Italian Society of Experimental Hematology and Italian Group for Bone Marrow Transplantation. Haematologica 2004; 89 : 215–232.
2. Brière JB. Essential thrombocythemia. Review. Orphanet J Rare Dis 2007; 2–3 : 1–17.
3. Carobbio A, Antoniovi E, Guglielmelli P et al. Leukocytosis and risk stratification assesment in essential trhobmocythemia. J Clin Oncol 2008; 26 : 2732–2736.
4. Falanga A, Marchetti M, Vignoli A et al. Leukocyte-platelet interaction in patients with essential thrombocythemia and polycythemia vera. Exp Hematol 2005; 33 : 523–530.
5. Gruppo Italiano Studio Policitemia. Polycythemia Vera: The NaTURAL History of 1 213 Patients Folowed for 20 Years. Ann Intern Med 1995; 123 : 656–664.
6. Jensen MK, de Nully Brown P, Lund BV et al. Increase platelet activation and abnormal membrane glycoprotein content and redistribution in myeloproliferative disorders. Br J Haematol 2000; 110 : 116–124.
7. Landolfi R, Cipriani MC, Novarese L. Thrombosis and bleeding in polycythemia vera and essential thrombocythemia: Pathogenetic mechanism and prevention. Best Pract Res Clin Haematol 2006; 19 : 617–633.
8. Landolfi R, Di Gennaro L, Falanga A. Thrombosis in myeloproliferative disorders: pathogenetic facts and speculation. Spotlight review. Leukemia 2008; 22 : 2020–2028.
9. Messa RA. Navigating the Evolving Paradigms in the Diagnosis and Treatment of Myeloproliferative Disorders. Hematology Am Soc Hematol Educ Program 2007; 92 : 355–362.
10. Michiels JJ, Berneman Z, Schroyens W et al. Platelet-mediated thrombotic complications in patients with ET: Reversal by aspirin, platelet reduction, and by coumadin. Blood Cells Mol Dis 2006; 36 : 199–205.
11. Passamonti F, Rumi E, Arcaini L et al. Prognostic factors for thrombosis, myelofibrosis, and leukemia in Essentials trhombocythemia: a study of 605 patients. Haematologica 2008; 93 : 1645–1651.
12. Penka M, Schwarz J, Pytlík R et al. Doporučený postup diagnostiky a terapie esenciální trombocytemie a trombocytemie provázející myeloproliferativní onemocnění. Vnitř Lék 2005; 51 : 741–751.
13. Penka M, Schwarz J, Ovesná P et al. Esenciální trombocytemie a jiné myeloproliferace s trombocytemií léčené Thromboreductinem. Výstupy z databáze Registru k prvnímu čtvrtletí roku 2010. Vnitř Lék 2010; 56 : 503–512.
14. Petrides PE, Siegel F. Thrombotic complications in essential trhombocythemia (ET): Clinical Facts and biochemical riddles. Blood Cells Mol Dis 2006; 36 : 379–384.
15. Rosendaal FR. Risk factors for venous thrombosis: prevalence, risk, and interactions. Semin Hematol 1997; 34 : 171–187.
16. Ruggeri M, Gisslinger H, Tosetto A et al. Factor V Leiden mutation carriership and venous thromboembolism in polycythemia vera and essential thrombocythemia. Am J Hematol 2002; 71 : 1–6.
17. Schwarz J, Hrachovinova I, Vorlova Z et al. Thromboembolism in thrombocythemia patients with an additional thrombophilic state. (Abstr. 974). Hematol J 2004; 5 (Suppl 2): S321.
18. StatSoft, Inc. (2009). STATISTICA (data analysis software system), version 9.0. www.statsoft.com
19. Vannucchi AM, Guglielmelli P, Teffer A. Advances in Understanding and Management of Myeloproliferative Neoplasms. Cancer J Clin 2009; 59 : 171–191.
20. Wu KK. Platelet hyperaggregability and thrombosis in patients with thrombocythemia. Ann Intern Med 1978; 88 : 7–11.
Labels
Diabetology Endocrinology Internal medicineArticle was published in
Internal Medicine

2011 Issue 3
Most read in this issue
- Mikroalbuminurie. Od diabetu ke kardiovaskulárnímu riziku
- Zevní vlivy podmiňující vznik nádorů nebo ochraňující před jejich vznikem
- Interna a kardiologie, internisté a kardiologové
- Systolický wall stress ľavej komory srdca pri antihypertenzívnej liečbe