
Přínos PET-CT vyšetření pro rozhodování o léčbě lokalizované nodulární formy plicní AL-amyloidózy
The role of PET-CT in decision making on the treatment of localized nodular form of pulmonary AL-amyloidosis
Depending on the extent of organism affected, there is a systemic (amyloid is deposited in the interstitial space of multiple tissues and organs) and localized (amyloid is deposited in one or a few solitary lesions) form of amyloidosis. Localized forms of amyloidosis have a significantly better prognosis than the systemic ones. The respiratory tract might be affected by diffuse interstitial involvement, associated with systemic AL-amyloidosis, as well as localised involvement of respiratory tract (localised laryngotracheobronchial amyloidosis) or pulmonary parenchyma called nodular form of localized pulmonary amyloidosis. Tracheobronchial form may affect larynx and bronchial tree, and forms plaques or nodules in the epithelium of the respiratory tract. Nodular form causes spherical or irregular lesions in the pulmonary parenchyma, indistinguishable from pulmonary parenchyma metastases. We describe a two-year follow up of a patient with nodular form of pulmonary amyloidosis. The patient had multiple lesions in both lungs, clearly visible on HRCT (High Resolution Computer Tomography) that intensively accumulated fluorodeoxyglucose (FDG) during the first PET-CT. At the time of diagnosis, the largest lesion SUV for FDG accumulation was 8.2. Histochemical analysis showed that amyloid consisted of the light λ chains, i.e. AL-amyloid. Investigations to detect a systemic form of amyloidosis, if present, were negative. The patient had no monoclonal immunoglobulin either in the urine or serum (negative immunofixation) and had normal levels of free light chains in the serum. Her symptoms were previously suggestive of the Sjögren’s syndrome. However, the rheumatologist consulted at the time of diagnosis of the nodular form of pulmonary amyloidosis did not find any signs of an active systemic connective tissue disorder. CRP was repeatedly normal. When systemic AL-amyloidosis was excluded, we decided to only monitor lesion development with no treatment intervention. The patient had 3 PET-CTs. CT showed that no lesions enlarged, some lesions decreased in size slightly. It should be emphasized that follow-up PET-CTs did not show increased FDG accumulation. We assume that the increased FDG accumulation in pulmonary lesions seen during the first PET-CT was due to the activity of the cells that formed this amyloid and that this activity spontaneously ceased, leading to normalization of FDG accumulation in pulmonary nodules. PET-CT is useful for monitoring of the development of pulmonary nodular amyloidosis. Normalization of originally increased FDG accumulation in amyloid lesions suggests cessation of the process of amyloid formation and is a positive prognostic sign.
Key words:
localized amyloidosis – nodular pulmonary amyloidosis – tracheobronchial amyloidosis – AL-amyloidosis – PET-CT imagination – multiple myeloma
Authors:
Z. Adam 1; M. Elleder 2; M. Moulis 3; M. Tichý 4; I. Červinková 5; Z. Řehák 6; R. Koukalová 6; Z. Fojtík 1; I. Hanke 7; L. Pour 1; M. Krejčí 1; L. Zahradová 1; P. Szturz 1; R. Hájek 1; Z. Král 1; J. Mayer 1
Authors‘ workplace:
Interní hematoonkologická klinika Lékařské fakulty MU a FN Brno, pracoviště Bohunice, přednosta prof. MUDr. Jiří Mayer, CSc.
1; Ústav dědičných metabolických poruch 1. lékařské fakulty UK a VFN Praha, přednosta doc. MUDr, . Viktor Kožich, CSc.
2; Radiodiagnostická klinika Lékařské fakulty MU a FN Brno, pracoviště Bohunice, přednostka doc. MUDr. Jarmila Skotáková, Ph. D.
3; Ústav patologie Lékařské fakulty MU a FN Brno, pracoviště Bohunice, přednosta doc. MUDr. Josef Feit, CSc.
4; Oddělení patologie, Lékařské fakulty UP a FN Olomouc, přednosta doc. MUDr. Martin Tichý, CSc.
5; Oddělení nukleární medicíny, centrum PET Masarykova onkologického ústavu Brno, přednosta prim. MUDr. Karol Bolčák
6; Chirurgická klinika Lékařské fakulty MU a FN Brno, pracoviště Bohunice, přednosta prof. MUDr. Zdeněk Kala, CSc.
7
Published in:
Vnitř Lék 2012; 58(3): 241-252
Category:
Case Reports
Overview
Z hlediska rozsahu postižení organizmu amyloidózou se rozlišuje systémová forma (amyloid je deponován v intersticiu více tkání a orgánů) a lokalizovaná forma (amyloid je deponován pouze v jednom či v několika solitárních ložiscích). Lokalizované (ložiskové) formy amyloidózy mají podstatně lepší prognózu než systémové formy. V rámci dýchacího traktu se vyskytuje jednak difuzní intersticiální postižení při systémové AL-amyloidóze a jednak lokalizované postižení dýchacích cest (lokalizovaná laryngo-tracheobronchiální forma amyloidózy) anebo lokalizované postižení plicního parenchymu, nazývané nodulární forma lokalizované plicní amyloidózy. Tracheobronchiální forma může postihnout larynx a bronchiální strom, tvoří plaky či noduly v epitelu dýchacích cest. Nodulární forma tvoří kulovitá či nepravidelná ložiska v plicním parenchymu, na CT zobrazení neodlišitelná od metastáz do plicního parenchymu. Popisujeme dvouleté sledování pacientky s nodulární formou plicní amyloidózy. V obou plicích této pacientky byla četná ložiska, dobře zřetelná na HRCT (high resolution computer tomography) vyšetření, která při prvním PET-CT vyšetření intenzivně akumulovala fluorodeoxyglukózu (FDG). Míra akumulace FDG v největším ložisku v době stanovení diagnózy dosahovala hodnoty SUV 8,2. Histochemická analýza prokázala, že amyloid byl složen z lehkých řetězců λ, šlo tedy o AL-amyloid. Vyšetření, která měla detekovat případnou systémovou formu amyloidózy, byla negativní. Pacientka neměla monoklonální imunoglobulin ani v moči ani v séru (negativní imunofixace) a měla normální hodnoty volných lehkých řetězců v séru. V předchozích letech měla příznaky odpovídající nejspíše Sjögrenovu syndromu. V době stanovení diagnózy nodulární formy plicní amyloidózy však konzultující revmatolog neshledal příznaky aktivity žádného systémového onemocnění pojiva. Hodnota CRP byla opakovaně v normě. Poté, co jsme vyloučili systémovou AL-amyloidózu, jsme se rozhodli pouze sledovat vývoj ložisek bez jakékoli léčebné intervence. Pacientka podstoupila celkem 3 PET-CT vyšetření. V CT obraze se velikost ložisek nezvětšila, některá ložiska se mírně zmenšila. Podstatné je, že při kontrolních PET-CT zobrazení již ložiska neakumulovala zvýšeně fluorodeoxyglukózu. Domníváme se, že podstatou zvýšeného vychytávání fluorodeoxyglukózy v plicních ložiscích amyloidu při prvním vyšetření byla aktivita buněk tvořících tento amyloid a že po roce tato aktivita spontánně vyhasla, což způsobilo normalizaci akumulace fluorodeoxyglukózy v místě plicních nodularit. PET-CT zobrazení je vhodné ke sledování vývoje plicní nodulární amyloidózy. Normalizace původně zvýšené akumulace fluorodeoxyglukózy v ložiscích amyloidu odpovídá vyhasnutí procesu tvorby amyloidu a je prognosticky příznivým znamením.
Klíčová slova:
lokalizovaná amyloidóza – nodulární plicní amyloidóza – tracheobronchiální amyloidóza – AL-amyloidóza – PET-CT zobrazení – mnohočetný myelom
Úvod
Systémová AL-amyloidóza je vzácné onemocnění, jeho incidence v ČR není přesně zmapována. Epidemiologické studie z jiných zemí uvádějí, že výskyt AL-amyloidózy je asi 5–10krát nižší než výskyt mnohočetného myelomu. V evropských zemích se pohybuje incidence všech forem amyloidóz kolem 0,8/100 000 obyvatel [1,2], což by znamenalo, že v ČR se ročně stanoví kolem 80 nových diagnóz této nemoci. Patofyziologickým podkladem AL-amyloidózy je klon plazmatických buněk, které produkují amyloidogenní lehké řetězce imunoglobulinů. Ty se pak deponují extracelulárně dle svého tropizmu, daného jejich molekulárním složením, v různých tkáních a orgánech těla [1–4].
Mimo systémové AL-amyloidózy existují však také velmi vzácné případy ložiskové AL-amyloidózy, při kterých se v organizmu nachází pouze jedno ložisko amyloidu. Předpokládá se, a někdy se i podaří histologickými vyšetřeními doložit, že v okolí ložiska AL-amyloidu jsou klonální buňky, které produkují amyloidogenní lehké řetězce imunoglobulinů. Ty se deponují v jejich okolí a vytvářejí izolovaná solitární ložiska amyloidu. Ložisková amyloidóza nemusí být tvořena jen AL-amyloidem, může to být i AA-amyloid a výjimečně i jiné formy amyloidu [5–7].
Co vše může být postiženo ložiskovou formou amyloidózy, dokládá publikace Biewendla (2006), v níž je popsán soubor pacientů s lokalizovanou amyloidózou, postihující izolovaně kůži, orofarynx, larynx, plíce, žlučník, tlusté střevo, spojivku a lymfatické uzliny, aniž by se při dalším sledování vyvinula systémová AL-amyloidóza [8].
Naší ambulancí prošlo za posledních 20 let 5 pacientů s ložiskovou formou amyloidózy:
- 2 pacienti měli jedno ložisko amyloidózy v tracheobronchiálním stromě, jsou stále ve sledování pulmologů;
- 1 pacient s ložiskem amyloidu v hrtanu, byl operačně vyléčen ORL lékaři;
- 1 nemocný s izolovaným ložiskem amyloidu v močovém měchýři, byl operačně vyléčen urology (operační odstranění ložiska);
- 1 pacientka s mnohočetnými denzními ložisky v plicním parenchymu, v nichž byl prokázán amyloid – nodulární forma plicní amyloidózy.
Všichni zmínění pacienti byli u nás vyšetřeni a někteří opakovaně kontrolováni s otázkou, zda se u nich nevyskytuje systémová forma amyloidózy. U žádného z nich jsme neprokázali další postižení, tedy systémovou amyloidózu, či pozdější přechod z ložiskové do systémové amyloidózy.
Cílem následujícího textu je uvést popis nodulární formy lokalizované plicní amyloidózy a informovat o přínosu PET-CT pro vyhodnocování aktivity procesu. Vzhledem k tomu, že se nám nepodařilo najít v české ani slovenské odborné literatuře publikace o nodulární plicní amyloidóze, předkládáme také podrobnější přehled informací o všech formách postižení plic amyloidózou.
Popis případu
Základní anamnestické údaje
Žena, narozená roku 1950, byla až do svých 50 let zcela zdráva. Od roku 2001 trpěla bolestmi a občasnými otoky drobných kloubů, zejména rukou, s maximem potíží po ránu, kdy pociťovala nutnost krátce se rozhýbat. Dále pacientka uváděla i pocity písku v očích a suchost očí. Stav onemocnění byl z pohledu revmatologa zhodnocen jako stav nejvíce podobný Sjögrenovu syndromu, s intermitentně hraničním až pozitivním antinukleárním faktorem, s pozitivitou anti-Ro a anti-La autoprotilátek, s mírně nadhraniční hodnotou sedimentace (FW), ale s dlouhodobě normální hodnotou CRP. Pacientka byla léčena injekčními kortikoidy (Diprophos) i.m. či intraartikulárně, přechodně byla zkoušena i léčba metotrexatem a leflunomidem (Arava). V době prvního vyšetření revmatologem na našem pracovišti v roce 2009 nebylo shledáno klinicky žádné zánětlivé kloubní postižení ani jiné jednoznačné známky svědčící pro systémové onemocnění pojiva. Pacientka se stran pohybového aparátu cítila dobře.
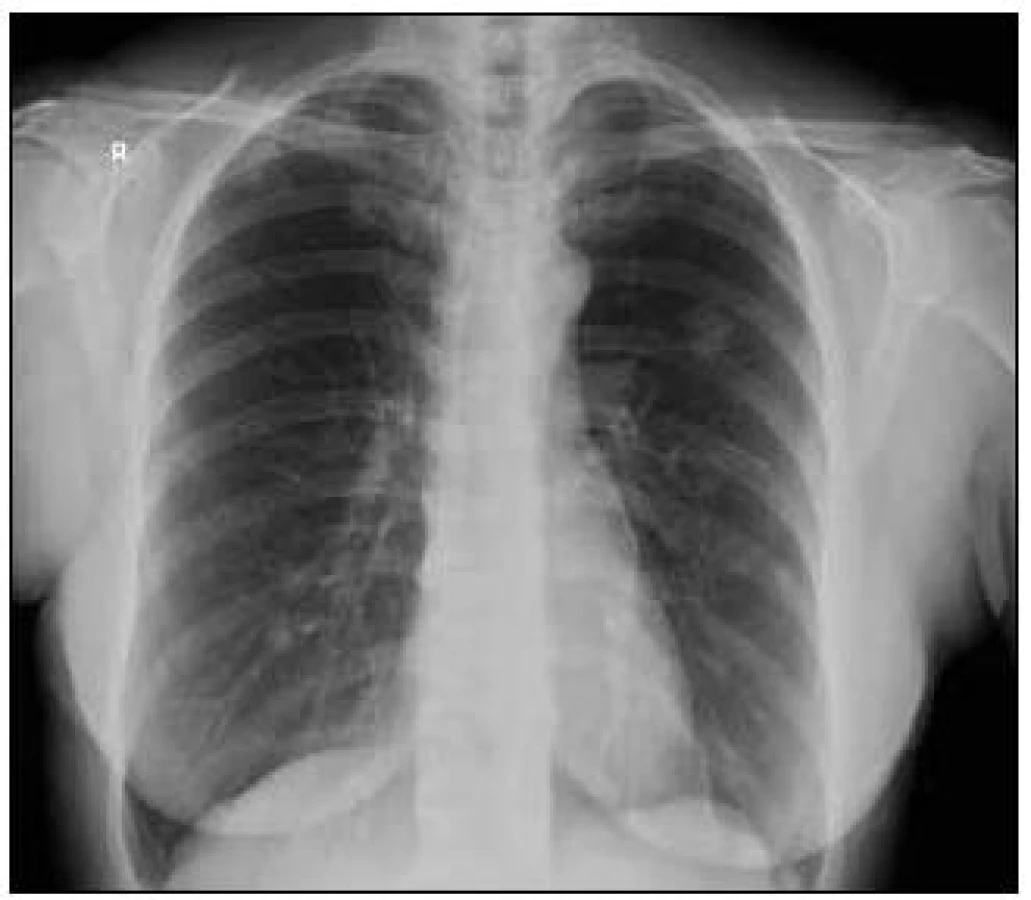
V roce 2008 si pacientka stěžovala na dušnost. Pro tuto dušnost byl proveden RTG snímek plic, který prokázal kulovitá ložiska v plicním parenchymu nejasného původu. Na jaře roku 2009 pacientka podstoupila první plicní operaci, resekci ložiska v plicích s diagnostickým cílem.
Histologické nálezy
První plicní ložisko bylo extirpováno chirurgy v Olomouci a tamtéž bylo provedeno histologické hodnocení. Zpracované vzorky plicní tkáně obsahovaly dobře ohraničená depozita téměř homogenního materiálu, dávajícího pozitivní reakci s konžskou červení. Byly přítomny okrsky depozit antrakopigmentu a kalcifikace. Bezprostředně navazující plicní tkáň vykazovala známky fibrózy a minimální kulatobuněčné celulizace intersticiálně. Plazmocyty roztroušené v okolí amyloidu nevykazovaly monoklonalitu.
Měli jsme před sebou tedy pacientku, u níž bylo extirpováno jedno z vícečetných ložisek v plicním parenchymu, v němž byly prokázány amyloidové hmoty. První histologické vyšetření však neprokázalo typ amyloidu, tedy neurčilo typ bílkoviny. Důležité také bylo, že v okolí ložiska nebyla prokázána infiltrace lymfoproliferativním onemocněním, které by mohlo produkovat amyloidogenní lehké řetězce.
Pro léčebné rozhodnutí však bylo zcela nezbytné znát typ amyloidogenních peptidů, z nichž je ložisko vytvořeno, zda je to AL-, nebo AA-, či zcela jiný typ amyloidu, protože od typu amyloidu se odvíjí léčebný postup. V případě AL-amyloidózy by bylo nutné potlačit klon tvořící amyloidogenní lehké řetězce imunoglobulinů, při AA-amyloidóze by bylo nutné potlačit zánětlivou reakci organizmu, v rámci níž je produkován amylopeptid-A, základ AA-amyloidu. Jednoduše řešeno, ověřit stav systémového onemocnění pojiva a zintenzivnit jeho léčbu.
Z tohoto důvodu byla provedena v pořadí druhá torakoskopie a bylo široce extirpováno největší a dle PET-CT nejaktivnější plicní ložisko. Operatér měl za úkol odebrat i široce zdravou tkáň v okolí ložiska, aby měl patolog dost materiálu k nelezení či vyloučení přítomnosti klonálního lymfocytárního či plazmocytárního onemocnění v okolí.
Morfologické hodnocení druhého extirpovaného plicního ložiska bylo provedeno jednak patology ve FN Brno a dále pak v Ústavu dědičných metabolických poruch VFN Praha. V okolí ložiska byly popsány rudimentální lymfocytární infiltráty. Dále byly patrny známky poměrně intenzivní resorpce amyloidu makrofágy, místy mnohojadernými. Klonální lymfocytární ani plazmocytární populace nebyla v histologických preparátech vyšetřujícími patology potvrzena.
Typ bílkoviny, která tvoří tento amyloid, prokázal prof. Elleder ve spolupráci s prof. P. Westermarkem ze Švédska. Jednalo se o amyloid tvořený lehkými řetězci λ. Historie tohoto závěru nebyla jednoduchá.
Při prvním vyšetření pomocí komerčně dostupných protilátek proti lehkým řetězcům κ a λ byl výsledek imunohistochemických barvení nejednoznačný, i když detekce λ řetězce byla o poznání silnější, ale nebyla uniformní. Naopak převládal, a to dost výrazně, signál pro ApoA1. Tento protein je sice sám amyloidogenní [116], ale zároveň je velmi často průvodním proteinem při depozici amyloidu různého typu [114,115 a Elleder – nepublikovaná pozorování]. Signál pro prealbumin byl slabý, signály pro ostatní amyloidogenní proteiny byly negativní nebo stopové. Přítomny byly i struktury vyššího řádu, které amyloidogenní fibrily vytvářely, a to formace palisádového typu. V souladu s tím dávalo silný signál i sérum amyloid-A protein, konstantní součást amyloidní fibrily.
O konzultaci byl požádán prof. Westermark, který poskytl monoklonální protilátku proti λ řetězcům, vyrobenou v jeho laboratoři. Reakce amyloidních depozit s monoklonální protilátkou proti λ řetězcům z laboratoře prof. Westermarka byla jednoznačná – silná a uniformní.
Závěr je tedy následující: jde o ložiskovou depozici fyzikálně „vyzrálých“ fibril AL-amyloidu.
I při tomto vyšetření byly prokazatelné četné makrofágy, i mnohojaderné, jako reakce na amyloidní depozita. Dále byla patrná i amyloidní infiltrace plicních cév v ložisku, která někdy způsobila obliteraci cévy nebo vedla k trombózám, které se organizovaly. Rozdíly v imunohistochemických barveních při použití běžné komerční protilátky proti lehkým řetězcům λ a při použití protilátky vyrobené v laboratoři prof. Westermarka zobrazuje obr. 1.
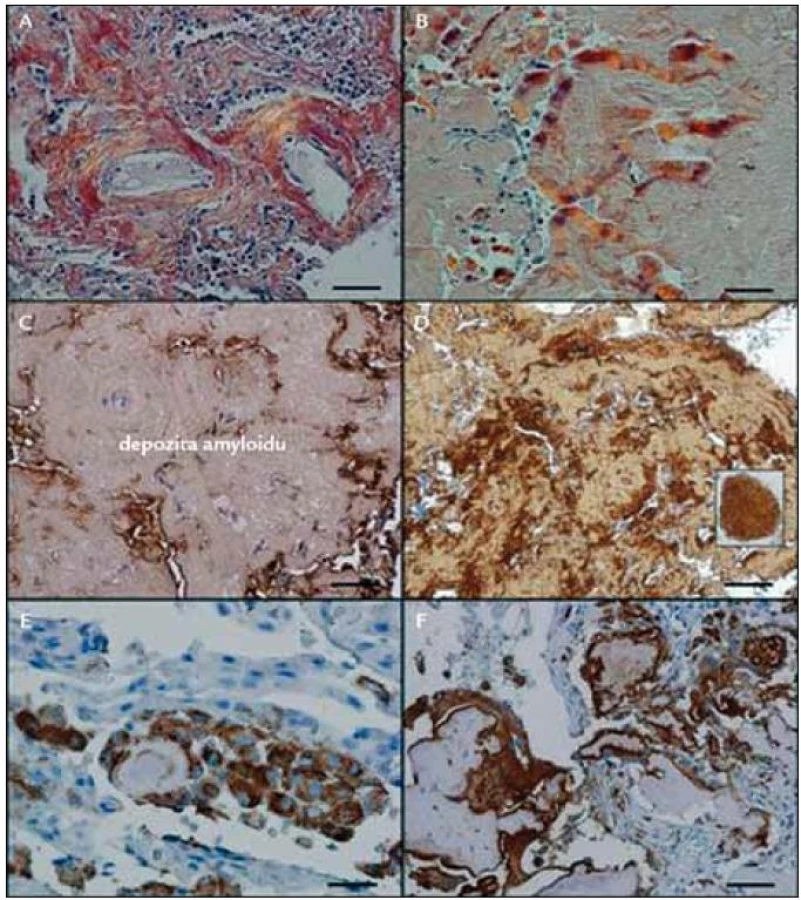
Měli jsme tedy prokázaná vícečetná ložiska v plicním parenchymu složená z AL-amyloidu. Histologická vyšetření, provedená v Olomouci, v Brně a posléze v Praze však nezachytila v okolí ložisek amyloidu klonální lymfocytární, lymfoplazmocytární či plazmocytární infiltráty, které by mohly produkovat klonální amyloidogenní lehké řetězce.
Vzhledem k průkazu lehkých řetězců λ je jasné, že tyto molekuly musí být tvořeny klonální lymfocytární či plazmocytární populací. A i když nebyla patology popsána, tak nejspíše v plicním parenchymu musí existovat. Ať je již původ λ řetězců jakýkoli, faktem je, že ve studovaném ložisku musí existovat podmínky pro amyloidogenezi na podkladě λ řetězce, tedy vytvoření kompletní β fibrily.
Stanovení rozsahu nemoci
U pacientky byla na našem pracovišti provedena vyšetření s cílem prokázat monoklonální gamapatii, nebo alespoň generalizovanou formu AL-amyloidózy. Průkaz monoklonálního imunoglobulinu metodou imunofixace byl v moči i v krvi negativní. Koncentrace imunoglobulinů IgG, IgM a IgA byla ve fyziologickém rozmezí. Koncentrace volných lehkých řetězců κ byla 14,2 mg/l, λ 20,7 mg/l, poměr κ/λ byl 0,69, tedy v mezích normy.
Histologické a cytologické hodnocení kostní dřeně neprokázalo žádný patologický nález, žádnou klonální expanzi plazmocytů či lymfocytů.
V rámci pátrání po dalších místech depozit AL-amyloidu byla provedena biopsie sliznice rekta, amyloid v ní však nebyl prokázán. Funkce ledvin byla neporušená, echokardiografie srdce bez patologického nálezu. Nenalezli jsme žádné další známky poškození organizmu amyloidem, vyjma ložisek v plicním parenchymu.
Takže jediným patologickým nálezem při zobrazovacích vyšetřeních byl nález v plicním parenchymu na HRCT vyšetření, které bylo u nás provedeno v rámci stanovení rozsahu choroby. V plicním parenchymu byla vícečetná denzní ložiska s nepravidelnými cípatými okraji. Většina ložisek byla umístěna subpleurálně, 2 vlevo v plicním parenchymu. Jejich velikost byla popsána následovně: vpravo v segmentu S3 ložisko velikosti 8 mm a 6 mm, v segmentu S2 ložisko 10 × 15 mm, v segmentu S5 ložisko velikosti 12 mm, v segmentu S4 ložisko velikosti 13 mm, v segmentu S10 ložisko velikosti 5 mm, vlevo v segmentu S3 ložisko 20 × 18 mm, další ložiska pak velikosti 8 a 6 mm, v segmentu S6 ložiska o průměru 5 a 11 mm a v segmentu S9 ložisko velikosti 5 mm. Oblast mediastina byla bez patologických změn. Vstupní nálezy v plicním parenchymu dokumentují obr. 2–7.
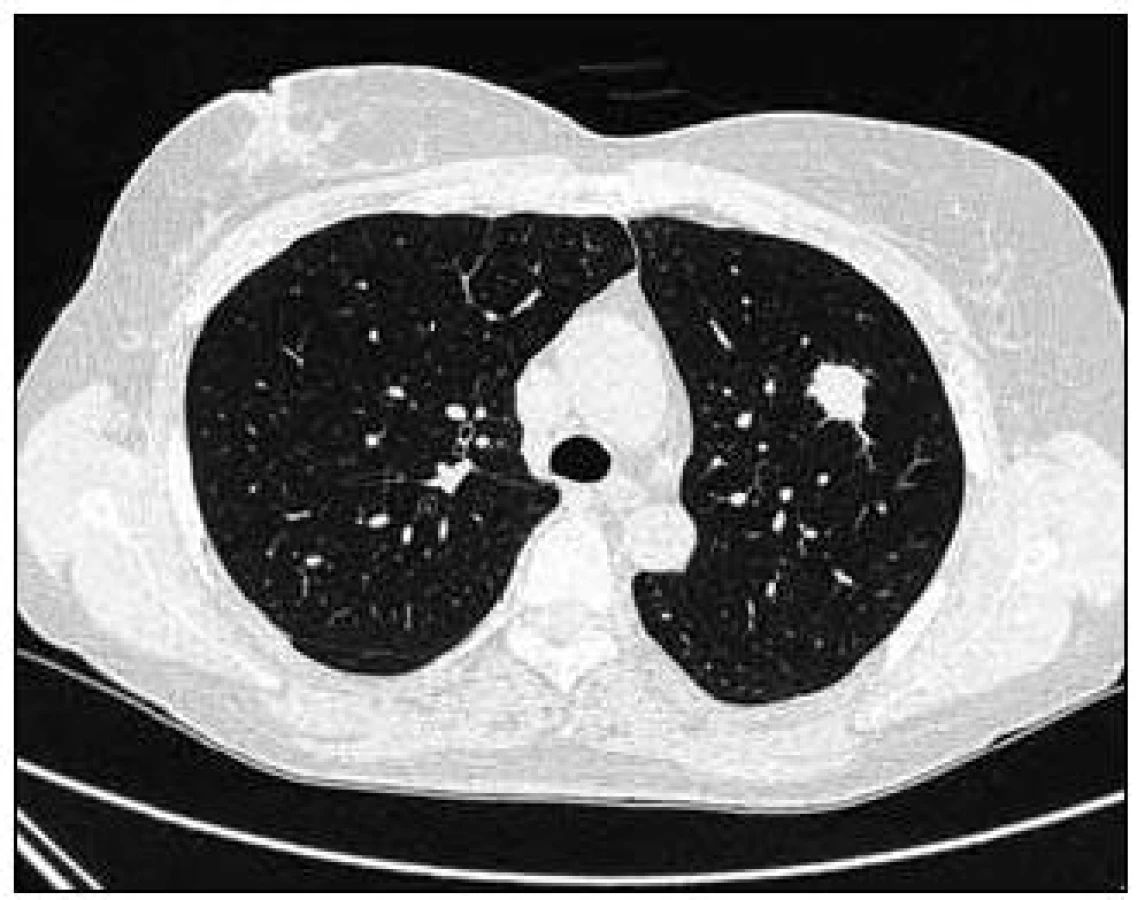
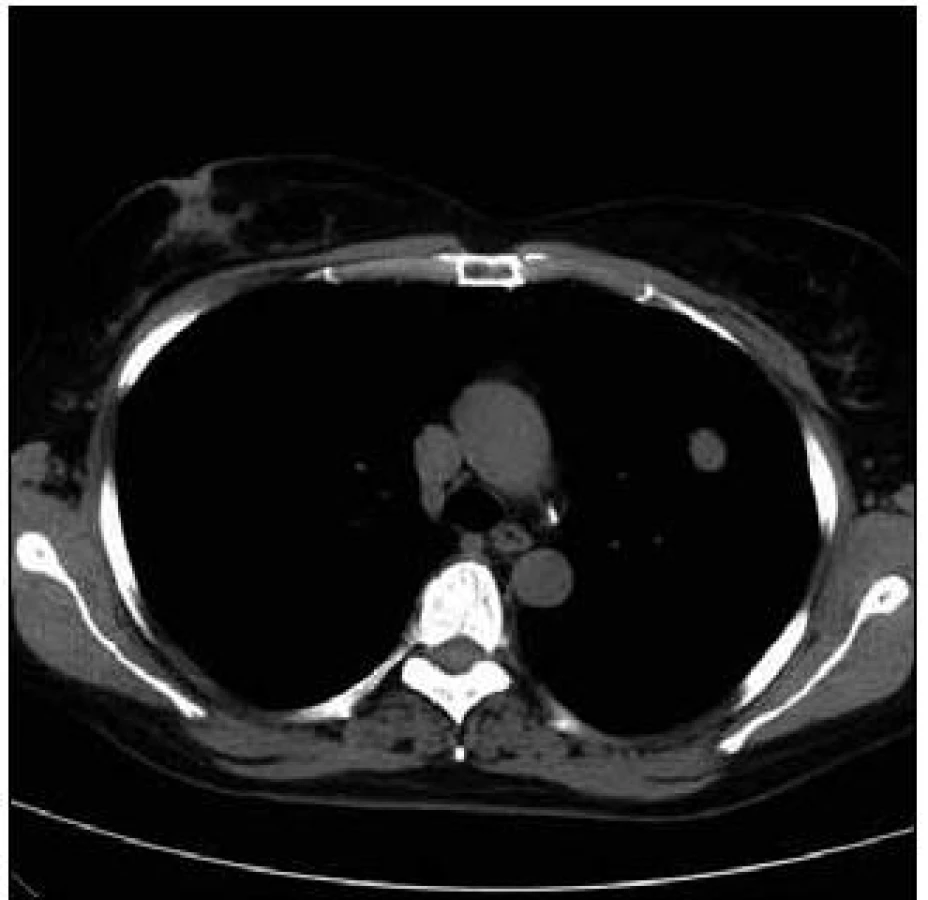

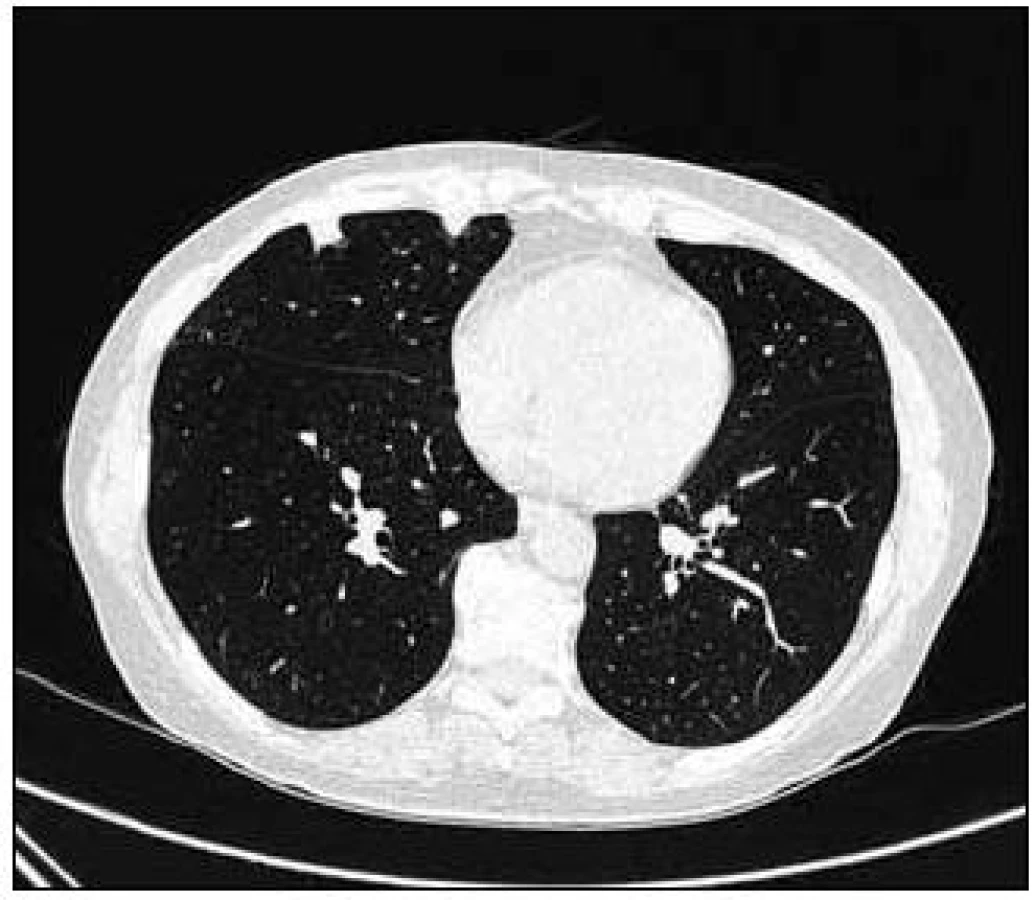
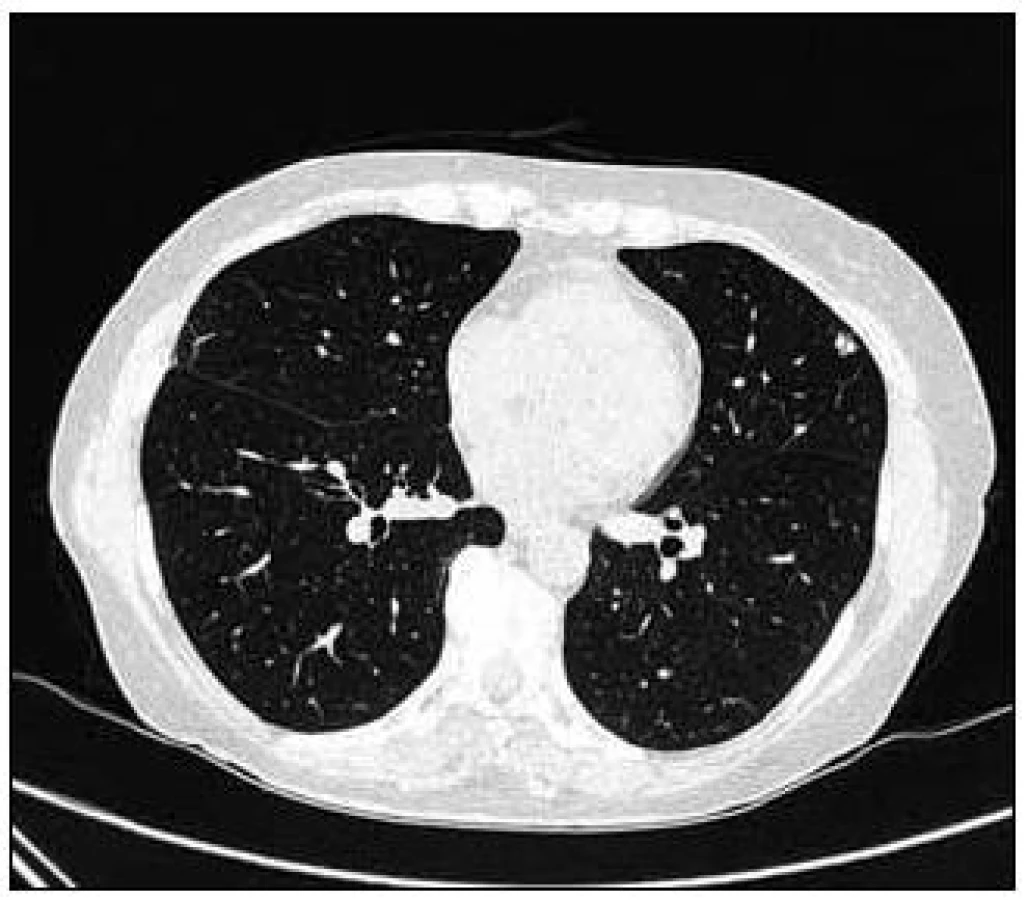
V rámci stanovení rozsahu nemoci jsme pacientku dále vyšetřili metodou PET-CT. První vyšetření bylo provedeno v září roku 2009 (obr. 8). PET komponenta vyšetření zachytila 2 ložiska patologicky zvýšené akumulace fluorodeoxyglukózy (FDG) v pravé a 2 v levé plíci. CT komponenta zachytila ložisek více, ale jejich velikost dle CT vyšetření byla již pod rozlišovací schopnost PET detektoru, takže u ložisek menších než 8 mm, zřetelných na CT vyšetření, nelze říci, zda měla také zvýšenou akumulace fluorodeoxyglukózu. Akumulace fluorodeoxyglukózy v největším ložisku v levé plíci dosáhla hodnoty SUV 8,02.

U pacientky jsme tedy, vyjma postižení plic amyloidem, neprokázali známky monoklonální gamapatie ani primární systémové AL-amyloidózy či mnohočetného myelomu s AL-amyloidózou.
Sledování aktivity nemoci
Protože pacientka byla bez dechových obtíží, funkční plicní vyšetření neprokázalo poruchu plicní ventilace, tak jsme nepodali žádnou léčbu a pacientku jsme pouze sledovali. Velikost ložisek byla hodnocena pomocí HRCT (vysoce senzitivní počítačová tomografie – high resolution computer tomography) zobrazení. Globální funkce plic byla sledována odpovídajícími funkčními vyšetřeními. Dále jsme průběžně provedli 2 kontrolní PET-CT vyšetření, protože na vstupním vyšetření nás překvapila vysoká akumulace v ložiscích amyloidu. Je jasné, že fluorodeoxyglukózu neakumuluje amyloid, ale buňky v okolí ložiska.
Při kontrolním HRCT vyšetření plic bylo konstatováno, že nedochází k žádné progresi velikosti ložisek. Dle kontrolních PET-CT zobrazení (poslední bylo provedeno v březnu roku 2011) byla v CT obraze, s výjimkou resekovaného největšího ložiska z levého plicního laloku, opět zřetelná sledovaná plicní ložiska, některá bez vývoje, některá s nepatrným zmenšením.
Při hodnocení akumulace fluorodeoxyglukózy bylo zjištěno, že ložiska, která při 1. vyšetření vykazovala zvýšenou akumulaci fluorodeoxyglukózy, ji při kontrolních PET-CT vyšetřeních již zvýšeně neakumulovala. Zpočátku zjištěná zvýšená akumulace fluorodeoxyglukózy v plicních nodulech amyloidu nebyla při 2 kontrolních vyšetřeních již vůbec patrná (obr. 9).
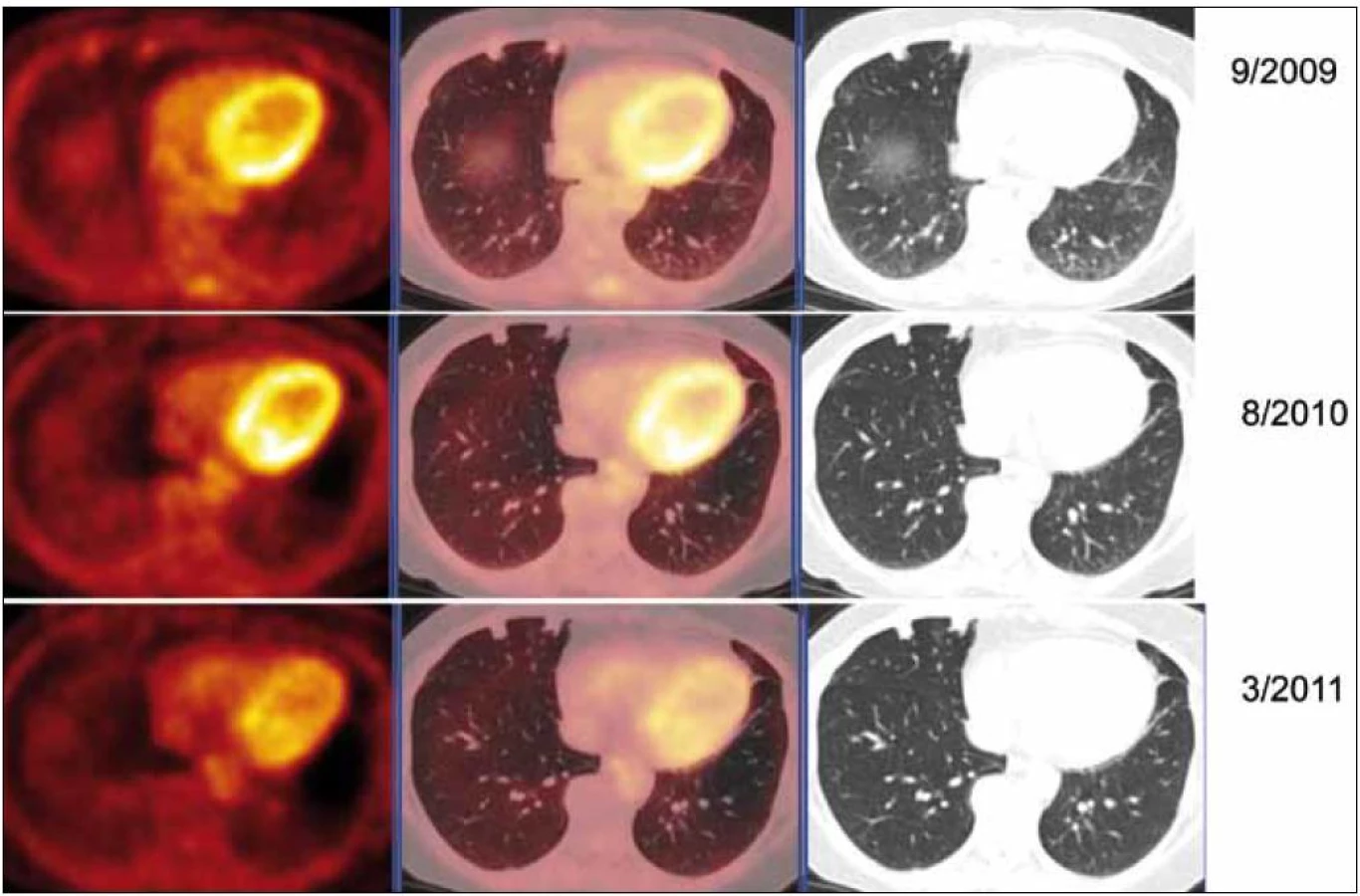
U pacientky jsme zatím nepřikročili k léčbě. Sledujeme velikost ložisek a jejich metabolickou aktivitu (HRCT a PET-CT) v jejich okolí. Léčbu plánujeme podat pouze v případě, že by velikost ložisek narůstala anebo že by narůstala metabolická aktivita v okolí ložisek.
Vstupně vysoká aktivita při PET zobrazení a vyhasnutí této vysoké akumulace při kontrole po roce signalizuje, že po roce ustala metabolická aktivita buněk v okolí ložiska, které se s vysokou pravděpodobností podílely na vzniku ložisek amyloidu.
Diskuze
Klasifikace AL-amyloidózy
Lehké řetězce imunoglobulinů se mohou ukládat v tkáních ve formě AL-amyloidu, ale i v jiných formách. Případy, kdy se lehké řetězce ukládají ve formě amorfních hmot, se nazývají „light chain deposition disease“, česky „nemoc z ukládání lehkých řetězců v amorfní podobě“. Lehké řetězce se mohou ukládat i v krystalech, které jsou fagocytovány histiocyty, anglický termín pro tento stav je „cristal storing histiocytosis“. I když v názvu je slovo histiocytóza, nejedná se o onemocnění z histiocytární řady, primární je vytvoření krystalků lehkých řetězců, které jsou špatně degradovatelné, a proto zůstávají viditelné v histiocytech, které je fagocytují [1,9].
AL-amyloidóza je termín pro onemocnění spojené s tvorbou extracelulárních depozit z lehkých řetězců. Pokud dochází k ukládání těchto depozit ve více tkáních či orgánech, tak se jedná o tzv. „systémovou AL-amyloidózu“.
Plazmocelulární proliferace, která tvoří amyloidogenní lehké řetězce imunoglobulinů, má buď benigní charakter, nebo charakter maligní, odpovídající mnohočetnému myelomu. Pokud má proliferace klonálních plazmocytů benigní charakter, tak se používá termín „primární systémová AL-amyloidóza“. Pokud má klonální proliferace maligní charakter, odpovídající mnohočetnému myelom, tak se pro tento stav používá termín „mnohočetný myelom s AL-amyloidózou“. Hranice mezi primární systémovou AL-amyloidózou a amyloidózou při mnohočetném myelomu je uměle vytvořená (arbitrární) a mění se průběžně dle definic přijímaných pro tyto nemoci a dle senzitivity vyšetření, která pro naplnění kritérií použijeme [1].
Opakem systémové amyloidózy s difuzním poškozením organizmu depozity amyloidu a špatnou prognózou jsou lokalizované amyloidózy, při kterých se v organizmu nachází pouze jedno ložisko nebo několik málo izolovaných (solitárních) ložisek amyloidu. Tyto lokalizované amyloidózy jsou mnohem vzácnější než systémové formy a mají podstatně lepší prognózu.
Amyloid v solitárním ložisku může svým složením odpovídat AL-amyloidu, ale může být vytvořen i z jiných amyloidogenních peptidů, druhý nejčastější je AA-amyloid.
Etiologie lokalizované formy amyloidózy
Etiologie lokalizované (ložiskové) amyloidózy nebývá vždy jasná. Mnohé publikace pouze popisují tento jev (přítomnost ložiska amyloidu) bez další specifikace typu amyloidu a nevyjadřují se k patofyziologii vzniku ložiska [10].
Problémem totiž bývá přesné stanovení složení amyloidu. To vyžaduje speciální postupy a nemusí se ve všech případech podařit. Dalším problémem je identifikovat v okolí ložiska ty buňky, které jsou zodpovědné za produkci amyloidogenních bílkovin a jejich ukládání v ložisku.
V některých případech lokalizované plicní formy AL-amyloidózy, či lokalizovaného ložiska amorfních hmot vytvořených z lehkých řetězců, byla v okolí nalezena nějaká forma lymfoproliferace typu nízce agresivního (indolentního) lymfomu či plazmocytomu [11–19].
V jiných případech byla v zdánlivě zánětlivé populaci kolem ložiska amyloidu popsána monoklonalita infiltrujících plazmatických buněk. Pro tyto závěry svědčily výsledky imunohistochemického vyšetření (průkaz exprese κ a λ řetězců na plazmocytech) a dále závěry analýzy plazmocytů v okolí ložiska pomocí polymerázové řetězové reakce [20–24].
Mnohé popisy případů však pouze konstatují zánětlivou buněčnou populaci v okolí ložiska bez jasně viditelného lymfomu či plazmocytomu. Tak je tomu i v našem případě.
V případě ložiskové formy AA-amyloidózy se předpokládá produkce amylopeptidu-A v nejbližším okolí ložiska AA-amyloidu.
V současnosti je tedy akceptována hypotéza, že ve všech případech lokalizované AL-amyloidózy či depozit lehkých řetězců ve formě amorfních hmot, anebo i ložiskové AA-amyloidózy je příčinou lokální syntéza amyloidogenního proteinu. Tato hypotéza se jeví více pravděpodobnou, než že by se ložisko vytvořilo z amyloidogenních proteinů syntetizovaných v jiné části organizmu. Pro tvorbu amyloidových proteinů v bezprostředním okolí ložiska svědčí i fakt, že v žádném ze sledovaných případů ložiskové AL-amyloidózy nebo AA-amyloidózy se nevyvinula systémová AL-amyloidóza nebo AA-amyloidóza [8].
To, zda se podaří prokázat patologickou buněčnou populaci v okolí ložiska, záleží na metodice zkoumání lymfocytární a plazmocytární populace v okolí ložiska. Je možné, že těchto klonálních buněk, tvořících amyloidové fibrily, není mnoho a že jejich průkaz bez speciálních postupů, popsaných v citovaných pracích, se nemusí vždy podařit. Dále je nutné přihlédnout k tomu, že nízce agresivní (indolentní) formy lymfomů mívají spontánní remise, takže je i možné, že klonální populace v době odběru vzorku na histologii již spontánně regredovala a zůstalo po ní vytvořené ložisko amyloidu.
Po histologickém zjištění, že ložisko je tvořeno amyloidem, je zásadní stanovení typu bílkoviny, která se ukládá v depozitech amyloidu, neboť od toho se odvíjí léčba.
A podobně jako u všech onkologických nemocí, zásadní je získání přesných informací o rozsahu nemoci, o rozsahu postižení organizmu, v tomto případě depozity amyloidu [25–28].
Formy postižení dýchacího systému amyloidem
Plicní parenchym může být postižen v rámci systémové AL-amyloidózy nebo pouze lokálními solitárními ložisky amyloidu. Tato druhá forma má podstatně lepší prognózu. Většina autorů [29–37] se shoduje na následující klasifikaci postižení plicního parenchymu a dýchacích cest.
Postižení plic při systémové formě amyloidózy
- intersticiální depozita AL-amyloidu v plicním parenchymu při systémové AL-amyloidóze,
- depozita AL-amyloidu v pohrudnici při systémové AL-amyloidóze.
Postižení plic při lokalizované formě amyloidózy
- lokalizovaná laryngo-tracheobronchiální amyloidóza,
- lokalizovaná nodulární plicní amyloidóza.
Intersticiální postižení plic při systémové amyloidóze
Intersticiální postižení plic při systémové AL-amyloidóze
Primární systémová AL-amyloidóza obvykle postihuje difuzně jednu či více tkání. To, která tkáň je postižena v rámci primární systémové AL-amyloidózy, záleží na tropizmu klonálních volných lehkých řetězců, z kterých je AL-amyloid utvořen.
Pokud primární systémová AL--amyloidóza postihuje plicní parenchym, tak tvoří depozita v septech alveolů difuzně po celé plíci. Postižení plicního parenchymu však bývá jen výjimečně diagnostikováno u žijících pacientů se systémovou AL-amyloidózou [38–40].
Naopak při sekčním zkoumání těl osob, zemřelých v důsledku systémové AL-amyloidózy, bylo prokázáno plicní postižení u 23 za 26 pitvaných [41] a v další studii u 11 ze 12 pitvaných [42].
Také postižení pleury při systémové AL-amyloidóze bylo popsáno [43]. Postižení pleury amyloidem může vyvolávat recidivující plicní výpotky [44–46].
Z těchto faktů lze odvodit, že postižení plicního parenchymu při systémové AL-amyloidóze je tedy velmi časté, ale obvykle dominují příznaky z postižení jiných orgánů depozity AL-amyloidu.
Celli et al se domnívali, že plicní postižení bylo důvodem smrti pouze u 1 ze 12 pitvaných pacientů s AL-amyloidózou [42], a Cordier se domníval, že plicní postižení vedlo ke smrti pouze u 10 % zemřelých pacientů s AL-amyloidózou [31].
Vzhledem k tomu, že míra postižení plic depozity AL-amyloidu korelovala s mírou postižení srdce AL-amyloidem, bylo pro přežití těchto nemocných limitujícím poškození myokardu amyloidem [41]. A tak průkaz plicního amyloidu signalizuje nepříznivou prognózu, protože současně je poškozen i myokard [41,47].
Podkladem difuzní plicní amyloidózy může být jak primární systémové AL-amyloidóza, tak systémová AL-amyloidóza, provázející mnohočetný myelom [47–54].
Plicní hypertenze
V některých případech byla popsána závažná plicní hypertenze při difuzní plicní amyloidóze, která limitovala život nemocného [55–57].
Krvácení z plic
Další vzácnou komplikací je krvácení z plicního parenchymu důsledkem amyloidózy se vznikem smrtící hemoptýzy [58].
Intersticiální plicní amyloidóza lokalizovaná pouze v plicním parenchymu
Zcela výjimečné jsou popisy případu intersticiálního plicního postižení bez prokazatelné systémové AL-amyloidózy [59–62].
Intersticiální postižení plicní při AA-amyloidóze
Sekundární AA-amyloidóza je méně častá než AL-amyloidóza, a tak je postižení plicního parenchymu touto formou amyloidózy vzácnější. Postižení plicního parenchymu zaznamenali v 1 případě v souboru 113 pacientů s AA-amyloidózou [41].
Lokalizované formy postižení plic amyloidózou
Lokalizovaná tracheobronchiální amyloidóza
Lokalizovaná tracheobronchiální amyloidóza, postihující dýchací cesty, tedy larynx a bronchiální strom, je relativně vzácná. Depozita amyloidu v dýchacích cestách tvoří relativně ploché plaky nebo nodularity. V literatuře lze najít popisy jak solitárních, tak vícečetných plaků amyloidu vyskytujících se v kterékoli části laryngu i bronchiálního stromu.
Příznakem těchto amyloidových plaků může být kašel, dušnost, hemoptýza nebo opakované pneumonie, či dokonce atelaktáza za překážkou tvořenou ložiskem amyloidu.
Lokalizovaná tracheobronchiální amyloidóza obvykle není projevem systémové AL-amyloidózy, jde opravdu o lokalizovaná depozita, proto má příznivou prognózu. Přesto ale může svého nositele ohrozit na životě opakovanými bronchopneumoniemi při bronchiální obstrukci anebo krvácením při narušení cév pod výstelkou dýchacích cest [63–73].
Nodulární amyloidóza plic
Nodulární plicní amyloidóza je nejvzácnější formou plicní amyloidózy. Podobně jako tracheobronchiální forma, také nebývá zpravidla provázena systémovou AL-amyloidózou. Vzácnost této formy může ilustrovat fakt, že na Mayo Clinic nalezli za 13leté období pouze 7 případů [36]. V plicním parenchymu může být přítomen jeden či více nodulů tvořených amyloidovými hmotami. Noduly mohou být umístěny v kterémkoli místě plicního parenchymu, mívají kulovitý vzhled, takže je možné je zaměnit s plicními metastázami. Jejich velikost se pohybuje od necelého centimetru do několika centimetrů, největší ložisko popisované literaturou mělo 15 cm v průměru.
Etiologicky je nodulární amyloidóza plicní dávána do souvislosti s lokalizovanou indolentní lymfocytární či plazmocytární proliferací s tvorbou amyloidotvorných klonálních lehkých řetězců imunoglobulinů. Další možností je lokální tvorba amylopeptidu-A. Ve většině publikovaných případů nebyla popsána progrese s nutností léčby [74–85] a tak je tomu i u naší pacientky.
V některých případech byla nodulární plicní amyloidóza popsána u pacientů se Sjögrenovým syndromem či jinými autoimunitními nemocemi pojiva [86–90].
Výjimečně mohou být v plicním parenchymu nalezena kulovitá ložiska z depozit lehkých řetězců v neamyloidové, amorfní podobě [91,92].
Zcela výjimečně je některá z forem plicní amyloidózy spojena se vznikem depozit amyloidu v uzlinách [93,94].
Terapie lokalizované amyloidózy
Lokalizovaná plicní amyloidóza se léčí pouze v případech, kdy dělá klinické potíže. Léčba je vždy lokální, to znamená odstranění ložiska s minimem okolní tkáně. A to je možné udělat jak chirurgicky přímým zásahem, tak endobronchiálním přístupem. V případě tracheobronchiální amyloidózy se používá destrukce ložiska laserovým paprskem či jiným způsobem, anebo se provádí cílená radioterapie (brachyradioterapie).
Pokud solitární ložisko nečiní potíže, pouze se sleduje. Vzhledem k tomu, že umístění ložiska v luminu dýchacích cest dělá větší potíže než nodulární plicní amyloidóza, je nutnost terapie častější u intraluminální, tracheobronchiální a laryngeální amyloidózy. Nenalezli jsme případ, v němž by byla popsána nutnost léčby nodulární plicní amyloidózy [95–100].
Možnosti sledování vývoje ložiskové plicní amyloidózy
V minulých letech byly rutině dostupné HRCT ke sledování velikosti a funkční plicní vyšetření ke sledování míry narušení plicních funkcí [101–105].
V posledních letech je zde k dispozici ještě PET-CT vyšetření. Toto vyšetření jednoznačně prokázalo svůj přínos pro pacienty s mnohočetným myelomem [106–108].
PET-CT vyšetření bylo přínosné i při lokalizované nodulární plicní formě AL - i AA-amyloidózy, provázející Sjögrenův syndrom. Např. v jedné publikaci se uvádí, že před zahájením léčby Sjögrenova syndromu byla vysoká akumulace fluorodeoxyglukózy v místě histologicky ověřeného ložiska, tvořeného AA-amyloidem, s hodnotou SUV 10,1. Po 6 letech léčby Sjögrenova syndromu došlo ke zmenšení velikosti ložiska dle CT hodnocení a zcela vymizela zvýšená akumulace fluorodeoxyglukózy [114].
Z toho plyne, že nodulární ložiska amyloidu se mohou zobrazit při PET-CT vyšetření jako vysoce aktivní, s intenzivním vychytáváním fluorodeoxyglukózy, podobně jako plicní metastázy.
PET-CT zobrazení neumožňuje rozpoznat, zda se jedná o zánětlivé nebo o nádorové ložisko, či o ložisko amyloidu. Umožní však jasně zviditelnit vývoj metabolické aktivity těchto ložisek.
Je jasné, že amyloid samotný nevychytává fluorodeoxyglukózu, za vychytávání fluorodeoxyglukózy jsou zodpovědné buňky v bezprostřední blízkosti ložiska, které se na tvorbě ložiska přímo podílejí. A jak uvádí literatura, při vyhasnutí amyloidotvorného procesu, ať již je to proces tvořící nodulární formu AA-amyloidu, nebo AL-amyloidu, dochází k vyhasnutí akumulace fluorodeoxyglukózy v ložisku a jeho bezprostředním okolí [109–114].
Podobný vývoj jsme pozorovali u naší pacientky – zpočátku byla aktivita vysoká, později při kontrolních PET-CT vyšetřeních došlo k vyhasnutí akumulace fluorodeoxyglukózy. Nezvětšování ložisek a ztrátu aktivity ložisek při opakovaném PET-CT vyšetření považujeme za prognosticky příznivé.
Vyhasnutí akumulace glukózy v oblasti nodulů amyloidu si vysvětlujeme jako vyhasnutí amyloidotvorného procesu.
Problémy se stanovením typu amyloidu v ČR
Rozpoznání extracelulárních depozit a po histochemické reakci s konžskou červení stanovení diagnózy amyloidu je běžně proveditelné ve všech morfologických laboratořích. Potvrzení, že se jedná o depozita amyloidu, pak přinese vyšetření elektronovou mikroskopií, které potvrdí lineární strukturu depozita, typickou pro amyloid.
Velkým problémem je však přesně stanovit typ proteinu, z něhož se amyloid vytvořil.
Stanovení typu amyloidu vyžaduje znalosti a praxi a dostatečné spektrum imunohistochemických vyšetření. Pro stanovení typu amyloidu se používají imunohistochemické reakce, jejichž cílem je přesně pojmenovat proteiny, které se v ložisku amyloidu uložily. Zde jen připomeneme, že reakce AL-amyloidu s protilátkami proti lehkým řetězcům může být falešně negativní, protože při přepracování amyloidogenního lehkého řetězce imunoglobulinu se pozmění antigenní determinanty, proti nimž jsou protilátky proti lehkým řetězcům zaměřené. A zde, jak je vidět z popisu prof. Elledera, hodně záleží na typu použité protilátky.
Diferenciální diagnostika hereditárních typů amyloidu spočívá v průkazu přítomnosti či nepřítomnosti dnes známých amyloidotvorných mutací bílkovin séra, z nichž se může vytvořit amyloid (např. mutovaný transthyretin). Vyšetření přítomností bílkovin séra, z nichž by se mohl vytvářet některý z hereditárních typů amyloidu, nejsou však běžně dostupná, proto je nutné se obrátit na specializované pracoviště [115–117].
Nejnovějším diagnostickým postupem je proteomické vyšetření s pomocí hmotností spektrometrie, která může nejpřesněji rozpoznat typ amyloidu [118]. Taková analýza se však zatím v ČR neprovádí. Bylo by velmi žádoucí, aby v ČR vzniklo alespoň pracoviště, které by se zabývalo diferenciální diagnostikou amyloidóz.
Závěr pro praxi
- Mnohočetná plicní ložiska při CT zobrazení i s vysokou aktivitou při PET-CT zobrazení nemusí vždy znamenat pouze metastatický proces, mohou být způsobena nodulární formou plicní amyloidózy. Jedině histologické ověření ložiska může rozpoznat izolovanou nodulární formu plicní amyloidózy.
- Nodulární forma plicní AL-amyloidózy obvykle nevyžaduje léčbu, léčba je oprávněná pouze při progresi ložiska, dokumentované na opakovaných CT zobrazeních plic a při zvyšující se aktivitě v PET zobrazení.
- Vymizení PET aktivity při sledování nodulární plicní amyloidózy signalizuje příznivou prognózu.
Práce byla vypracována v rámci aktivity následujících grantů: výzkumného záměru MZ ČR: FUNDIN MZ0MOU2005, výzkumného záměru MŠMT MSM0021622434, specifického výzkumu MUNI/A/0784/2011 a grantů IGA MZd NT11154, NT12130 a NT12215.
prof. MUDr. Zdeněk Adam, CSc.
www.fnbrno.cz
e-mail: z.adam@fnbrno.cz
Doručeno do redakce: 14. 7. 2011
Přijato po recenzi: 5. 9. 2011
Sources
1. Ščudla V, Pika T. Současné možnosti diagnostiky a léčby systémové AL-amyloidózy. Vnitř Lék 2009; 55 (Suppl 1): S77–S84.
2. Sideras K, Gerz MA. Amyloidosis. Adv Clin Chem 2009; 47 : 1–44.
3. Adam Z, Ščudla V. Clinical manifestations of AL-amyloidosis and some othertypes of amyloidosis. Vnitř Lék 2001; 47 : 36–45.
4. Adam Z, Ščudla V, Tomíska M. Léčba AL--amyloidózy a některých dalších typů amyloidóz. Vnitř Lék 2001; 47 : 46–52.
5. Džingozovová M, Skričková J, Mačák J et al. Lokalizovaná forma tracheobronchiální amyloidózy AA-typu, a její odlišení od systémové formy mayloidózy v klinické praxi. Vnitř Lék 2009; 55 : 593–598.
6. Kuchyňka P, Paleček T, Šimek S et al. The isolated form of cardiac amyloidosis in the form of beginning infiltrative cardiomyopathy without restrictive physiology. Vnitř Lék 2008; 54 : 1010–1013.
7. Krejčí J. Cardiac amyloidosis – an underestimated threat? Vnitř Lék 2008; 54 : 950–951.
8. Biewend ML, Menke DM, Calamia KT. The spectrum of localized amyloidosis: a case series of 20 patients and review of the literature. Amyloid 2006; 13 : 135–142.
9. Adam Z, Pour L, Krejcí M et al. Poškození ledvin u mnohočetného myelomu a u dalších monoklonálních gamapatií. Vnitř Lék 2008; 54 : 847–861.
10. Westlake S, Edwards CJ. Pulmonary nodular amyloidosis and systemic lupus erythematosus. Lupus 2005; 14 : 639–640.
11. Georghiou GP, Boikov O, Vidne BA et al. Primary pulmonary amyloidosis due to low-grade B cell lymphoma. Asian Cardiovasc Thorac Ann 2007; 15 : 69–71.
12. Lim JK, Lacy MQ, Kurtin PJ et al. Pulmonary marginal zone lymphoma of MALT type as a cause of localised pulmonary amyloidosis. J Clin Pathol 2001; 54 : 642–646.
13. Michaels L, Hyams VJ. Amyloid in localised deposits and plasmacytomas of the respiratory tract. J Pathol 1979; 128 : 29–38.
14. Ihling C, Weirich G, Gaa A et al. Amyloid tumors of the lung – an immunocytoma? Pathol Res Pract 1996; 192 : 446–452.
15. King CS, Holley AB, Sherner JH. Severe bullous lung disease due to marginal-zone-lymphoma-associated amyloidosis. Respir Care 2008; 53 : 1495–1498.
16. Satani T, Yokose T, Kaburagi T et al. Amyloid deposition in primary pulmonary marginal zone B-cell lymphoma of mucosa-associated lymphoid tissue. Pathol Int 2007; 57 : 746–750.
17. Wieker K, Röcken C, Koenigsmann M et al. Pulmonary low-grade MALT-lymphoma associated with localized pulmonary amyloidosis. A case report. Amyloid 2002; 9 : 190–193.
18. Nakamura N, Yamada G, Itoh T et al. Pulmonary MALT lymphoma with amyloid production in a patient with primary Sjögren’s syndrome. Intern Med 2002; 41 : 309–311.
19. Sugai S. Mucosa-associated lympoid tissue (MALT) lymphoma and primary amyloidosis in the lung in Sjögren’s syndrome. Intern Med 2002; 41 : 251–252.
20. Khoor A, Myers JL, Tazelaar HD et al. Amyloid-like pulmonary nodules, including localized light-chain deposition: clinicopathologic analysis of three cases. Am J Clin Pathol 2004; 121 : 200–204.
21. Miyamoto T, Kobayashi T, Makiyama M et al. Monoclonality of infiltrating plasma cells in primary pulmonary nodular amyloidosis: detection with polymerase chain reaction. J Clin Pathol 1999; 52 : 464–467.
22. Ross P Jr, Magro CM. Clonal light chain restricted primary intrapulmonary nodular amyloidosis. Ann Thorac Surg 2005; 80 : 344–347.
23. Stokes MB, Jagirdar J, Burchstin O et al. Nodular pulmonary immunoglobulin light chain deposits with coexistent amyloid and nonamyloid features in an HIV-infected patient. Mod Pathol 1997; 10 : 1059–1065.
24. Kaplan B, Martin BM, Boykov O et al. Co-deposition of amyloidogenic immunoglobulin light and heavy chains in localized pulmonary amyloidosis. Virchows Arch 2005; 447 : 756–761.
25. Yanagawa N, Ogata SY, Motoyama T. Pulmonary localized AA type amyloidosis with cyst-like structures and marginal zone B-cell lymphoma of the MALT type coexisting independently in the left upper lung. Intern Med 2008; 47 : 1529–1533.
26. Shah PL, Gillmore JD, Copley SJ et al. The importance of complete screening for amyloid fibril type and systemic disease in patients with amyloidosis in the respiratory tract. Sarcoidosis Vasc Diffuse Lung Dis 2002; 19 : 134–142.
27. Rostagno A, Frizzera G, Ylagan L et al. Tumoral non-amyloidotic monoclonal immunoglobulin light chain deposits (“aggregoma”): presenting feature of B-cell dyscrasia in three cases with immunohistochemical and biochemical analyses. Br J Haematol 2002; 119 : 62–69.
28. Okuda M, Okuda Y, Ogura T et al. Primary lung involvement with amyloid deposition in Waldenstöm’s macroglobulinemia: observations from over 20 years. Respirology 2004; 9 : 414–418.
29. Beji M, Louzir B, Haouet S et al. Primary localized bronchopulmonary amyloidosis. Rev Pneumol Clin 1996; 52 : 36–38.
30. Berk JL, O’Regan A, Skinner M. Pulmonary and tracheobronchial amyloidosis. Semin Respir Crit Care Med 2002; 23 : 155–165.
31. Cordier JF. Pulmonary amyloidosis in hematological disorders. Semin Respir Crit Care Med 2005; 26 : 502–513.
32. Lachmann HJ, Hawkins PN. Amyloidosis and the lung. Chron Respir Dis 2006; 3 : 203–214.
33. Miura K, Shirasawa H. Lambda III subgroup immunoglobulin light chains are precursor proteins of nodular pulmonary amyloidosis. Am J Clin Pathol 1993; 100 : 561–566.
34. Pitz MW, Gibson IW, Johnston JB. Isolated pulmonary amyloidosis: case report and review of the literature. Am J Hematol 2006; 81 : 212–213.
35. Xu L, Cai BQ, Zhong X et al. Respiratory manifestations in amyloidosis. Chin Med J (Engl) 2005; 118 : 2027–2033.
36. Utz JP, Swensen SJ, Gertz MA. Pulmonary amyloidosis. The Mayo Clinic experience from 1980 to 1993. Ann Intern Med 1996; 124 : 407–413.
37. Howard ME, Ireton J, Daniels F et al. Pulmonary presentations of amyloidosis. Respirology 2001; 6 : 61–64.
38. Šterclová M, Matěj R. Difuzní plicní amyloidóza jako příčina recidivujícího fluidothoraxu. Kazuistiky v pneumologii 2006; 3 : 4–8.
39. Cordier JF, Loire R, Brune J. Amyloidosis of the respiratory tract. Clinical and pathologic features in a series of 21 patients. Chest 1989; 90 : 827–231.
40. Lewinsohn G, Bruderman I, Bohdana A. Primary difuse pulmonary amyloidosis with monoclonal gammopathy. Chest 1976; 69 : 682–685.
41. Smith RR, Hutchins GM, Moore GW et al. Type and distribution of pulmonary parenchymal and vascular anuloid. Correlation with cardiac amyloidosis. Am J Med 1979; 66 : 96–104.
42. Celli BR, Rubinow A, Cohen AS et al. Patterns of pulmonary involvement in systemic amyloidosis. Chest 1978; 74 : 543–547.
43. Romero Candeira S, Martín Serrano C, Hernandez Blasco L. Amyloidosis and pleural disease. Chest 1991; 100 : 292–293.
44. Berk JL, Keane J, Seldin DC et al. Persistent pleural effusions in primary systemic amyloidosis: etiology and prognosis. Chest 2003; 124 : 969–977.
45. Berk JL, Wiesman JF, Skinner M et al. Diaphragm paralysis in primary systemic amyloidosis. Amyloid 2005; 12 : 193–196.
46. Berk JL. Pleural effusions in systemic amyloidosis. Curr Opin Pulm Med 2005; 11 : 324–328.
47. Duygu H, Akilli A, Ozerkan F et al. Primary cardiac amyloidosis mimicking interstitial lung disease and bleeding diathesis: a case report. Heart Surg Forum 2007; 10: E177–E179.
48. Cottin V, Cordier JF. Interstitial pulmonary amyloidosis. Respiration 2008; 75 : 210.
49. Doshi A, Rodrigues M, Deshpande R et al. Spontaneous resolution of diffuse alveolar septal amyloidosis. Indian J Chest Dis Allied Sci 2001; 43 : 177–179.
50. Ege E, Uzaslan E, Ursavaş A et al. Primary pulmonary amyloidosis associated with multiple myeloma. Tuberk Toraks 2006; 54 : 65–70.
51. Haydar AA, Li A, Hilton R et al. Eosinophilia and symptomatic pulmonary amyloidosis. Postgrad Med J 2004; 80 : 738–739.
52. Hess T, Egerer G, Kasper B et al. Atypical manifestations of multiple myeloma: radiological appearance. Eur J Radiol 2006; 58 : 280–285.
53. Chim CS, Wong M, Fan Y. Pulmonary interstitial amyloidosis complicating multiple myeloma. J Clin Oncol 2008; 26 : 504–506.
54. Vervaet V, Smeets P, Verstraete K. Diffuse amyloidosis of the lung associated with multiple myeloma. JBR-BTR 2003; 86 : 176–177.
55. Dingli D, Utz JP, Gertz MA. Pulmonary hypertension in patients with amyloidosis. Chest 2001; 120 : 1735–1738.
56. Eder L, Zisman D, Wolf R et al. Pulmonary hypertension and amyloidosis – an uncommon association: a case report and review of the literature. J Gen Intern Med 2007; 22 : 416–419.
57. Lehtonen J, Kettunen P. Pulmonary hypertension as a dominant clinical picture in a case of amyloidosis and smoldering multiple myeloma. Int J Cardiol 2007; 115: e29–e30.
58. Sterlacci W, Veits L, Moser P et al. Idiopathic systemic amyloidosis primarily affecting the lungs with fatal pulmonary haemorrhage due to vascular involvement. Pathol Oncol Res 2009; 15 : 133–136.
59. Boyd King A, Sharma O, Stevenson K. Localized interstitial pulmonary amyloid: a case report and review of the literature. Curr Opin Pulm Med 2009; 15 : 517–520.
60. Fujimoto N, Masuoka H, Kosaka H et al. Primary amyloidosis with pulmonary involvement which presented exudative pleural effusion and high fever. Intern Med 2003; 42 : 756–760.
61. Orriols R, Aliaga JL, Rodrigo MJ et al. Localised alveolar-septal amyloidosis with hypersensitivity pneumonitis. Lancet 1992; 339 : 1261–1262.
62. Poh SC, Tria TS, Seah HC. Primary difuse alveolar septal amyloidosis. Torax 1975; 30 : 186–191.
63. Cazalets C, Belleguic C, Sost G et al. Tracheobronchial amyloidosis: apropos of 2 cases. Rev Med Interne 2002; 23 : 317–321.
64. Daniels JT, Cury JD, Diaz J. An unusual cause of postobstructive pneumonia. Chest 2007; 131 : 930–933.
65. Deutschmann HA, Schwarz T, Schoellnast H et al. Isolated tracheobronchial amyloidosis: a rare cause of a hilar space-occupying lesion. Rofo 2006; 178 : 1264–1266.
66. Hanon S, De Keukeleire T, Dieriks B et al. Primary tracheobronchial amyloidosis: a series of 3 cases. Acta Clin Belg 2007; 62 : 56–60.
67. Hui AN, Koss MN, Hochholzer L et al. Amyloidosis presenting in the lower respiratory trakt. Clinicopathologic, radiologic, immunohistochemical, and histochemical studies on 48 cases. Arch Pathol Lab Med 1986; 110 : 212–218.
68. Minogue SC, Morrisson M, Ansermino M. Laryngo-tracheo-bronchial stenosis in a patient with primary pulmonary amyloidosis: a case report and brief review. Can J Anaesth 2004; 51 : 842–845.
69. Peterman W, Barth J, Schlüter E. Localised amyloidosis of central airweys. Eur J Respir Dis 1987; 71 : 210–212.
70. Racil H, Ben Amar J, Rouhou SC et al. Haemoptysis revealing tracheobronchial amyloidosis. Rev Pneumol Clin 2011; 67 : 109–112.
71. Rubinow A, Celli BR, Cohen AS et al. Localised amyloidosis of the respiratory tract. Am Rev Respir Dis 1978; 118 : 212–218.
72. Thomson PJ, Citron KM. Amyloid and the lower respiratory tract. Torax 1983; 38 : 84–87.
73. Yamazaki S, Kanda S, Yasuo M et al. Laryngo-tracheo-bronchial amyloidosis presenting severe airway stenosis. Intern Med 2006; 45 : 1021–2022.
74. Breitenfelder M, Kusch E, Leichsenring M et al. Pulmonary nodular amyloidosis mimicking multiple pulmonary metastases of carcinoma of the corpus uteri. Chirurg 2001; 72 : 1062–1066.
75. Cotton RE, Jackson JW. Localized amyloid tumour’s of the lung simulating malignant neoplasmasms. Thorax 1964; 19 : 97–103.
76. Gaurav K, Panda M. An uncommon cause of bilateral pulmonary nodules in a long-term smoker. J Gen Intern Med 2007; 22 : 1617–1620.
77. Holme S Desal JBN, Sapsford RN. Nodular pulmonary amyloidosis. A case report and review of the literature. Br J Dis Chest 1988; 82 : 414–417.
78. Kos S, Savic S, Steinbrich W. Nodular form of primary pulmonary amyloidosis. Rofo 2007; 179 : 1277–1279.
79. Papla B, Rudnicka L. Primary amyloid tumors of the lungs – six cases. Pol J Pathol 2005; 56 : 197–202.
80. Pusztaszeri M, Kamel EM, Artemisia S et al. Nodular pseudotumoral pulmonary amyloidosis mimicking pulmonary carcinoma. Thorax 2005; 60 : 440.
81. Seith A, Kumar J, Hari S et al. Isolated nodular pulmonary amyloidosis. Intern Med J 2008; 38 : 293–294.
82. Siong Chuan L, Johnson HA. Multiple nodular pulmonary amyloidosis. Torax 1975; 30 : 178–185.
83. Skagseth E Jr, Normann T. Localized amyloid tumour of the lung. Scand J Thorac Cardiovasc Surg 1970; 4 : 135–138.
84. Suzuki H, Matsui K, Hirashima T et al. Three cases of the nodular pulmonary amyloidosis with a longterm observation. Intern Med 2006; 45 : 283–286.
85. Tokunaga T, Takeda S, Sawabata N et al. Nodular pulmonary amyloidosis. Jpn J Thorac Cardiovasc Surg 2006; 54 : 399–401.
86. Calatayud J, Candelas G, Gómez A et al. Nodular pulmonary amyloidosis in a patient with rheumatoid arthritis. Clin Rheumatol 2007; 26 : 1797–1798.
87. Niepolski L, Grzegorzewska AE, Szymaś J. Nodular pulmonary amyloidosis and Sjögren’s syndrome in a patient treated with intermittent hemodialysis. Hemodial Int 2007; 11 : 406–410.
88. Rodrigues K, Neves FS, Stoeterau KB et al. Pulmonary amyloidosis in Sjögren’s syndrome: a rare diagnosis for nodular lung lesions. Int J Rheum Dis 2009; 12 : 358–360.
89. Srinivas P, Liam CK, Jayaram G. Localised nodular pulmonary amyloidosis in a patient with sicca syndrome. Med J Malaysia 2000; 55 : 385–387.
90. Kambouchner M, Godmer P, Guillevin L. Low grade marginal zone B cell lymphoma of the breast associated with localised amyloidosis and corpora amylacea in a woman with long standing primary Sjögren’s syndrome. J Clin Pathol 2003; 56 : 74–77.
91. Khoor A, Myers JL, Tazelaar HD et al. Amyloid-like pulmonary nodules including localised light chain deposition: clinicopathologic analysis of three cases. Amer J Clin Pathol 2004; 121 : 200–204.
92. Bhargava P, Rushin JM, Rusnock EJ et al. Pulmonary light chain deposition disease: report of five cases and review of the literature. Am J Surg Pathol 2007; 31 : 267–276.
93. Yong HS, Woo OH, Lee JW et al. Primary localized amyloidosis manifested as supraclavicular and mediastinal lymphadenopathy. Br J Radiol 2007; 80: e131–e133.
94. Dalton HR, Featherstone T, Athanasou N. Organ limited amyloidosis with lymphadenopathy. Postgrad Med J 1992; 68 : 47–50.
95. Kalra S, Utz JP, Edell ES et al. External-beam radiation therapy in the treatment of diffuse tracheobronchial amyloidosis. Mayo Clin Proc 2001; 76 : 853–856.
96. Madden BP, Lee M, Paruchuru P. Successful treatment of endobronchial amyloidosis using Nd: YAG laser therapy as an alternative to lobectomy. Monaldi Arch Chest Dis 2001; 56 : 27–29.
97. Monroe AT, Walia R, Zlotecki RA et al. Tracheobronchial amyloidosis: a case report of successful treatment with external beam radiation therapy. Chest 2004; 125 : 784–789.
98. Neben-Wittich MA, Foote RL et al. External beam radiation therapy for tracheobronchial amyloidosis. Chest 2007; 132 : 262–267.
99. Piazza C, Cavaliere S, Foccoli P et al. Endoscopic management of laryngo-tracheobronchial amyloidosis: a series of 32 patients. Eur Arch Otorhinolaryngol 2003; 260 : 349–354.
100. Rubinow A, Celli BR, Cohen AS et al. Localized amyloidosis of the lower respiratory tract. Am Rev Respir Dis 1978; 118 : 603–611.
101. Aylwin AC, Gishen P, Copley SJ. Imaging appearance of thoracic amyloidosis. J Thorac Imaging 2005; 20 : 41–46.
102. Kim HY, Im JG, Song KS et al. Localized amyloidosis of the respiratory system: CT features. J Comput Assist Tomogr 1999; 23 : 627–631.
103. Konietzko N. Amyloidoses of the lung. Pneumologie 2004; 58 : 339–343.
104. Urban BA, Fishman EK, Goldman SM et al. CT evaluation of amyloidosis: spectrum of disease. Radiographics 1993; 13 : 1295–1308.
105. Horger M, Lengerke C, Pfannenberg C et al. Significance of the “halo” sign for progression and regression of nodular pulmonary amyloidosis: radiographic-pathological correlation (2005 : 6b). Eur Radiol 2005; 15 : 2037–2040.
106. Mysliveček M, Bačovský J, Ščudla V et al. 18F-FDG PET/CT and 99mTc-MIBI scintigraphy in evaluation of patients with multiple myeloma and monoclonal gammopathy of unknown significance: comparison of methods. Klin Onkol 2010; 23 : 325–331.
107. Bačovský J, Mysliveček M, Ščudla V et al. Tc-99m MIBI scintigraphy in multiple myeloma: prognostic value of different Tc-99m MIBI uptake patterns. Clin Nucl Med 2010; 35 : 667–670.
108. Umeda Y, Demura Y, Takeda N et al. FDG-PET findings of nodular pulmonary amyloidosis with a long-term observation. Nihon Kokyuki Gakkai Zasshi 2007; 45 : 424–429.
109. Currie GP, Rossiter C, Dempsey OJ et al. Pulmonary amyloid and PET scanning. Respir Med 2005; 99 : 1463–1464.
110. Grubstein A, Shitrit D, Sapir EE et al. Pulmonary amyloidosis: detection with PET-CT. Clin Nucl Med 2005; 30 : 420–421.
111. Kung J, Zhuang H, Yu JQ et al. Intense fluorodeoxyglucose activity in pulmonary amyloid lesions on positron emission tomography. Clin Nucl Med 2003; 28 : 975–976.
112. Ollenberger GP, Knight S, Tauro AJ. False-positive FDG positron emission tomography in pulmonary amyloidosis. Clin Nucl Med 2004; 29 : 657–658.
113. Yadav S, Sharma S, Gilfillan I. Unusual positron emission tomography findings in pulmonary amyloidosis: a case report. J Cardiothorac Surg 2006; 1 : 32.
114. Tan H, Guan Y, Zhao J et al. Findings of pulmonary amyloidosis on dual phase FDG PET/CT imaging. Clin Nucl Med 2010; 35 : 206–207.
115. Sakata N, Hoshii Y, Nakamura T et al. Colocalization of apolipoprotein AI in various kinds of systemic amyloidosis. J Histochem Cytochem 2005; 53 : 237–242.
116. Booth DR, Tan SY, Booth SE et al. A new apolipoprotein Al variant, Trp50Arg, causes hereditary amyloidosis. QJM 1995; 88 : 695–702.
117. Röcken C, Shakespeare A. Pathology, diagnosis and pathogenesis of AA amyloidosis. Review. Virchows Arch 2002; 440 : 111–122.
118. Sethi S, Theis JD, Leung N et al. Mass spectrometry-based proteomic diagnosis of renal immunoglobulin heavy chain amyloidosis. Clin J Am Soc Nephrol 2010; 5; 2180–2187.
Labels
Diabetology Endocrinology Internal medicineArticle was published in
Internal Medicine

2012 Issue 3
Most read in this issue
- Castlemanova choroba
- Přínos PET-CT vyšetření pro rozhodování o léčbě lokalizované nodulární formy plicní AL-amyloidózy
- Gen pro FTO a jeho role v genetické determinaci obezity
- Zmeny krvného tlaku u chronicky hemodialyzovaných pacientov