
Péče o pacienty s hemofilií a jejich rodiny v ÚHKT
Care for patients with haemophilia and their families at The Institute of Haematology and Blood Transfusion in Prague
Beginning of the treatment of patients with hemophilia in IHBT (Institute of Hematology and Blood Transfusion) dates back to the 60th the last century. To date it was created a comprehensive care; from diagnostic to preventive and therapeutic. Diagnostic is concentrated in the NRL for hemostasis, there got all the available methodologies, protein and molecular-genetic. Therapeutic and preventive care is concentrated in the Center for Thrombosis and Hemostasis (CTH IHBT) and clinical departments of IHBT. In collaboration with orthopedic, surgical and other specialized clinics they provide care during invasive procedures. IHBT was appointed as one of the two CCC (Comprehensive Care Centers) for the diagnosis and treatment of adult patients with hemophilia in the Czech Republic. CTH is taking care about more than 200 patients with hemophilia A/B. One third of patients have a severe type of hemophilia. Part of the preventive care it is the active search for female carriers of hemophilia and the collaboration with centers of medical genetics in prenatal diagnosis of hemophilia. About half of the patients of Czech Republic with a complicated form of hemophilia – with the presence of F VIII/IX inhibitor are followed up in the center. CTH is also taking care about patients with acquired hemophilia A.
Key words:
hemophilia A/B – coagulation factor VIII/IX – F VIII inhibitor – carrier of hemophilia – mutations in gene for F VIII/FIX – substitutive treatment
Authors:
I. Hrachovinová; P. Salaj
Authors‘ workplace:
Ústav hematologie a krevní transfuze Praha, ředitel prof. MUDr. Marek Trněný, CSc.
Published in:
Vnitř Lék 2012; 58(Suppl 2): 65-69
Category:
Overview
Počátek péče o pacienty s hemofilií v ÚHKT se datuje do 60. let minulého století. Do dnešní doby byla vytvořena komplexní péče od diagnostické po preventivní a terapeutickou. Diagnostika je soustředěna do NRL pro poruchy hemostázy, disponuje všemi dostupnými metodikami proteinovými i molekulárně genetickými. Terapeutická a preventivní péče je soustředěna do Centra pro trombózu a hemostázu ÚHKT a na klinická oddělení ÚHKT. Ve spolupráci s ortopedickými, chirurgickými a jinými klinikami zajišťujeme specializovanou péči při invazivních výkonech. ÚHKT byl ustanoven jako jeden ze dvou CCC (Comprehensive Care Centres) pro diagnostiku a léčbu dospělých pacientů s hemofilií v České republice. V péči ÚHKT je více než 200 pacientů s hemofilií A/B. Jednu třetinu tvoří pacienti s těžkou formou hemofilie. Součástí preventivní péče je aktivní vyhledávání přenašeček hemofilie a spolupráce s centry lékařské genetiky na prenatální diagnostice hemofilie. V centru je dispenzarizována polovina pacientů České republiky s komplikovanou formou hemofilie – s přítomností inhibitoru F VIII/F IX. Centrum se stará také o pacienty se získanou formou hemofilie A.
Klíčová slova:
hemofilie A/B – faktor VIII/IX – inhibitor F VIII – přenašečka hemofilie – mutace v genu pro F VIII/IX – substituční léčba
Úvod
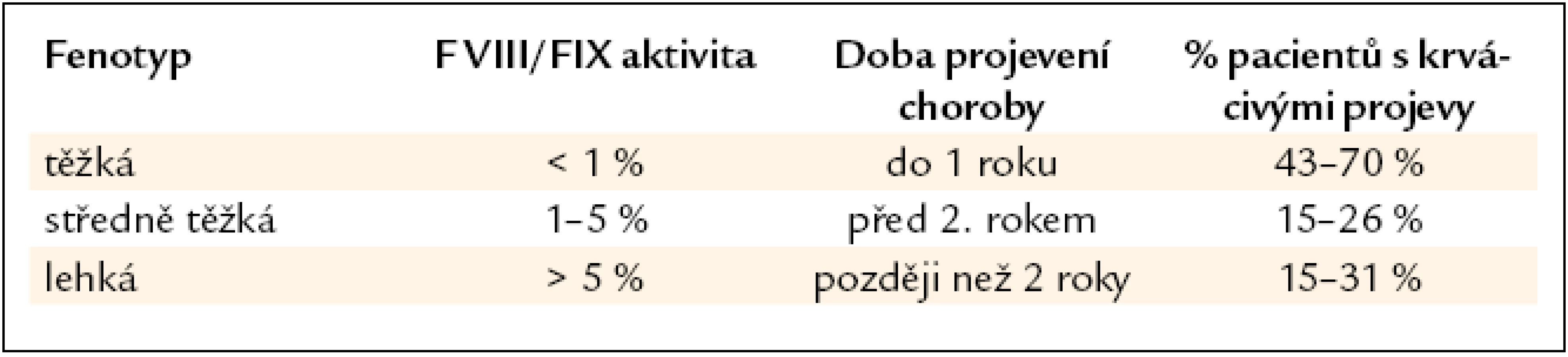
Hemofilie A je vrozené krvácivé koagulační onemocnění, které postihuje přibližně 1 z 10 000 mužů. Vedle hemofilie A, která je 6krát četnější a je způsobena deficitem F VIII v cirkulující krvi, existuje také hemofilie B, která je způsobena deficitem F IX. Stejná klinická manifestace obou onemocnění je způsobena rolí F VIII a F IX v plazmatické koagulaci. Podle hloubky deficitu klasifikujeme hemofilii na těžkou (< 1 %), středně těžkou (1–5 %) a lehkou (5–40 %) formu. Tíže krvácivých projevů většinou koreluje s hloubkou deficitu faktorů. Nemocní s těžkou formou hemofilie zpravidla krvácejí i několikrát do měsíce, většinou do nosných kloubů a do svalů. Nemocní s lehčími formami nadměrně krvácejí při chirurgických a stomatologických výkonech nebo po menším traumatu. Závažným problémem hemofilie je vznik protilátek inhibujících F VIII (F IX). Vyskytuje se u 5–35 % pacientů s těžkou až středně těžkou formou onemocnění. Častěji se objevuje u hemofilie A, což je dáno výraznějšími riziky vzniku inhibitoru u tohoto onemocnění. Lehká až středně těžká forma hemofilie A může být zaměněna za von Willebrandovu chorobu (a naopak). Diferenciální diagnostika obou onemocnění je založena na rozdílném způsobu dědičnosti a specifickém funkčním testu, který hodnotí vazbu F VIII na von Willebrandův faktor (vWF) jakožto nosičovou bílkovinu F VIII.
Historie
Počátky péče o pacienty s hemofilií v ÚHKT se datují do 60. let minulého století a jsou spjaté s vývojem laboratorních metodik, které stanovovaly aktivitu koagulačních faktorů (prof. Pudlák, 1966). Ke specifikaci onemocnění přispěla další vyšetření, která byla v ÚHKT zavedena, např. jednofázová metoda stanovení F VIII a F IX (RNDr. Sikorová, 1976), stanovení antigenu vWF (RNDr. Sikorová, 1981). Na konci 80. let 20. století byla zavedena molekulární genetika hemofilií (MUDr. Křepelová, 1988). V té době byl také ÚHKT pověřen metodickým vedením krajských center pro léčbu poruch hemostázy, centrální evidencí a centrální distribucí speciálních diagnostických a léčebných přípravků (koncentrátů F VIII, F IX a FEIBA). Kliničtí lékaři ÚHKT se podíleli na 1. uceleném Metodickém listu o komplexní péči o nemocné s hemofilií, který je v planosti od 1. 1. 1990.
Současnost
Péče o hemofiliky
V dnešní době je péče o hemofiliky soustředěna do Centra pro trombózu a hemostázu (CTH), které má laboratorní zázemí v NRL (národní referenční laboratoř) pro poruchy hemostázy.
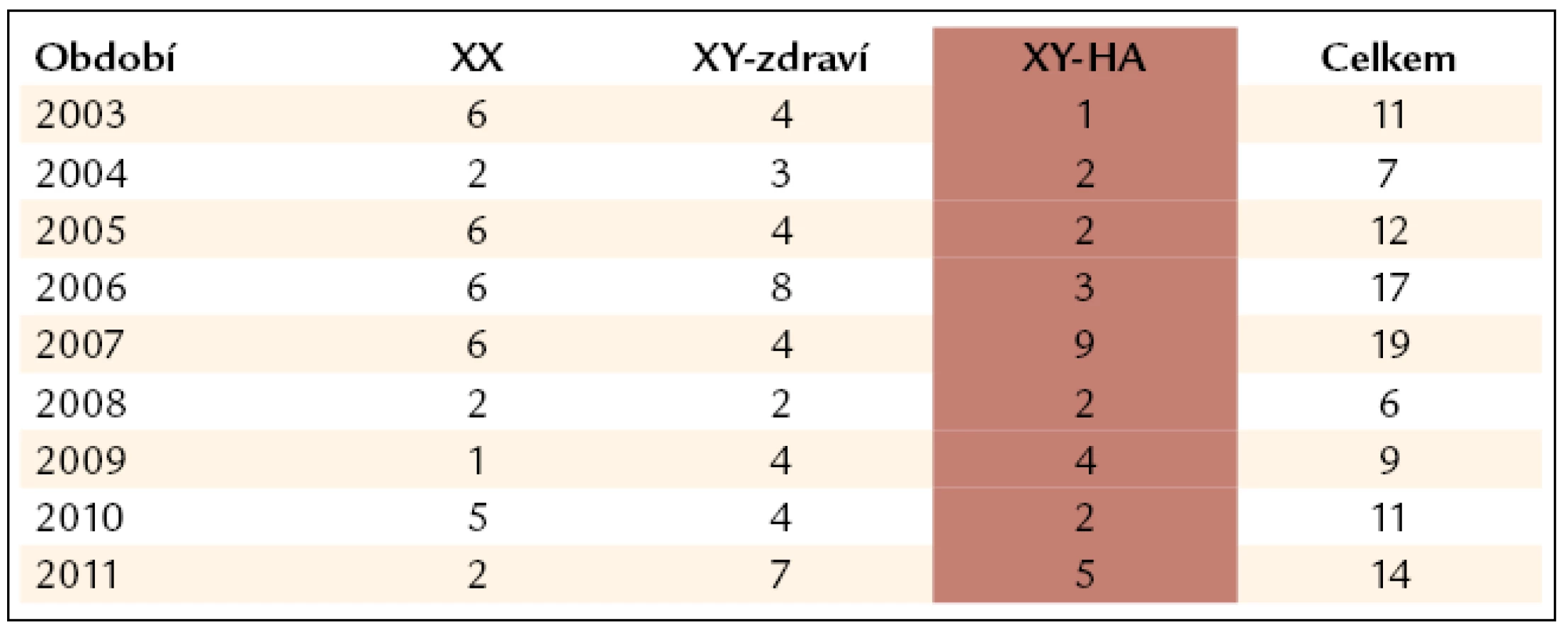
V CTH je dispenzarizováno celkem 204 žijících hemofiliků. Jejich rozdělení mezi hemofilii A a B, těžkou, středně těžkou a lehkou formu je v tab. 1. Víc než 1/3 pacientů má těžkou formu hemofilie. Za poslední 4 roky přibylo do našeho centra 23 pacientů s hemofilií. Část z nich byla předána po dovršení 18–20 let z Kliniky dětské hemato--onkologie v Praze-Motole. Snahou Centra je, aby pacienti měli celoživotní komplexní péči od stanovení diagnózy přes vyšetření členů rodiny u vrozených chorob po všestrannou léčebnou i preventivní podporu. Jako součást klinického úseku ÚHKT má pro to veškeré předpoklady. ÚHKT byl ustanoven jako jeden ze dvou CCC (Comprehensive Care Centres) pro diagnostiku a léčbu dospělých pacientů s hemofilií v České republice. V Centru je sledována a léčena více než 1/2 pacientů ČR s hemofilií a s inhibitorem F VIII/F IX (8 pacientů). CTH se také stará o pacienty se získanou formou inhibitoru F VIII/F IX, v dispenzarizaci má t.č. 10 pacientů. V průměru diagnostikujeme a na klinickém oddělení léčíme 4 pacienty se získaným inhibitorem F VIII ročně.

Spolupráce
S ortopedickou klinikou Nemocnice Na Bulovce (MUDr. R. Kubeš) spolupracujeme na konziliárním zajištění většiny endoprotetických výkonů kloubů u pacientů s hemofilií z celé ČR. V průměru se realizuje 8–15 TEP (totálních endoprotéz) ročně. Od roku 2006 ÚHKT jako jediný v ČR hematologicky zajišťuje i provádění nejrizikovějších elektivních chirurgických a ortopedických výkonů u pacientů s hemofilií a inhibitorem F VIII/F IX. V roce 2009 byla u jednoho z vysoce rizikových pacientů s inhibitorem F VIII provedena ve spolupráci našeho Centra a ortopedické kliniky Nemocnice Na Bulovce unikátní operace – simultánní endoprotéza obou kolenních kloubů. Ve spolupráci s ortopedickou klinikou 2. LF UK a FN Motol-Praha (MUDr. P. Teyssler) se v posledních letech úspěšně rozvíjí program radionuklidové synovektomie kloubů u pacientů s menším kloubním postižením. Od roku 2008 je v ÚHKT pacientům k dispozici i ambulantní rehabilitační program. Kromě ortopedických zákroků se realizuje ročně několik desítek menších i větších chirurgických zákroků, ke kterým je nutné koagulačně připravit pacienty. Jsou prováděny ve spolupráci s VFN Praha, FN Královské Vinohrady Praha nebo IKEM Praha. Dosud nejsložitějším chirurgickým zákrokem byla celosvětově unikátní exstirpace rozsáhlého pseudotumoru v retroperitoneu u pacienta s inhibitorem F VIII (Salaj, 2009). Vlastní chirurgický výkon byl proveden na pracovišti IKEM Praha profesorem MUDr. R. Gürlichem.
Léčba
Základem léčby hemofiliků je v současné době pouze léčba substituční. Spočívá v podávání chybějících/defektních faktorů k dosažení jejich hemostaticky dostatečné hladiny. Cílem je zástava krvácení či prevence jeho vzniku.
Všichni spolupracující pacienti s těžkou a středně těžkou formou hemofilie a s rizikem krvácení jsou léčeni formou domácí terapie, tj. aplikací chybějícího koagulačního faktoru pacientem bezprostředně po zakrvácení.
Dle způsobu zahájení dělíme substituční terapii na léčbu „on demand“, kdy koncentrát F VIII/F IX je podáván až při objevení se krvácení, a na léčbu profylaktickou, která je doporučeným standardem léčby v dětském věku u všech těžších forem hemofilií. Cílem je držet hladiny faktorů minimálně nad 1 %, ideálně nad 2 %. U dospělých hovoříme někdy o krátkodobé profylaxi, což je přechodná profylaktická aplikace koncentrátů F VIII/F IX vyžádaná klinickým stavem (po operaci, po úrazu, při rehabilitaci).
Koncentráty koagulačního F VIII/F IX
V současné době jsou v ČR k dispozici plazmatické, vysoce čištěné a protivirově ošetřené koncentráty F VIII/F IX i rekombinantní koncentráty F VIII. Obecně se předpokládá, že u F VIII zvýší 1 jednotka (j)/kg hladinu v průměru o asi 2 %, zatímco u F IX 1 j/kg asi o 1 %. Plazmatický poločas F VIII je přibližně 8–12 hod, F IX kolem 18 hod. Tyto údaje sice platí pro většinu pacientů, ale reakce na podaný F VIII či F IX je značně individuální a je vhodné mít u každého hemofilika ověřeno alespoň „recovery“ (dochází-li k přiměřenému procentuálnímu vzestupu plazmatické hladiny F VIII/F IX po aplikaci určitého množství koncentrátu udaného v j/kg), zejména před chirurgickým zákrokem či při změně preparátu.
Diagnostika hemofilie
Diagnostika hemofilie je kombinací klinické praxe a laboratorního vyšetření.
Krvácivé projevy hemofilie jsou často první známkou hemofilie u dětí z rodin se sporadickou formou hemofilie (tab. 2). Průkazem přítomnosti hemofilie je snížená aktivita F VIII spolu s normální funkční i antigenní hladinou vWF, respektive sníženou aktivitou F IX u hemofilie B. Správnou diagnostiku hemofilie A komplikuje pouze podobnost laboratorních výsledků u vW choroby, subtyp Normandy, a lehké formy hemofilie A. Diferenciální diagnostika je možná pouze speciální metodikou, která kvantifikuje vazbu F VIII k vWF. Na základě našich zkušeností byl záchyt vW choroby, se subtypem Normandy, přibližně u 1 % pacientů s domnělou lehkou formou hemofilie A. Velmi zajímavé bylo zjištění, že u téměř 70 % žen, které byly považovány za sporadické přenašečky hemofilie A (měly nízkou aktivitu F VIII a normální antigen vWF), byla prokázána vW choroba, subtyp Normandy.
Součástí vyšetření těžké až středně těžké hemofilie je stanovení přítomnosti autoprotilátek proti F VIII nebo F IX. V lékařské veřejnosti se pro tento jev používá výraz „inhibitor“ F VIII/F IX. Průkaz inhibitoru výrazně komplikuje léčbu pacientů. Musí být léčeni přípravky s takzvanou by-passovou aktivitou, jako je rF VIIa, nebo směsí různě aktivovaných faktorů koagulační kaskády (F X, F II, F IX atd.). Sledování léčby těmito přípravky si vyžádalo zavedení nových metodik, jako jsou trombinový generační čas nebo tromboelastografie.
Kompletním průkazem hemofilie je molekulárně genetické nalezení kauzální mutace v genu pro F VIII/F IX. U našich pacientů jsme nalezli kauzální mutaci u 80 % nemocných s hemofilií A a 91 % s hemofilií B. Rozdělení jednotlivých mutací je na obr. 1a, b.


Diagnostika přenašečství hemofilie
Stanovení přenašečství hemofilie u žen v rodinách hemofiliků je důležité pro plánování rodiny, partnerský vztah, prenatální vyšetření a vedení porodu hemofilického dítěte.
Přenašečka hemofilie na rozdíl od hemofilika zpravidla nemá žádné krvácivé příznaky a aktivita F VIII/F IX bývá v normě. Přenašečství lze stanovit z rodokmene. Jistá přenašečka je:
- a) dcera hemofilika,
- b) matka dvou hemofiliků,
- c) matka jednoho hemofilika v rodině s výskytem hemofilie.
Tohoto genetického pravidla lze použít u velmi malého procenta pravděpodobných přenašeček. Rodokmen rodiny je důležitou informací při stanovení přenašečství, protože je jiný postup u sporadické formy hemofilie, kde není možné vyloučit de novo vzniklou mutaci, a u rodinné formy hemofilie, kde je dědičnost prokázána u několika generací. Ke stanovení přenašečství se využívá polymorfních intragenových markerů při nepřímé diagnostice v kombinaci s koagulačním vyšetřením (Sikorová, 1984), nebo přímé diagnostiky tam, kde je již známa kauzální mutace (obr. 2a, b). Nepřímá diagnostika je použitelná pouze u rodin s familiárním výskytem hemofilie a navíc u tzv. „informativních“ rodin, kde je možné u žen odlišit alelu hemofilickou od normální (asi 85 % rodin). Tato metoda je rychlá. Výsledek je znám do týdne až 14 dní, proto se využívá v časovém tlaku (těhotenství). Vyžaduje ale genetický materiál více členů rodiny a zejména genetický materiál hemofilika. Je spojena s 0,01–0,05% rizikem chyby z možné rekombinace genu. Přímá diagnostika je vázaná na průkaz kauzální mutace, její zjištění je technicky a časově náročné. Dovoluje však vyloučit, nebo prokázat přenašečství i u vzdálených příbuzných hemofilika. V rodinách se sporadickým výskytem hemofilie je doporučeno zahájit vyšetření suspektních přenašeček z hemofilických rodin již v 10 letech jejich věku.


Prenatální diagnostika
Prenatální vyšetření se provádí u těžké formy hemofilie. Přehled prenatálních vyšetření u hemofilie A v našem Centru je v tab. 3. Standardně se provádí ze vzorku choriové biopsie (CVS) 11.–13. týden gravidity, když pacientka uvažuje o přerušení těhotenství. Je možné provést vyšetření též z amniové tekutiny 15.–16. týden. To je v případě, že se prenatální diagnostika hemofilie provádí jen informativně pro správné vedení porodu a amniocentéza je prováděna ještě z jiné indikace (věk, jiné onemocnění). Rychlou metodou je zjišťována přítomnost chromozomu Y. U plodů mužského pohlaví je stanovována diagnóza hemofilie buď přímým důkazem kauzální mutací, nebo nepřímou diagnostikou polymorfními markery. Většina metod funguje na principu PCR, a proto je výsledek vyšetření do týdne, avšak jen v případě, že přenašečka byla předem vyšetřena a byl připraven postup vyšetření. Prenatální vyšetření je invazivní výkon s rizikem potratu 1–2 % (na expertních pracovištích), proto se nově zavádí vyšetření pohlaví plodu z periferní krve budoucí matky v 10. týdnu těhotenství. Spolehlivost vyšetření dosahuje 99,9 %. Tím se vyloučí zbytečný zákrok u plodů ženského pohlaví. V některých centrech asistované reprodukce se začíná nabízet preimplantační genetická diagnostika (PGD) hemofilie. Je založena na panelu mikrosatelitních markerů pro multiplexní analýzu na jedné buňce. V případě znalosti kauzální mutace je vyšetření doplněno o primer-alelově specifické vyšetření mutace. Vyšetření není dosud dostatečně spolehlivé u velkých delecí nebo inverzí, což vylučuje přibližně 50 % rodin s těžkou hemofilií A.
RNDr. Ingrid Hrachovinová, Ph.D.
www.uhkt.cz
e-mail: ingrid.hrachovinova@uhkt.cz
Doručeno do redakce: 31. 5. 2012
Sources
1. Auerswald G, Salek SZ, Benson G et al. Beyond patient benefit: clinical development in hemophilia. Hematology 2012; 17 : 1–8.
2. Habart D, Kalabova D, Hrachovinova I et al. Significant prevalence of the intron 1 factor VIII gene inversion among patients with severe hemophilia A in the Czech Republic. J Thromb Haemost 2003; 1 : 1323–1324.
3. Habart D, Kalabova D, Novotny M et al. Thirty-four novel mutations detected in factor VIII gene by multiplex CSGE: modeling of 13 novel amino acid substitutions. J Thromb Haemost 2003; 1 : 773–781.
4. Habart D, Kleibl Z, Hrachovinová I. Evaluation of DHPLC analysis for mutation detection in haemophilia A. Čas Lék Česk 2006; 145 : 484–487.
5. Hrachovinová I, Vorlová Z. Detection of carriers of hemophilia B. Čas Lék Česk 1992; 131 : 761–763.
6. Hrachovinová I, Vorlová Z. Identification of seven novel factor IX mutations in Czech hemophilia B patients. Blood 1997; 90 : 96.
7. Krepelová A, Vorlová Z, Zavadil J et al. Factor VIII gene deletions in haemophilia A patients in Czechoslovakia. Br J Haematol 1992; 81 : 271–276.
8. Salaj P, Gurlich R, Svorcová V et al. Prophylactic preparation and surgical extirpation of a very large abdominal blood cyst in a severe haemophilia A patient with inhibitors managed by rFVIIa. Haemophilia 2009; 15 : 380–382.
9. Salaj P, Brabec P, Penka M et al. Effect of rFVIIa dose and time to treatment on patients with haemophilia and inhibitors: analysis of HemoRec registry data from the Czech Republic. Haemophilia 2009; 15 : 752–759.
10. Salaj P, Penka M, Smejkal P et al. Economic analysis of recombinant activated factor VII versus plasma-derived activated prothrombin complex concentrate in mild to moderate bleeds: haemophilia registry data from the Czech Republic. Thromb Res 2012; 129: e233–e237.
11. Sikorová J, Zvárová J, Paluska E et al. Processing data for predicting hemophilia transmission. Vnitr Lek 1984; 30 : 764–770.
Labels
Diabetology Endocrinology Internal medicineArticle was published in
Internal Medicine

2012 Issue Suppl 2
Most read in this issue
- Imunohematologie – historie, současný stav poznání a role ÚHKT
- Naléhavé stavy v hematologii
- Hemaferéza – vysoce účinná technika v terapii nemocných
- Chronická myeloidní leukemie – zásadní změna prognózy nemocných po zavedení léčby inhibitory tyrozinových kináz