
Myelodysplastický syndrom. Pokroky v diagnostice a léčbě během 30 let trvání registru nemocných s myelodysplastickým syndromem v ÚHKT
Myelodysplastic syndromes. Progress in diagnosis and treatment within 30 years of myelodysplastic syndromes registry in Institute of Hematology in Prague
An analysis of data obtained from 367 patients with primary myelodysplastic syndromes (MDS) treated in Institute of Hematology and Blood Transfusion in the years 1980–2009 revealed substantional changes in diagnostic and therapeutic approaches within past 30 years. Molecular biology methods have been recently incorporated into the set of diagnostic methods and the importance of results obtained by molecular genetic methods for prognosis and optimal treatment is intensively studied. The treatment approaches switched from supportive care and palliative chemotherapy to new drugs that may effectively target the molecular basis of the disease as lenalidomide in early disease and hypomethylating agents in advanced MDS. Introduction of stem cell transplantation (SCT) significantly improved outcome especially in younger patients with advanced MDS and SCT still represents the only curative treatment approach.
Key words:
myelodysplastic syndromes – diagnosis – treatment – prognosis – registry
Authors:
J. Čermák
Authors‘ workplace:
Ústav hematologie a krevní transfuze Praha, ředitel prof. MUDr. Marek Trněný, CSc.
Published in:
Vnitř Lék 2012; 58(Suppl 2): 8-15
Category:
Overview
Analýza dat získaných od 367 nemocných s primárním myelodysplastickým syndromem (MDS) sledovaných v ÚHKT v letech 1980–2009 ukázala zásadní změny v přístupu k diagnostice a k léčbě tohoto onemocnění během posledních 30 let. V diagnostice MDS se zejména v poslední době uplatňují molekulárně genetické metody a význam nálezů získaných těmito metodami pro prognózu nemocných, a tím i pro způsob jejich léčby, je intenzivně zkoumán. Při zhodnocení vývoje léčby vidíme přechod od podpůrné léčby a paliativní terapie k moderním léčebným metodám užívajícím léky, jež mohou cíleně ovlivnit právě molekulárně genetickou podstatu onemocnění. Takovými léky jsou lenalidomid u nízce rizikových nemocných a hypometylační látky u pokročilé fáze onemocnění. Zavedení transplantace krvetvorných buněk (SCT) výrazně zlepšilo délku přežití zejména u mladších nemocných s pokročilými formami choroby s nadbytkem blastů. SCT představuje stále jediný kurativní přístup k této chorobě.
Klíčová slova:
myelodysplastický syndrom – diagnostika – léčba – prognóza – registr
Úvod
Již dlouho je známo, že část akutních leukemií nezačíná u nemocných náhle z plného zdraví, ale po různě dlouhém období charakterizovaném sníženým počtem jednoho či více typů krvinek a z toho vyplývajícími komplikacemi, nejčastěji opakovanými infekty či krvácením. Popisy těchto případů lze najít již v odborných článcích publikovaných mezi dvěma světovými válkami, první nozologickou jednotku charakterizovanou výše uvedenými příznaky popsal v polovině 50. let minulého století Björkman jako získanou sideroblastickou anémii [1]. O 25 let později byly blíže definovány jako samostatné jednotky refrakterní anémie s nadbytkem blastů a chronická myelomonocytární leukemie, nazývané dříve paušálně preleukemie či doutnající leukemie. V roce 1982 pak byly výše uvedené jednotky zahrnuty do společné skupiny tzv. myelodysplastických syndromů (MDS), jenž byly definovány jako heterogenní skupina onemocnění kmenové krvetvorné buňky s odlišnou prognózou týkající se jak přežití, tak rizika progrese do akutní leukemie [2]. Na patogenezi onemocnění se podle současných názorů podílí v časné fázi mutace kmenové krvetvorné buňky, jež vede k abnormální imunitní odpovědi organizmu, k aktivaci cytotoxických T-lymfocytů a ke zvýšené produkci cytokinů, jež indukují apoptózu zejména zralejších forem krvetvorby [3]. Poškozený genom je náchylný k akumulaci dalších mutací, v tomto procesu hrají významnou roli jednak mutace onkogenů vedoucí k abnormální proliferaci klonu vznikajícího z poškozené kmenové krvetvorné buňky, jednak mutace tzv. tumor supresorických genů, což vede k poruše diferenciace prekurzorů do zralejších forem krvetvorby a celkové regulace krvetvorby [4]. Konečným výsledkem je postupný rozvoj akutní myeloidní leukemie (AML).
Diagnostika
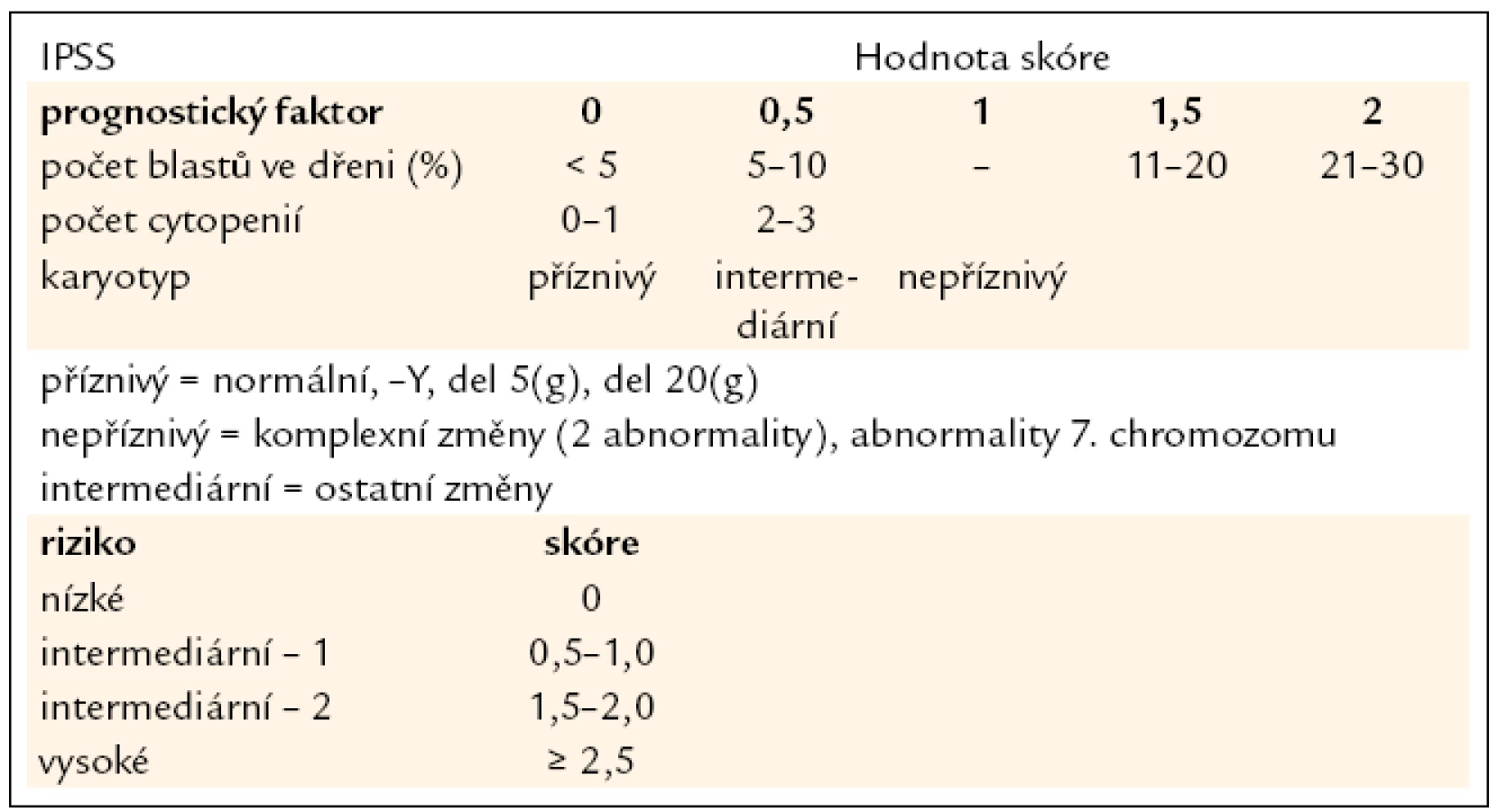
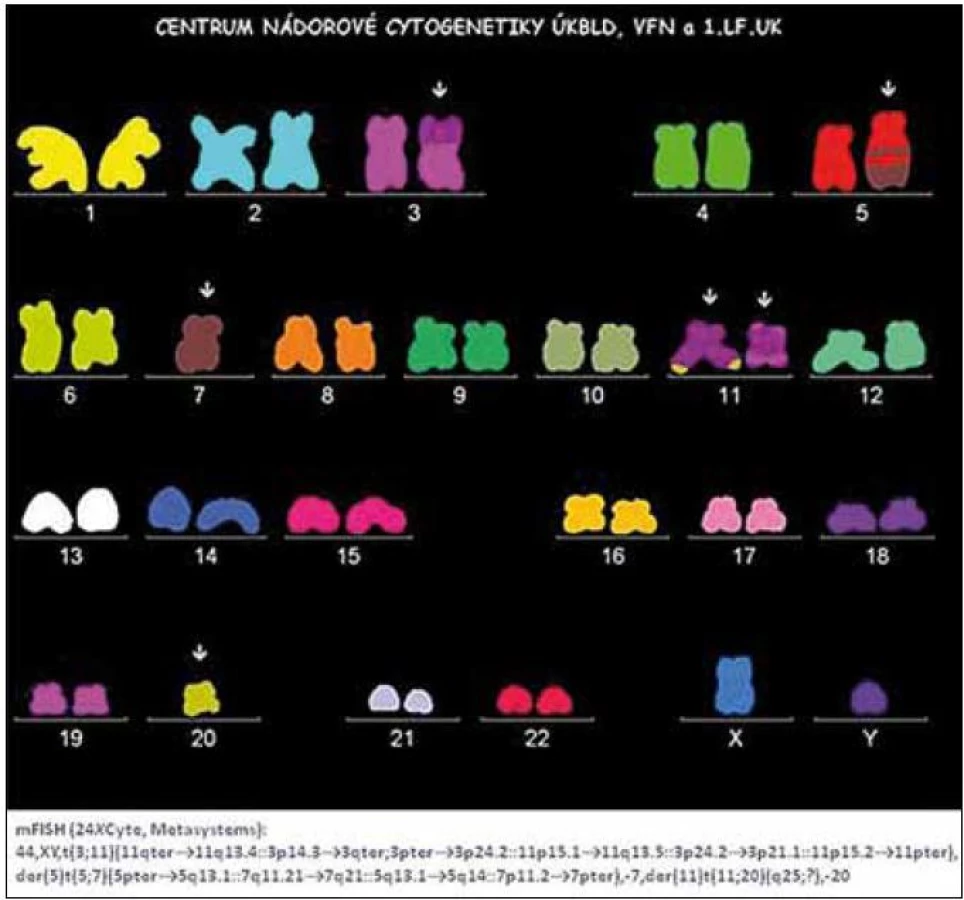
V diagnostice MDS hraje stále základní roli pečlivé morfologické vyšetření kostní dřeně vzhledem k tomu, že jak předešlá, tak současná klasifikace MDS definuje přesně a poměrně striktně kritéria, jaké morfologické změny jsou hodnoceny jako projev dysplazie a jaké procento buněk v jednotlivých krevních řadách musí tyto změny vykazovat (tab. 1) [5]. Krom vyšetření punktátu kostní dřeně je při podezření na MDS nutno provést histologické vyšetření dřeně z trepanobioptického vzorku, z něhož je možno krom potvrzení dysplastických změn lépe posoudit i buněčnost dřeně, některé topické změny a event. přítomnost fibrózy. Zhruba u 40 % nemocných s MDS jsou při vyšetření klasickými cytogenetickými metodami přítomny přestavby chromozomů, nález abnormit karyotypu může pomoci při diagnostice MDS, řada studií však prokázala zásadní význam typu a počtu chromozomálních změn pro prognózu nemocných a cytogenetický nález je jedním ze základních parametrů při hodnocení prognózy nemocných s MDS pomocí tzv. Mezinárodního prognostického skórovacího systému (IPSS) (tab. 2) [6]. Tento cytogenetický prognostický systém, stejně jako jeho recentní revize [7] je postaven na vyšetření karyotypu pomocí standardních pruhovacích metod. Rozvoj molekulární cytogenetiky a metod, jakými jsou fluorescenční hybridizace in situ (FISH) a komparativní genomová hybridizace (aCGH), přinesl možnost komplexního vyšetření genomu a upřesnění lokalizace postižení daného chromozomu (obr. 1). Nicméně jejich přínos pro revizi stupně rizika u jednotlivých nemocných není zcela jednoznačný [8]. Vyšetření exprese povrchových buněčných antigenů pomocí průtokové cytometrie může upřesnit procento CD34+ blastů v kostní dřeni, v současné době se ukazuje, že změny exprese povrchových antigenů u nemocných s MDS mají krom přínosu pro diagnostiku i význam prognostický [9]. Řada molekulárně genetických metod dnes slouží ke zkoumání mutací a změn exprese nejrůznějších genů u MDS. Vzhledem k řadě faktorů, které se podílejí na vzniku a progresi MDS, se však dosud nepodařilo identifikovat gen či skupinu genů, jež by měly univerzální diagnostický význam. Studium možného prognostického významu molekulárně genetických změn je zmíněno níže. V současné době je intenzivně studován i význam proteinového profilu séra u nemocných s MDS pomocí proetomických metod [10].

Prognóza
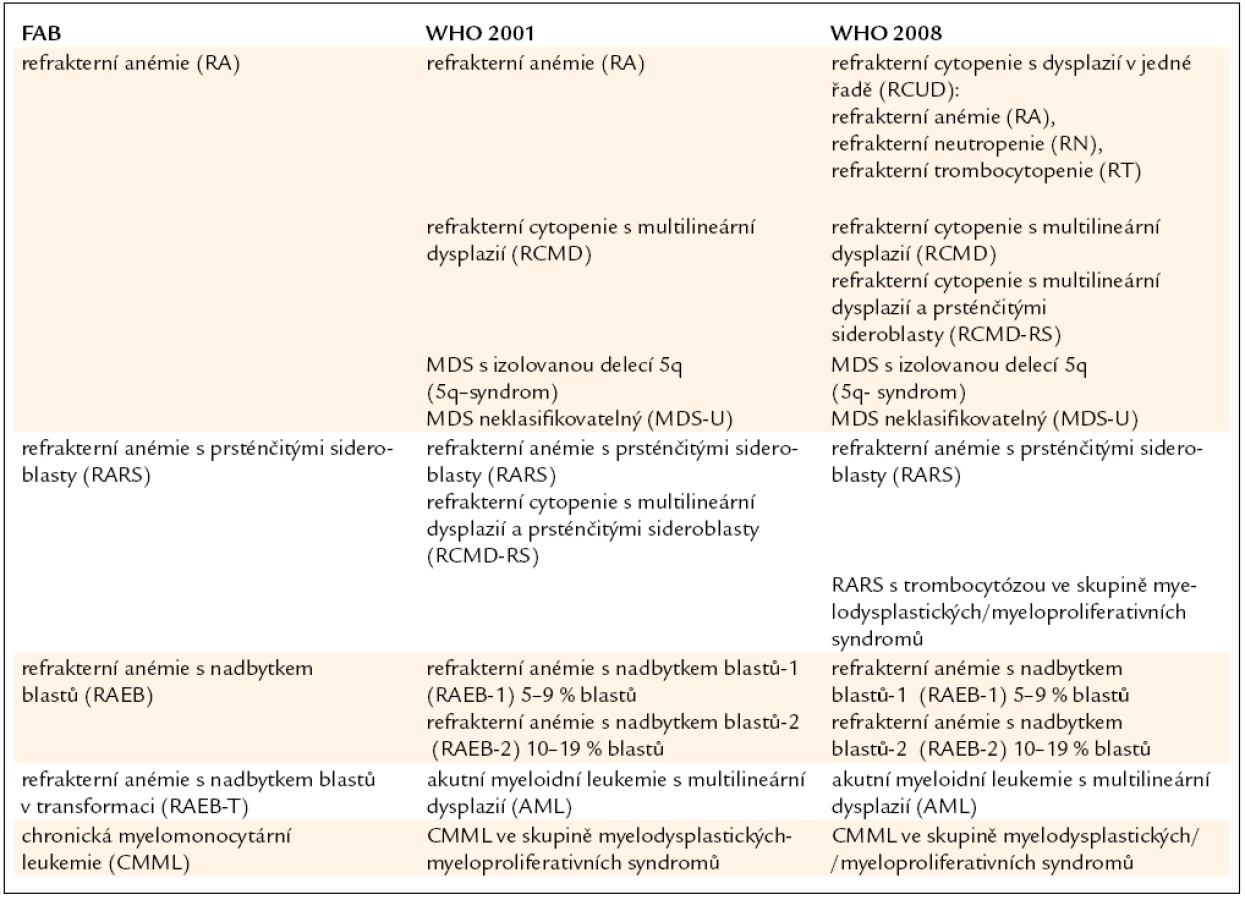
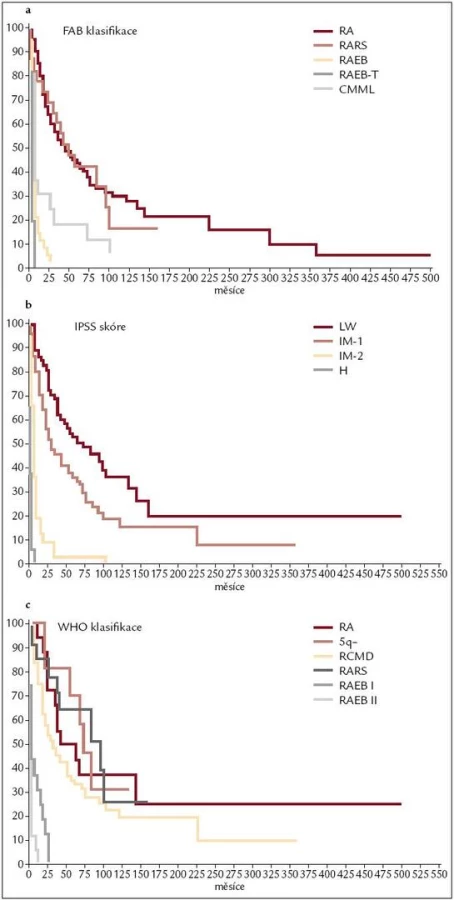
Již výše zmíněná první klasifikace MDS (tzv. FAB – francouzsko/americko/britská klasifikace) z roku 1982 (tab. 3) [2] rozděluje nemocné do podskupin s relativně lepší prognózou, nižší incidencí komplikací, delší dobou přežití a malým rizikem leukemizace choroby (RA, RARS), a podskupin se zmnožením blastů v kostní dřeni (RAEB, RAEB-T a CMML) s nepříznivou prognózou a vysokým rizikem progrese do AML. Na obr. 2a je znázorněna délka přežití 182 neléčených nemocných z celkového počtu 367 nemocných s primárním MDS z registru ÚHKT sledovaných v letech 1980–2009 a rozdělených do jednotlivých podskupin podle FAB klasifikace. Během 80. let minulého století bylo vytvořeno několik prognostických schémat hodnotících jako prognosticky významné různé faktory, zejména počet cytopenií v periferní krvi a změny karyotypu [11], a v roce 1997 byl publikován již výše zmíněný tzv. Mezinárodní prognostický skórovací systém (IPSS) [6], jenž na základě počtu blastů v kostní dřeni, počtu cytopenií v periferní krvi a počtu a typu abnormit karyotypu dělí nemocné s primárním MDS do 4 podskupin s různou délkou přežití a rizikem leukemické transformace. Obr. 2b ukazuje délku přežití našich nemocných rozdělených podle rizikových podskupin IPSS. V roce 2001 byla publikována tzv. WHO klasifikace MDS [12], která na základě rozdílné prognózy rozděluje nemocné v původní skupině refrakterních anémií do podskupiny s čistou refrakterní anémií (RA), refrakterní cytopenií s dysplazií ve více řadách (RCMD) a tzv. 5q-syndromem. Vzhledem k prakticky identickému průběhu a prognóze byli nemocní s diagnózou RAEB-T přeřazeni mezi akutní myeloidní leukemie a CMML byla na základě svého biologického chování zařazena do skupiny smíšených myelodysplastických/myeloproliferativních onemocnění (MDS/MPS). Obr. 2c ukazuje délku přežití našich nemocných rozdělených podle WHO klasifikace z roku 2001. Nová revize WHO klasifikace z roku 2008 [5] pak detailněji rozděluje nemocné s cytopenií v jedné řadě a nemocné s RAEB dělí do 2 podskupin dle počtu blastů v kostní dřeni (tab. 2). V roce 2007 byl publikován tzv. WHO-based prognostic scoring system (WPSS) využívající podskupiny dle WHO klasifikace (2001), prognostické skupiny karyotypu dle IPSS a přidávající jako významný prognostický parametr závislost na podávání transfuzí erytrocytů [13]. Nepříznivý vliv opakovaného podávání transfuzí erytrocytů na délku přežití může být důsledkem toxického působení nadbytečného železa dodaného transfuzemi, zejména na myokard, nicméně transfuzní dependence může odrážet i prognosticky nepříznivé rozsáhlé postižení krvetvorby. V naší práci bylo podávání transfuzí erytrocytů nezávislou proměnnou negativně ovlivňující délku přežití pouze u skupiny nemocných s izolovanou dysplazií v červené řadě a dependencí na více než 2 TU erytrocytů měsíčně. U nemocných s RCMD byl negativní dopad podávání transfuzí přítomen pouze u nemocných s nepříznivými změnami karyotypu (dle IPSS) a závislostí na ≥ 3 TU erytrocytů měsíčně [14].
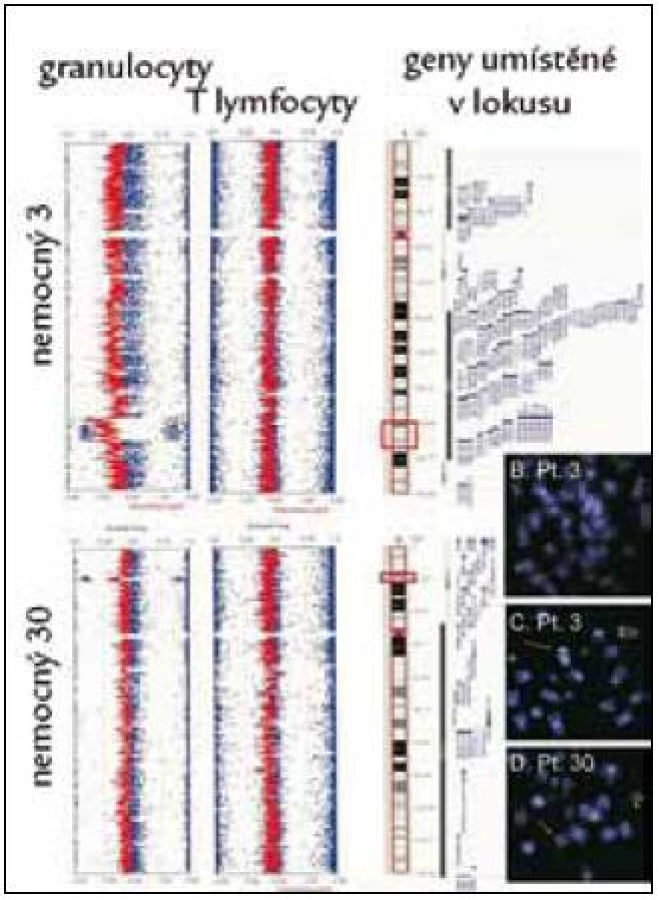
Na rozdíl od do určité míry omezeného přínosu molekulární cytogenetiky pro klasifikaci a prognózu nemocných s MDS ukazují recentní studie na možný prognostický význam detekce změn genomu pomocí molekulárně genetických metod. Detekce polymorfizmu jednotlivých nukleotidů pomocí tzv. SNP array karyotypování představuje citlivou metodu k odhalení diskrétních nebalancovaných translokací u nemocných s normálním nálezem při užití konvenční cytogenetické analýzy. Může jít o variaci počtu kopií alel (copy number variation – CNV), nebo o ztrátu heterozygozity daného úseku genomu (LOH) kvůli tzv. uniparentální disomii (UPD), při níž jde o ztrátu genetického materiálu jednoho chromozomu s následným vyplněním ztraceného úseku duplikací zbývajícího chromozomu, takže nedochází ke ztrátě genetického materiálu (a proto změny nemohou být detekovány klasickou cytogenetikou). Počet kryptických chromozomálních aberací může mít vztah k délce přežití nemocných [8], kromě toho může tato metodika odhalit některé diskrétní změny karyotypu s prognostickým významem [15]. Na obr. 3 je příklad detekce kryptické delece dlouhého raménka 5. chromozomu, která byla následně ověřena metodou FISH s užitím lokus specifické sondy. V kombinaci s detekcí změn exprese genů pomocí studia expresního profilu genomu v úseku identifikovaném pomocí SNP karyotypování pak můžeme určit kandidátní geny, jejichž změny exprese jsou důsledkem zjištěné diskrétní změny genomu [15,16]. V poslední době se podařilo pomocí velmi citlivých metodik, jakými jsou genotypování pomocí hmotnostní spektrometrie a tzv. sekvenace „příští generace“ (next generation sequencing) identifikovat somatické bodové mutace některých genů, jejichž postižení může mít prognostický význam [17]. Řada z těchto genů (ASXL1, EZH2, TET2) se podílí na procesu regulace exprese zejména diferenciačních genů cestou ovlivnění aktivity koncových nepřepisovaných regulačních úseků daného genu změnou stupně metylace genových promoterů a acetylace histonů. Některé geny mají zřejmě klíčovou roli v regulaci buněčné proliferace a regulace reparace poškození (TP53, RUNX1), nejnověji byl popsán možný prognostický význam mutací genů podílejících se na procesu sestřihu nukleových kyselin z proformy do přepisované formy (SFR3B1) [18].
Léčba
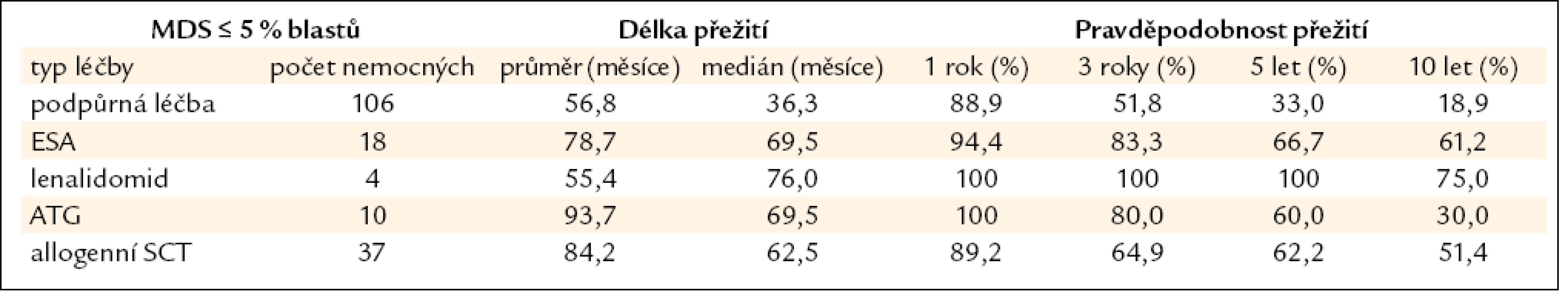
Všechny výše uvedené klasifikace MDS i prognostické skórovací systémy ukázaly na významný rozdíl v délce přežití mezi jednotlivými podskupinami nemocných, zejména mezi nemocnými s normálním počtem mladých prekurzorů v kostní dřeni a nemocnými s narůstajícím procentem blastů (zejména > 10 % v kostní dřeni) – viz obr. 2a–c. Z této zkušenosti vychází i současný přístup k léčbě nemocných s MDS. U nemocných s nízkým či středním skóre dle IPSS (tab. 2), resp. u nemocných bez nadbytku blastů v kostní dřeni, převažují konzervativní přístupy k léčbě. V tab. 4 jsou uvedeny výsledky různých léčebných přístupů k 175 nemocným s časným MDS s nižším rizikem (≤ 5 % blastů v kostní dřeni) z registru ÚHKT. Vidíme, že část nemocných může profitovat pouze z podpůrné léčby, na níž 33 % nemocných přežívá 5 let, a pravděpodobnost přežití 10 let od diagnózy činí 19 %. U těchto nemocných se může rozvinout přetížení železem v důsledku opakovaného podávání transfuzí erytrocytů a je u nich namístě včasné zahájení efektivní chelatační léčby [14,19]. U části nemocných může mít účinek podávání erytropoézu stimulujících látek (ESA), většinou jde o nemocné s určitým deficitem endogenního erytropoetinu (EPO). Nejlepší efekt byl pozorován u nemocných s hladinou EPO v séru < 200 IU/l a závislých na ≤ 2 TU erytrocytů měsíčně [20]. Podávání imunosupresiv, zejména antithymocytárního globulinu (ATG), je indikováno především u nemocných s hypoplastickou formou MDS [21], 5 let přežívalo 60 % našich nemocných, kterým byl podáván ATG. Nicméně u 6 z 10 nemocných došlo během let k progresi choroby směrem k akutní leukemii, u 4 z nich byla v období progrese nalezena delece 7q či úplná ztráta 7. chromozomu. Tyto nálezy souhlasí s pozorováním jiných autorů [22], kteří přítomnost změn na 7. chromozomu v době podávání ATG hodnotí jako důležitý nepříznivý faktor vedoucí k potenciální progresi choroby, pravděpodobně kvůli defektu funkce některých tumor supresorických genů lokalizovaných na 7. chromozomu. V současné době je proto doporučováno u všech nemocných indikovaných k intenzivní imunosupresi vyloučit přítomnost změn na 7. chromozomu pomocí metod molekulární cytogenetiky či molekulární genetiky.
Efekt lenalidomidu na korekci anémie u nemocných s delecí 5q je vysvětlován ovlivněním funkce některých proliferačních genů a regulačního efektu tzv. mikroRNA (mRNA) [23]. U nemocných odpovídajících na léčbu je v našem registru pravděpodobnost přežití 10 let 75 %, počet takto léčených nemocných je ale zatím poměrně malý. Před léčbou je nutno všechny nemocné vyšetřit na možný výskyt mutace genu p53, její přítomnost je obecně nepříznivým prognostickým faktorem [17] a při léčbě lenalidomidem je spojena s vysokým rizikem progrese choroby [24]. Alogenní transplantace krvetvorných buněk je efektivní metodou u nemocných s časným MDS, 5 let po SCT přežívá 62 % našich nemocných a pravděpodobnost přežití 10 let je 51 %. Nicméně mortalita spojená s transplantací není u této skupiny nemocných zanedbatelná (22 % v našem souboru), což ukazuje na to, že u nemocných s MDS s nízkým rizikem je třeba indikaci SCT vždy pečlivě zvážit, tato léčba přichází v úvahu zejména u nemocných s těžkou cytopenií a z ní vyplývajícími život ohrožujícími komplikacemi, s komplexními změnami karyotypu či při současné přítomnosti vyššího stupně fibrózy v kostní dřeni (MF-2) [25].
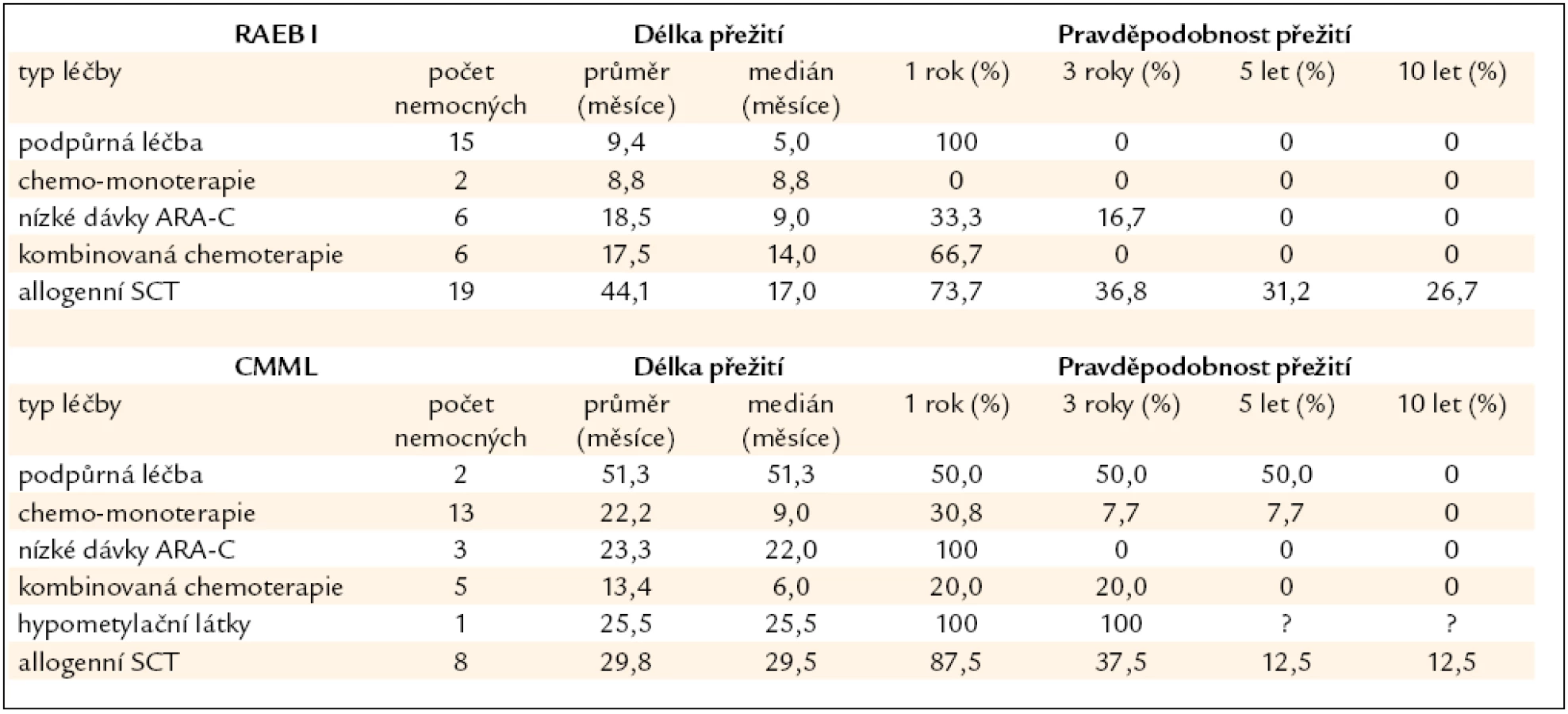
Nemocní s MDS s 6–10 % blastů (RAEB I dle WHO klasifikace z roku 2008) v kostní dřeni tvoří skupinu s nejednotnou a většinou významně horší prognózou než nemocní s MDS a ≤ 5 % blastů v kostní dřeni (obr. 2). Nepříznivý průběh je dán poměrně častou progresí směrem k akutní leukemii (27 % v naší sestavě) a častými závažnými komplikacemi rezultujícími z těžké cytopenie (21 % našich nemocných). Příčina není zcela jasná, uvažuje se o vlivu dlouhodobě se rozvíjejícího procesu, jenž vede k hrubé poruše krvetvorné buňky i hemopoetického stromatu. V tab. 5 jsou shrnuty výsledky léčby našich nemocných s MDS typu RAEB I. Jedinou efektivní metodou se ukazuje být alogenní SCT, ale i zde ve srovnání s nízcerizikovými nemocnými přežívá 5 let pouze 32 % nemocných a peritransplantační mortalita činí 42 %. Nicméně přítomnost progredujícího počtu blastů v kostní dřeni je indikací k provedení alogenní SCT [26]. Chronická myelomonocytární leukemie je dnes řazena mezi MDS/MPS. V tab. 5 jsou uvedeny i výsledky léčby této jednotky u našich nemocných, někteří mohou poměrně dlouho přežívat pouze s podpůrnou léčbou či při podávání hydroxyurey v monoterapii, transplantace bývá spojena s častými relapsy, určitý efekt může mít podávání hypometylačních látek (azacytidinu), ale počty nemocných na této léčbě jsou zatím poměrně malé.
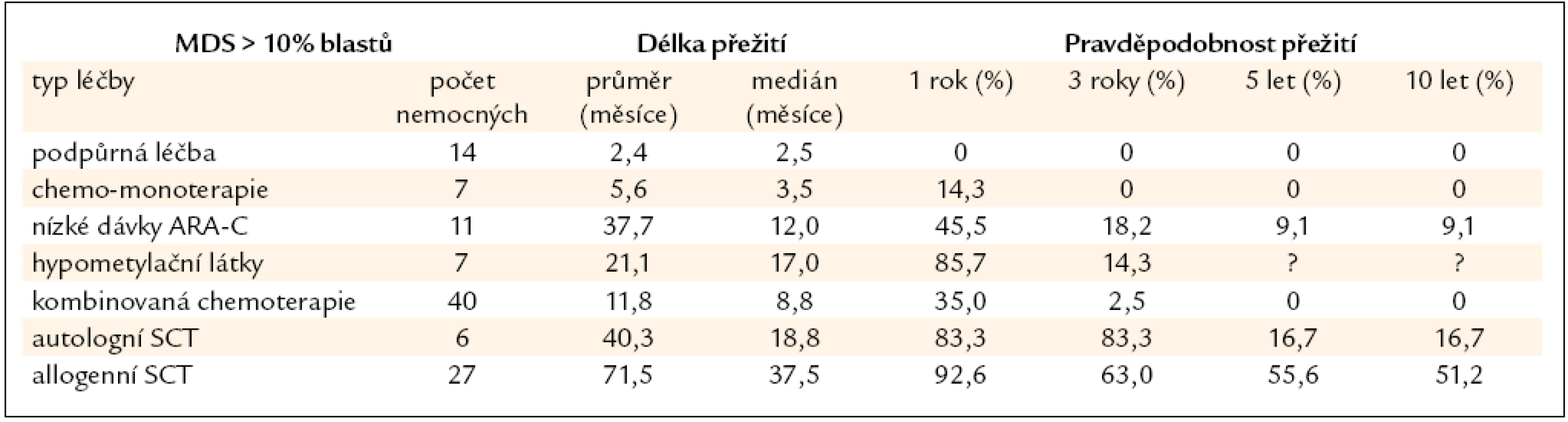
Nemocní s MDS s > 10 % blastů v kostní dřeni jsou považováni za vysoce rizikovou skupinu a je u nich indikována intenzivní léčba včetně alogenní SCT. Vzhledem k tomu, že bez léčby prakticky všichni nemocní zemřou na komplikace či na progresi do AML, nemá pouhá podpůrná léčba či perorální monoterapie smysl. To tab. 6 ukazuje i výsledky získané v souboru 112 nemocných léčených v ÚHKT s diagnózou RAEB II či RAEB-T. Z konzervativních typů léčby používaných zejména u starších nemocných má určitý efekt podávání nízkých dávek cytarabinu a v poslední době pak hypometylačních látek, jež svým účinkem mohou revertovat vyhasínání funkce některých důležitých genů hrajících roli zejména v buněčné diferenciaci a regulaci krvetvorby [27]. Autologní SCT nemá větší význam vzhledem k tomu, že se často jen velmi obtížně dosáhne molekulárně genetické remise choroby a zbytková nemoc je pak zdrojem relapsů choroby. Z naší skupiny 6 autologně transplantovaných nemocných s MDS přežívá dlouhodobě pouze 1 nemocná. U 27 nemocných byla provedena alogenní SCT, 5 let přežívá 56 % nemocných, což je srovnatelné s výsledky u nízce rizikových nemocných, pravděpodobnost přežití 10 let je 52 %, peritransplantační mortalita činí 30 %. Nebyl pozorován významnější rozdíl ve výsledcích při užití standardních a redukovaných přípravných režimů a při transplantaci od příbuzného či nepříbuzného dárce, stejně jako v zahraničních studiích [28,29]. U skupiny našich nemocných byl přítomen signifikantní vliv na délku přežití, pokud bylo před SCT dosaženo redukce počtu blastů chemoterapií pod 10 % [30], jinak samotná kombinovaná chemoterapie bez následné transplantace měla jen omezený efekt (3leté přežití pouze u 3 % nemocných).
Závěr
Při zhodnocení vývoje léčby nemocných s MDS během posledních 30 let na datech z registru ÚHKT vidíme přechod od podpůrné léčby a paliativní terapie k moderním léčebným metodám užívajícím léky, jež mohou cíleně ovlivnit molekulárně genetickou podstatu onemocnění. Takovými léky jsou lenalidomid u nízce rizikových nemocných a hypometylační látky u pokročilé fáze onemocnění. Zavedení transplantace krvetvorných buněk výrazně ovlivnilo délku přežití zejména mladších nemocných s pokročilými formami choroby s nadbytkem blastů a představuje stále jediný kurativní přístup k této chorobě.
Poděkování
Autor děkuje za spolupráci Mgr. Monice Beličkové, RNDr. Michaele Dostálové-Merkerové, Mgr. Alžbětě Vašíkové, prof. Ing. Kyře Michalové, CSc., MUDr. Janě Březinové, CSc., MUDr. Daně Mikulenkové, MUDr. Petře Kačírkové a všem lékařům klinického a výzkumného úseku ÚHKT, kteří se podíleli na diagnostice MDS a na léčbě nemocných.
doc. MUDr. Jaroslav Čermák, CSc.
www.uhkt.cz
e-mail: cermak@uhkt.cz
Doručeno do redakce: 19. 6. 2012
Sources
1. Björkman SE. Chronic refractory anemia with sideroblastic bone marrow: a study of four cases. Blood 1956; 11 : 250–259.
2. Bennett J, Catovsky D, Daniel MT et al. Proposals for the classification of the myelodysplastic syndromes. Br J Haematol 1982; 51 : 189–199.
3. Raza A, Gezer S, Mundle S et al. Apoptosis in bone marrow biopsy samples involving stromal and hematopoietic cells in 50 patients with myelodysplastic syndromes. Blood 1995; 86 : 268–276.
4. Parker JE, Mufti GJ. Ineffective haemopoiesis and apoptosis in myelodysplastic syndromes. Br J Haematol 1998; 101 : 220–230.
5. Brunning RD, Orazi A, Germing U et al. Myelodysplastic syndromes/neoplasms, overview. In: Swerdlow SH, Campo E, Harris NL et al (eds). WHO Classification of Tumours of Haematopoietic nad Lymphoid Tissues. 4th ed. Lyon: IARC Press 2008 : 88–93.
6. Greenberg P, Cox C, LeBeau MM et al. International scoring system for evaluating prognosis in myelodysplastic syndromes. Blood 1997; 89 : 2079–2088.
7. Schanz J, Tüchler H, Solé F et al. A new comprehensive cytogenetic scoring system for primary myelodysplastic syndromes (MDS) and oligoblastic AML after MDS derived from an international database merge. J Clin Oncol 2012; 30 : 820–829.
8. Tiu RV, Gondek LP, O’Keefe CL et al. Prognostic impact of SNP array karyotyping in myelodysplastic syndromes and related myeloid malignancies. Blood 2011; 117 : 4552–4560.
9. van de Loosdrecht AA, Alhan C, Béné MC et al. Standardization of flow cytometry in myelodysplastic syndromes: report from the first European LeukemiaNet working conference on flow cytometry in myelodysplastic syndromes. Haematologica 2009; 94 : 1124–1134.
10. Májek P, Reicheltová Z, Suttnar J et al. Plasma proteome changes associated with refractory cytopenia with multilineage dysplasia. Proteome Sci 2011; 9 : 64.
11. Sanz GF, Sanz MA, Vallespí T et al. Two regression models and a scoring system for predicting survival and planning treatment in myelodysplastic syndromes: a multivariate analysis of prognostic factors in 370 patients. Blood 1989; 74 : 395–408.
12. Harris NL, Jaffe ES, Diebold J et al. World Health Organization classification of neoplastic diseases of the hematopoietic and lymphoid tissues: report of the Clinical Advisory Committee meeting – Airlie House, Virginia, November 1997. J Clin Oncol 1999; 17 : 3835–3849.
13. Malcovati L, Germing U, Kuendgen A et al. Time-dependent prognostic scoring system for predicting survival and leukemic evolution in myelodysplastic syndromes. J Clin Oncol 2007; 25 : 3503–3510.
14. Čermák J, Kacirkova P, Mikulenkova D et al. Impact of transfusion dependency on survival in patients with early myelodysplastic syndrome without excess of blasts. Leuk Res 2009; 33 : 1469–1474.
15. Dostálová Merkerová M, Bystrická D, Belicková M et al. From cryptic chromosomal lesions to pathologically relevant genes: integration of SNP-array with gene expression profiling in myelodysplastic syndrome with normal karyotype. Genes Chromosomes Cancer 2012; 51 : 419–428.
16. Vasikova A, Belickova M, Budinska E et al. A distinct expression of various gene subsets in CD34+ cells from patients with early and advanced myelodysplastic syndrome. Leuk Res 2010; 34 : 1566–1572.
17. Bejar R, Stevenson K, Abdel-Wahab O et al. Clinical effect of point mutations in myelodysplastic syndromes. N Eng J Med 2011; 364 : 2496–2506.
18. Malcovati L, Papaemmanuil E, Bowen DT et al. Clinical significance of SFR3Bl mutations in myelodysplastic syndromes and myelodysplastic/myeloproliferative neoplasms. Blood 2011; 118 : 6239–6246.
19. Bennett JM. Consensus statement on iron overload in myelodysplastic syndromes. Am J Hematol 2008; 83 : 858–861.
20. Hellström-Lindberg E. Efficacy of erythropoietin in the myelodysplastic syndromes: a meta-analysis of 205 patients from 17 studies. Br J Haematol 1995; 89 : 67–71.
21. Passweg JR, Giagounidis AA, Simcock M et al. Immunosuppressive therapy for patients with myelodysplastic syndrome: a prospective randomized multicenter phase III trial comparing antithymocyte globulin plus cyclosporine with best supportive care – SAKK 33/99. J Clin Oncol 2010; 29 : 303–309.
22. Sloand EM, Wu CO, Greenberg P et al. Factors affecting response and survival in patients with myelodysplasia treated with immunosuppressive therapy. J Clin Oncol 2008; 26 : 2505–2511.
23. Bejar R, Levine R, Eberet BJ. Unraveling the molecular pathophysiology of myelodysplastic syndromes. J Clin Oncol 2011; 29 : 504–515.
24. Kuendgen A, Lauseker M, List AM et al. Lenalidomid treatment is not related to AMl progression risk but is associated with survival benefit in RBC-dependent transfusion patients with IPSS Low - or Int1-Risk MDS with del5q. Blood 2011; 118. Abstract 119.
25. Cermák J, Vítek A, Michalová K. Combined stratification of refractory anemia according to both WHO and IPSS criteria has a prognostic impact and improves identification of patients who may benefit from stem cell transplantation. Leuk Res 2004; 28 : 551–557.
26. Cutler CS, Lee SJ, Greenberg P et al. A decision analysis of allogeneic bone marrow transplantation for the myelodysplastic syndromes: delayed transplantation for low-risk myelodysplasia is associated with improved outcome. Blood 2004; 104 : 579–585.
27. Fenaux P, Mufti GJ, Hellstrom-Lindberg E et al. Efficacy of azacitidine compared with that of conventional care regimens in the treatment of higher-risk MDS: a randomized, open-label, phase III study. Lancet Oncology 2009; 10 : 223–232.
28. Martino R, Iacobelli S, Brand R et al. Retrospective comparison of reduced-intensity conditioning and conventional high-dose conditioning for allogeneic hematopoietic stem cell transplantation using HLA-identical sibling donors in myelodysplastic syndromes. Blood 2006; 108 : 836–846.
29. Schmid C, Schleuning M, Ledderose G et al. Sequential regimen of chemotherapy, reduced-intensity conditioning for allogeneic stem-cell transplantation, and prophylactic donor lymphocyte transfusion in high-risk acute myeloid leukemia and myelodysplastic syndrome. J Clin Oncol 2005; 23 : 5675–5687.
30. Cermak J, Vitek A, Markova M et al. Combination chemotherapy leading in advanced MDS patients in a rapid clearence of bone marrow blasts prior stem cell transplantation (SCT) is superior to up-front SCT even with intensified conditiong for long-term survival. Blood 2010; 116. Abstract 4020.
Labels
Diabetology Endocrinology Internal medicineArticle was published in
Internal Medicine

2012 Issue Suppl 2
Most read in this issue
- Imunohematologie – historie, současný stav poznání a role ÚHKT
- Naléhavé stavy v hematologii
- Hemaferéza – vysoce účinná technika v terapii nemocných
- Chronická myeloidní leukemie – zásadní změna prognózy nemocných po zavedení léčby inhibitory tyrozinových kináz