
Familiárna stredomorská horúčka – klinický obraz, diagnóza a liečba
Familial Mediterranean Fever – clinical picture, diagnosis and treatment
Familial mediterranean fever (FMF) is the most prevalent genetically determined autoinflammatory disease. FMF significantly decreases the quality of life and limits life expectancy due to the development of amyloidosis in affected individuals. Prevalence of FMF is highest in the south-eastern Mediterraneans. In other parts of the world, its occurance is often restricted to high-risk ethnic goups. In Central Europe, experience with FMF is scarse. As for Slovakia, we have reported the first cases of FMF in ethnic Slovaks only recently. Along with their complicated fates, this has lead us to compile a comprehensive overview of the clinical picture, diagnosis and treatment of this elusive disease. Hereby we hope to be able to promote the awareness about this disease and possibly aid the diagnosis in new patients.
Key words:
familial mediterranean fever – amyloidosis – colchicin – overview of the clinical picture
:
Tomáš Dallos; Denisa Ilenčíková; László Kovács
:
II. detská klinika LF UK a DFNsP, Bratislava, Slovenská republika, prednosta prof. MUDr. László Kovács, DrSc., MPH
:
Vnitř Lék 2014; 60(1): 30-37
:
Reviews
Familiárna stredomorská horúčka (FMF) je najčastejšie geneticky podmienené autoinflamačné ochorenie. Významne zhoršuje kvalitu života pacientov a rozvojom amyloidózy limituje dĺžku ich života. Najvyššia prevalencia je v juhovýchodnom Stredomorí, v iných častiach sveta je výskyt väčšinou viazaný na etnický pôvod z týchto oblastí. V strednej Európe sú skúsenosti s FMF obmedzené, na Slovensku až donedávna chýbali úplne. Len nedávno potvrdené prvé prípady FMF u etnických Slovákov a ich zložité osudy nás viedli k zostaveniu prehľadu o klinickom obraze, diagnostike a liečbe tohto ochorenia, ktoré pravdepodobne uniká našej pozornosti. Týmto chceme prispieť k zlepšeniu povedomia odbornej verejnosti o tomto ochorení a odhaleniu nových prípadov.
Kľúčové slová:
familiárna stredomorská horúčka – amyloidóza – kolchicín – prehľad o klinickom obraze
Úvod
Autoinflamačné ochorenia sú výsledkom spontánnej a nekontrolovanej aktivácie mechanizmov prirodzenej imunity v dôsledku genetickej poruchy v kľúčových mechanizmoch zápalovej kaskády [1]. Familiárna stredomorská horúčka (Familial Mediterranean Fever – FMF, OMIM 249100) je prototypom autoinflamačného ochorenia. V juhovýchodnom Stredomorí a na Blízkom východe dosahuje u Arménov, Turkov, Židov a Arabov vysokú prevalenciu (1 : 200 – 1 : 500) a jej klinický obraz je preto známy už od začiatku 20. storočia [2–4]. Avšak až o takmer 90 rokov neskôr sa pri FMF ako u prvého autoinflamačného ochorenia podarilo odhaliť genetickú podstatu FMF [5,6]. Tak sa otvorila cesta k pochopeniu až do vtedy neznámych porúch vykazujúcich zvýšenú aktivitu prirodzenej imunity.
FMF sa typicky prejavuje rekurentnými neprovokovanými epizódami horúčok sprevádzanými bolesťami brucha alebo hrudníka. Avšak v závislosti od genotypu, etnika a prostredia, v ktorom postihnutý žije, je klinický obraz variabilný a najmä u príslušníkov nerizikových etník netypický. Celkovo nízka prevalencia (1 : 465 500) v strednej Európe [7] a z toho vyplývajúci nedostatok skúseností s FMF sa podieľajú na významnom oneskorení diagnózy a pravdepodobne aj na nezanedbateľnom počte nediagnostikovaných pacientov. Túto skutočnosť sme demonštrovali aj na osudoch 5 pacientov nedávno diagnostikovaných na Slovensku [8].
Aj keď klinické príznaky FMF významne znižujú kvalitu života pacienta [9], bezprostredne ho neohrozujú na živote a umožňujú dlhodobé prežívanie aj s nediagnostikovaným ochorením. Avšak až u 60 % neliečených pacientov sa rozvinie amyloidóza, ktorá je život limitujúcou komplikáciou. Rozpoznanie ochorenia a dôsledná celoživotná liečba sú preto rozhodujúce pre prognózu pacienta s FMF.
Ako sme nedávno zistili, FMF sa môže vyskytnúť nielen u etnicky disponovaných jedincov, ale aj u etnických Slovákov [8], a preto vnímame potrebu oboznámiť širokú odbornú verejnosť s touto nozologickou jednotkou. I keď manifestácia ochorenia je vždy v detskom veku, naše vlastné skúsenosti demonštrujú, že s nediagnostikovaným pacientom sa v našich podmienkach rovnako často môže stretnúť aj lekár pre dospelých. V nasledovnom texte ponúkame aktuálny prehľad poznatkov z odbornej literatúry a veríme, že takto prispejeme k zvýšeniu povedomia o tomto ochorení a odhaleniu ďalších až donedávna nediagnostikovaných prípadov.
Autoinflamačné syndrómy
Imunitný systém chráni organizmus pred infekciou a malignitou pomocou mechanizmov vrodenej nešpecifickej a získanej špecifickej imunity. Nedostatočná funkcia oboch typov imunity vedie k imunodeficitu, neprimeraná aktivita špecifickej imunity k autoimunite. Autoinflamačné ochorenia vyplnili chýbajúcu časť mozaiky porúch imunitného systému a predstavujú stavy spôsobené nadmernou funkciou nešpecifickej imunity [1]. V ich patogenéze sa uplatňuje spontánna aktivácia alebo nedostatočná inhibícia jej kľúčových mechanizmov.
Patrí k nim inflamazóm – multiproteínový cytoplazmový komplex myeloidných buniek zodpovedný za rýchlu, ale prísne regulovanú aktiváciu kaspázy 1 (IL1β konvertujúceho enzýmu), a tým aj produkciu IL1β – kľúčového zápalového cytokínu. Súčasne sa zvyšuje aktivita ďalšej zápalovej kaskády regulovanej nukleárnym faktorom NFκB. Oboje sa aktivujú rozpoznaním konzervovaných molekulových štruktúr typických pre patogény (pathogen-associated molecular pattern – PAMP) ako sú lipopolysachrid, peptidoglykan, flagelín a kyselina lipoteichová pomocou povrchových TLR (toll-like) alebo cytoplazmových NLR (Nod-like) receptorov, alebo molekulovými štruktúrami typickými pre poškodenie vlastných buniek (damage-associated molecular pattern – DAMP). Vrodené syndrómy v dôsledku nadmernej funkcie týchto mechanizmov prirodzenej imunity môžu byť prejavom zvýšenej produkcie IL1β inflamazómom (kryopyrinopatie, FMF, deficit antagonistu receptora IL1), aktivácie NFκB (FMF, PAPA syndróm, Blauov syndróm, resp. detská granulomatózna artritída) a TNFα (periodický syndróm asociovaný s receptorom TNFα).
Patogenéza FMF
Prvého pacienta s FMF zdokumentovali Janeway a Mosenthal už v roku 1908 [2]. O takmer pol storočia neskôr Siegal et al zvolili výstižné popisné pomenovanie „benígna paroxyzmálna peritonitída“, resp. „familárna paroxyzmálna polyserozitída“ [3,4]. Už vtedy zaznamenali familiárny výskyt FMF [4], ale až v roku 1997 dve nezávislé skupiny potvrdili asociáciu ochorenia s génom MEFV (MEditerranean FeVer) (16p13.3) [5,6]. I keď je dedičnosť autozómovo recesívna, v dôsledku dominantnej dedičnosti niektorých alel, variabilnej penetrancie a vplyvu epigenetických faktorov pôsobí niekedy pseudodominantne [10].
FMF je prototypom autoinflamačného ochorenia. Dnes je známych už takmer 200 mutácií lokalizovaných prevažne v 2. a 10. exóne génu MEFV, ktoré spôsobujú nedostatočnú funkciu 781-aminokyselinovej molekuly pyrínu/marenostrínu [11]. Pyrín sa tvorí v neutrofiloch, eozinofiloch, dendritových bunkách a fibroblastoch. Jeho N-terminálna pyrínová doména je súčasťou viacerých proteínov. Aktivuje transkripčný faktor NFκB a patrí aj do molekulového komplexu inflamazómu, kde pôsobí inhibične. Nedostatočná funkcia pyrínu vyústi do neprovokovanej a nekontrolovanej tvorby IL1β [12], produkcie endogénnych pyrogénov, proteínov akútnej fázy (CRP) a aktivácie neutrofilov. Vzniká tak klinický a laboratórny obraz akútneho bakteriálneho zápalu bez prítomnosti infekcie.
Epidemiológia FMF
Celosvetovo je ochorením postihnutých asi 100 000 ľudí. Výskyt FMF má charakteristickú geografickú distribúciu s prevalenciou 1 : 250 – 1 : 1 000 v krajinách Blízkeho východu a juhovýchodnej mediteránnej oblasti (Arménsko, Turecko, Libanon, Izrael). Predpokladá sa, že 3 najčastejšie mutácie vznikli pred približne 2 500 rokmi v Mezopotámii [13] a pretrvali, lebo heterozygoti profitovali zo zvýšenej odolnosti voči patogénom. Z týchto oblastí sa mutácie šírili do ostatných častí sveta, kde je v súčasnosti prevalencia podstatne nižšia, s výskytom najmä u uvedených etník (napr. Turci v Nemecku, Arabi vo Francúzsku). Etnický pôvod pacienta je preto dôležitým anamnestickým údajom. Avšak viaceré historické vlny migrácie a miešanie s domácim obyvateľstvom vysvetľujú aj výskyt v krajinách, ako je Taliansko (obchodovanie Feničanov), Španielsko (maurská okupácia v 8. storočí), Etiópia (židovská populácia), ale aj Japonsko (obchodovanie pozdĺž hodvábnej cesty). Nedá sa však vylúčiť, že sa môže jednať aj o mutácie de novo, keďže práve exóny 2 a 10 obsahujú veľa hot-spotov. V celej strednej Európe je dokumentovaných 11 geneticky potvrdených a 49 suponovaných prípadov ochorenia, prevažne v balkánskych krajinách s prevalenciou odhadovanou na 1 : 465 500 [7], pričom až donedávna neboli publikované prípady FMF na území Slovenska.
Klinický obraz FMF
Klinickému obrazu FMF dominujú:
- a) recidivujúce epizódy febrilít
- b) prejavy serozitídy
- c) zvýšené zápalové parametre
Charakteristický je rýchly nástup všetkých príznakov, ktoré v priebehu niekoľkých hodín dosiahnu vysokú intenzitu, krátko pretrvávajú (12–72 hod) a náhle úplne ustupujú [14]. Horúčky väčšinou presahujú 38 °C, ale sú známi aj pacienti so subfebrilitami. Najmä u detí môžu byť jediným príznakom FMF [15], i keď s trvaním ochorenia pribúdajú ďalšie. Atypické ataky môžu pretrvávať dlhšie ako 72 hod. K manifestácii ochorenia dochádza u 90 % pacientov do 20. roku života, ale až u 30 % už pred 2. rokom života [16].
Sterilný zápal seróznych blán sa manifestuje ako peritonitída, pleuritída, synovitída alebo zápal tunica vaginalis skróta u 90 %, 75 %, 40 %, resp. 5 % pacientov. Bolesti brucha sú difúzne, veľmi intenzívne až neznesiteľné. Počas epizódy býva sklon k obstipácii, u detí sú častejšie hnačky. Príznaky peritoneálneho dráždenia a rádiologický obraz subileózneho stavu imitujú náhlu brušnú príhodu a až 30–40 % pacientov podstúpi zbytočný chirurgický zákrok [17].
Synovitída prebieha ako rekurentná monoartritída septického charakteru s bolestivosťou, zateplením, začervenaním a obmedzením funkcie kĺbu [18]. Postihuje najmä veľké kĺby dolných končatín. Kĺbový aspirát má septický charakter s výraznou neutrofíliou, ale zachovanou viskozitou. U detí môže byť artritída prvou manifestáciou FMF a asymetricky postihovať aj viacero kĺbov. Rýchlo ustupuje, ale u 5 % pacientov býva chronická a pretrváva aj niekoľko mesiacov [19]. Je väčšinou bez trvalých následkov, i keď boli popísané aj prípady s masívnou synovitídou a potrebou protetickej náhrady. Artritídu je potrebné odlíšiť od polyartralgií, ktoré sa vyskytujú ako konštitučný príznak pri epizódach FMF.
Bolesti na hrudníku sú spôsobené pleuritídou alebo perikarditídou. Pleuritída je väčšinou jednostranná, vyvolá intenzívnu, ostrú bolesť, ktorá spôsobí tachypnoe alebo plytké dýchanie. Zriedkavo je prítomné zatienenie kostofrenického uhla. Prechodné zväčšenie tieňa srdca sa vyskytuje pri perikarditíde, ktorá je zriedkavá, ale môže byť jediným rekurentným príznakom FMF [20]. Často ju odhalí len echokardiografické vyšetrenie.
Najmä v detskom veku sa vyskytuje jednostranný zápal tunica vaginalis, ktorý sa prejaví ako opuch, bolestivosť a začervenanie hemiskróta. Imituje iné príčiny akútneho skróta (torkvácia testes, epidydimitída, orchitída), ktoré sú u detí časté. K zriedkavým manifestáciám patria recidivujúce sterilné meningitídy.
Charakteristickým, ale zriedkavým (5 %) kožným prejavom FMF je rozsiahle, dobre ohraničené, mierne prominujúce, horúce a bolestivé začervenanie kože oboch predkolení presahujúce cez členky až na dorzum nohy – eryzipeloidný exantém. Ojedinele sa na predkoleniach vyskytujú bolestivé podkožné uzly do 1 cm so začervenaním kožného krytu, ktoré sa ponášajú na erythema nodosum [21].
Postihnutie svalov môže nadobudnúť viacero podôb:
- a) myalgie počas febrilnej ataky
- b) ponámahové bolesti lýtok
- c) protrahovaná febrilná myalgia
- d) kolchicínová myopatia
Ponámahové bolesti lýtok sú dôležitý diagnostický znak, lebo sa vyskytujú až u 30 % pacientov. Protrahovaná febrilná myalgia je niekoľko týždňov pretrvávajúci stav extrémnej bolesti svalov oboch dolných končatín sprevádzaný horúčkami a prípadne aj rašom [22]. Ani napriek intenzite príznakov nie sú prítomné biochemické ani elektromyografické známky myogénnej lézie; pravdepodobne sa jedná o formu vaskulitídy. Vysokodávková kortikoterapia poskytne pacientovi úľavu.
Laboratórny obraz FMF
Laboratórny obraz FMF zodpovedá zápalovému procesu s leukocytózou s neutrofíliou, zvýšenou sedimentáciou erytrocytov a proteínov akútnej fázy (CRP, sérový amyloid, fibrinogén). Podobne ako klinické príznaky, aj zápalová aktivita spontánne ustupuje s koncom febrilnej epizódy. Medzi epizódami bývajú zápalové parametre vo fyziologickom rozmedzí, avšak až u 2/3 pacientov pretrvávajú zvýšené aj v asymptomatickom období ako prejav trvalej aktivácie zápalovej kaskády [23].
Uvedené príznaky vytvárajú pestrý a individuálny obraz recidivujúcich a krátko trvajúcich epizód horúčky s výraznými bolesťami. Ataky FMF sa vyskytujú spontánne a nepredvídateľne v nepravidelných časových intervaloch raz za týždeň, alebo len raz za rok. Niekedy môžu byť provokované únavou, stresom, cestovaním cez časové pásma, liekmi, infekciou, prítomnosťou Helicobacter pylori či mastným jedlom. Na základe koincidencie aták s menštruačným krvácaním sa predpokladá ochranný efekt estrogénov [24]. Pacienti niekedy pociťujú prodrómy ako iritabilitu, cefaleu, myalgie, artralgie, nauzeu, vracanie, dyspnoe a lumbalgie [25]. Medzi epizódami sú väčšinou bez ťažkostí, niekedy trpia muskuloskeletálnymi prejavmi (myalgie, artralgie a chronické artritídy). Klinický obraz je často ovplyvnený etnickým pôvodom, environmentálnymi a epigenetickými faktormi. Počas života sa klinické ťažkosti často zmierňujú.
Komplikácie FMF
Dôsledkom rekurentnej či trvalej aktivácie zápalovej kaskády pri FMF je zvýšená tvorba sérového amyloidu A (SAA), prekurzora amyloidu A, ktorý sa ukladá do orgánov (obličky, črevo, pečeň, štítna žľaza, srdce). Vedie k najzávažnejšej komplikácii FMF – systémovej amyloidóze [26]. Jej najčastejším prejavom pri FMF je nefropatia, ktorá cez asymptomatické, proteinurické, nefrotické a azotemické štádium prechádza do chronickej renálnej insuficiencie s urémiou. Do 10–15 rokov od manifestácie ochorenia sa vyskytuje až u 60 % pacientov s neliečenou FMF a vedie k zlyhaniu obličiek pred 40. rokom života. Prežívanie pacientov s neliečenou FMF po 60. roku života je raritné [26]. Ak sa u pacienta zistí perzistujúca proteinúria > 0,5 g/24 hod, je diagnóza amyloidózy takmer istá [27]. Potvrdiť ju možno biopsiou obličky alebo rektálnej sliznice. Amyloidóza ostatných orgánov sa môže prejaviť malabsorpciou, hepatomegáliou s ikterom, arytmiou alebo kongestívnym zlyhaním srdca.
Výskyt amyloidózy je závislý od mnohých faktorov napr. genetických a environmentálnych; Turci a sefardský Židia trpia amyloidózou častejšie ako Arménci a aškenázski Židia. Ak žijú v západnej Európe alebo v USA, je u nich rozvoj amyloidózy menej častý. Vyššie riziko majú muži, pacienti s artritídou a amyloidózou u príbuzných s FMF [28]. I keď zložený heterozygotný stav nevylučuje amyloidózu, homozygotná mutácia M694V sa spája s včasnou manifestáciou FMF a oboje sú asociované s vyšším rizikom amyloidózy [29]. Včasná diagnóza má zásadný význam pre pacientov s rozvojom amyloidózy v detskom veku. Rozhodujúcim faktorom úspešnej prevencie amyloidózy je kompliancia s liečbou. Tá je sťažená aj tým, že riziko ani závažnosť amyloidózy nesúvisia s frekvenciou ani intenzitou aták FMF. Výskyt amyloidózy je preto len ťažko predvídateľný a je s ním potrebné vždy rátať. Ako užitočné sa ukazuje monitorovanie hladiny SAA, ktorého vysoké hladiny a izotyp SAA a/a sú spojené s vysokým rizikom tejto komplikácie [30].
Pri FMF je častejší výskyt
- a) vaskulitíd
- b) glomerulonefritíd
- c) niektorých ďalších autoimúnnych ochorení
- d) komplikácií recidivujúcej serozitídy
Z vaskulitíd je najmä v detskom veku najčastejšia Henochova-Schönleinova purpura, ktorá sa mnohými príznakmi môže podobať na atak FMF [31] a polyarteriitis nodosa. Vyskytuje sa aj spolu Behçetovou chorobou, ktorá navyše dobre reaguje na liečbu kolchicínom. Nefritický syndróm, ktorý je potrebné odlíšiť od nefrotického pri amyloidóze, môže byť taktiež prejavom polyarteriitis nodosa, ale aj mezangioproliferatívnej a zriedkavej fibrilárnej glomerulonefritídy. Boli popísaní pacienti s FMF so súčasnou juvenilnou idiopatickou artritídou [32], spondylartropatiou a nešpecifickými zápalovými ochoreniami čreva [33]. Podobne ako pri autoimunitných ochoreniach sa u 30 % vyskytuje fibromyalgický syndróm, ktorý môže byť náročné odlíšiť od afebrilných aták FMF. Opakované serozitídy podporujú vznik adhézií s následnými nezápalovo podmienenými bolesťami brucha, obštrukčným ileom či konstriktívnou perikarditídou. V rizikových etnikách sa preto u detí s vznikom invaginácie čreva počas Henochovej-Schönleinovej purpury odporúča myslieť aj na FMF.
Diagnóza FMF
Diagnóza FMF vychádza predovšetkým z klinického obrazu, čo odzrkadľuje aj viacero diagnostických kritérií pre dospelých [34] a deti [35,36] (tab). Všetky boli vyvinuté v rizikových populáciách a s cieľom predísť závažným následkom nediagnostikovaného ochorenia. Nie je k dispozícii žiaden laboratórny parameter, ktorý by podporil diagnózu FMF a dostupné laboratórne markery ako CRP, SAA, fibrinogén sú vhodné len na sledovanie aktivity ochorenia. V endemických populáciách sa ako súčasť diagnostického procesu odporúča terapeutický pokus kolchicínom, ktorý je aj súčasťou diagnostických kritérií: po 6–12-mesačnom užívaní kolchicínu sa liečba ukončí a v prípade, že dôjde k ústupu a následnému návratu klinických ťažkostí, sa diagnóza považuje za potvrdenú. Vzhľadom na nižšiu pravdepodobnosť výskytu sa v nerizikových populáciách kolchicínový test bežne neodporúča.
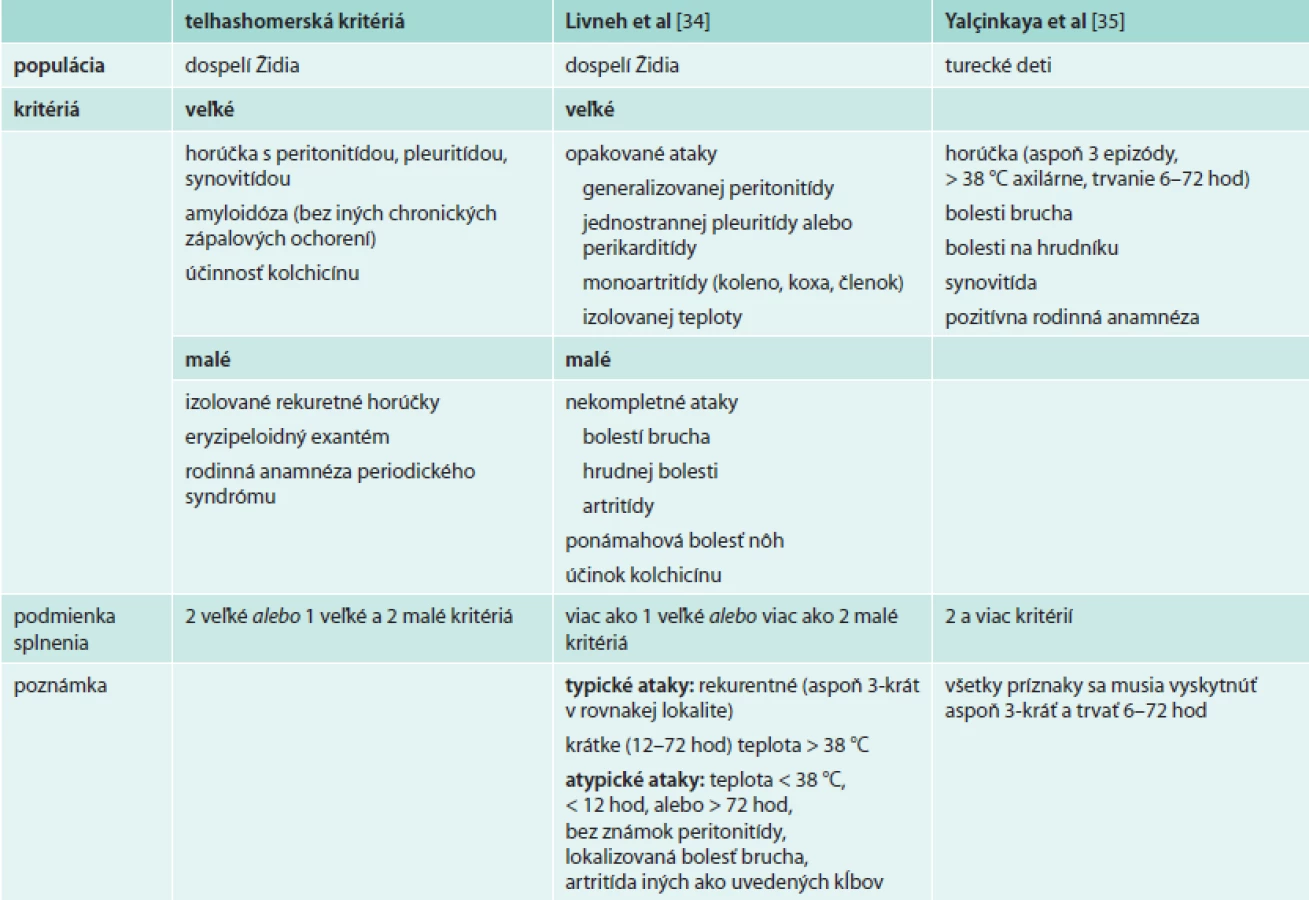
Dôkaz mutácie v géne MEFV nie je podmienkou diagnózy FMF. Navyše genetická analýza nepriniesla významné zlepšenie diagnostiky FMF a jej postavenie je zložité a neuspokojivé. Päť najčastejších mutácií (M694I, M680I, M694V, E148Q a V726A) je v homozygotnom stave zodpovedných za 63–74 % prípadov FMF v rizikových etnikách [37], a preto sa pri klinickom podozrení na FMF aj používajú v rutinnom skríningu. Ostatné mutácie sú zriedkavejšie a vyžadujú technicky a finančne náročnejšiu, a teda ťažšie dostupnú analýzu. Interpretácia výsledkov sa ďalej komplikuje neúplnou genotypovo-fenotypovou koreláciou. Hoci homozygoti M694V a nosiči komplexnej alely V726A-E148Q mávajú ťažšie priebehy aj vyššie riziko amyloidózy, môžu byť aj bez klinických príznakov. Naopak aj u zložených heterozygotov či dokonca heterozygotov, ktorí predstavujú až 25–40 % postihnutých v nerizikových populáciách, sa môže vyskytovať plný klinický obraz FMF [38,39]. Navyše sú známi pacienti s typickým klinickým obrazom FMF, u ktorých ani sekvenácia nepreukáže mutáciu v géne MEFV. Ak teda pacient z endemickej oblasti splní klinické kritériá, genetické vyšetrenie podľa niektorých autorov nie je potrebné [40].
Uvedené skutočnosti sťažujú indikáciu aj interpretáciu výsledkov vyšetrenia génu MEFV v niektorých klinických situáciách. Vo všeobecnosti sa dodržiavajú nasledovné zásady:
- a) pacient s jasným klinickým nálezom sa aj pri chýbaní mutácie musí liečiť kolchicínom
- b) u pacientov z rizikového etnika s nejasným klinickým obrazom je u heterozygotov aj pri negatívnom genetickom vyšetrení indikovaný terapeutický pokus
- c) homozygotov bez klinických ťažkostí je potrebné dispenzarizovať
- d) heterozygoti bez klinických ťažkostí nevyžadujú sledovanie [40]
Diferenciálna diagnóza
Spektrum ochorení, ktoré môžu FMF imitovať a navyše sú v našich podmienkach nepomerne častejšie, je veľmi široké. Bolesti brucha pri FMF svojou intenzitou napodobňujú náhle brušné príhody a ich odlíšenie u pacienta bez známej diagnózy FMF je takmer nemožné. Vysoký počet vykonaných chirurgických zákrokov pre nepotvrdené náhle brušné príhody patrí k typickej anamnéze pacienta s FMF. Koincidencia bolesti brucha s febrilitami je charakteristická aj pre iné autoinflamačné ochorenia (deficit mevalonátkinázy – MKD a periodický syndróm asociovaný s TNF receptorom – TRAPS / tumor necrosis factor receptor associated periodic syndrome). Pri MKD bolesti brucha neimitujú peritonitídu, je prítomná krčná lymfadenopatia a makulózny exantém a oligoartritída je symetrická. TRAPS sa odlišuje najmä výrazne dlhšími epizódami febrilít. V detskom veku môže byť problematické odlíšenie „skrotálneho ataku“ od častejších príčin akútneho skróta (orchitída, epidydimitída, torzia testes). „Hrudná forma“ FMF je taktiež častá u detí, u dospelých môže imitovať iné príčiny bolestí na hrudníku. Akútnu exacerbáciu artritídy pri FMF je potrebné odlíšiť od záchvatu dny, infekčnej artritídy, ataku Henochovej-Schönleinovej purpury alebo artritídy pri Behçetovej chorobe. Aj pri FMF sa vyskytuje sakroileitída, zápalová bolesť chrbtice, monoartritída nebo oligoartritída veľkých kĺbov dolných končatín a entezitída. Na rozdiel od spondylartropatie sa pri nej nevyskytuje uveitída, „bambusová“ chrbtica ani antigén HLA-B27. Polyartikulárna migrujúca artritída a postihnutie malých kĺbov by mali upozorňovať na možnosť inej etiológie artritídy. Patologické močové nálezy vyžadujú dôkladnú analýzu. Pretrvávajúca proteinúria je pravdepodobne prejavom obličkovej amyloidózy. Nefritícký syndróm môže byť prejavom glomerulonefritídy, systémového lupus erythematodes, prípadne nefropatie pri často sa vyskytujúcej Henochovej-Schönleinovej purpure. Netreba zabúdať ani na tubulointersticiálnu nefritídu vyvolanú užívaním nesteroidových antiflogistík. Napokon je častá aj izolovaná benígna erytrocytúria. Z autoimúnnych ochorení môže obraz FMF dokonale imitovať Behçetova choroba. Vyskytuje sa v rovnakých etnikách, môže byť spojená s bolesťami brucha a artritídou a podobne ako FMF dobre reaguje na liečbu kolchicínom. Preto je potrebné pátrať po orogenitálnych aftách, uveitíde a erythema nodosum, ktoré sú pre ňu typické.
Liečba FMF
Liečbou voľby FMF je celoživotné profylaktické užívanie kolchicínu [41], ktorého účinok u dospelých aj detí bol preukázaný už v roku 1972 [42]. Kolchicín je lipofilný alkaloid, ktorý pochádza z Jesienky obyčajnej (Colchicum autumnale) – krókusu, ktorý rastie aj v našich zemepisných šírkach. Jedná sa o jedno z najstarších známych liečiv vôbec, ktoré sa spomína už v Ebersovom papyruse zo 16. storočia pred našim letopočtom. Kumuluje sa v neutrofiloch, v ktorých kolokalizuje s mikrotubulmi. Predpokladá sa, že destabilizuje cytoskelet, a tak bráni chemotaxii, diapedéze, fagocytóze a degranulácii neutrofilov, ale inhibuje aj syntézu oxidu dusného (NO), apoptózu, aktiváciu NFκB a produkciu IL1β.
Kolchicín sa dospelým pacientom podáva v dávke 1–2 mg/deň, ale maximálne 2,5 mg/deň. U detí do 5 rokov sa odporúča dávka 0,03–0,07 mg/kg/deň, resp. 0,5 mg/deň, od 5 do 10 rokov 1 mg/deň. V akútnom ataku sa neodporúča zvyšovať dávku kolchicínu, pridávajú sa nesteroidové antiflogistiká. Kolchicín je účinný u viac ako 90 % pacientov s FMF (dospelých aj detí) [43]; u 60 % úplne vymiznú klinické príznaky, u 20–30 % sa zníži ich frekvencia a/alebo intenzita, čím sa výrazne zlepší kvalita života [44]. Je nevyhnutné jeho každodenné a celoživotné užívanie, keďže aj vynechanie jednej dávky môže umožniť atak FMF. U 5–10 % pacientov dávka 2 mg/deň a viac nenavodí ústup febrilných aták. Ďalšie zvyšovanie dávky sa neukázalo ako prínosné a bolo spojené s vysokým rizikom závažnej toxicity. Rezistencia na liečbu je dôsledkom geneticky podmienenej farmakorezistencie znižujúcej koncentrácie kolchicínu v mononukleárnych bunkách.
Ani chýbanie účinku na klinické príznaky by nemalo pacienta odradiť od pokračovania v liečbe, keďže aj za týchto okolností pravidelné neprerušované užívanie kolchicínu účinne bráni rozvoju amyloidózy [45]. Predpokladá sa, že sa jedná o priamy účinok na produkciu SAA v pečeni [45]. Aj vynechanie jednotlivých dávok zvyšuje riziko amyloidózy. Kompliancia s liečbou je preto rozhodujúca pre prognózu pacienta. U pacientov s renálnou amyloidózou sa aj napriek nefrotoxickému potenciálu kolchicínu pokračuje v liečbe v maximálne tolerovanej dávke, keďže kolchicín rýchlo zníži proteinúriu a až na 10 rokov navodí remisiu nefrotického syndrómu [46].
Dlhodobé užívanie kolchicínu je bezpečné. V terapeutických dávkach kolchicín nemá onkogénny ani teratogénny či toxický účinok na plod, resp. dojčené dieťa [47]. Naopak, v gravidite znižuje riziko spontánneho abortu a predčasného pôrodu. Pacientky pokračujú v liečbe počas gravidity aj dojčenia bez úpravy dávky. Ustupuje sa od vyšetrenia plodovej vody, pokiaľ nie je indikované z iných dôvodov. U detí kolchicín neznižuje rast; po začatí liečby dôjde k vzostupu IGF1 [48] a rastovému špurtu [49].
Z nežiadúcich účinkov je najčastejšia hnačka, ktorej sa dá predísť rozdelením denných dávok, bezmliečnou a nízkotukovou diétou. Tolerancia je lepšia, ak sa kochicín zavádza v postupne sa zvyšujúcich dávkach a môže byť závislá aj od typu preparátu. K potenciálnym závažným, ale vzácnym nežiaducim účinkom patrí myelotoxicita, nefrotoxicita, hepatotoxicita a rozvoj neuromyopatie, ktoré je potrebné monitorovať v polročných intervaloch (krvný obraz, AST, ALT, LD, CK, urea, kreatinín, močový sediment). Bol popísaný aj negatívny vplyv na spermatogenézu. Všetky nežiadúce účinky sú reverzibilné redukciou dávky alebo vysadením liečby.
Pacientov je dôležité upozorniť, že v prípade predávkovania hrozí fatálna intoxikácia, ktorá sa ponáša na otravu arzénom a je neliečiteľná. Liečivá, ktoré sú metabolizované pečeňovým enzýmom CYP3A4, patriacim k cytochrómu P450 (diltiazem, cimetidín, etokonazol, rifampicin, fenobarbital, fenytoín a statíny) môžu zvýšiť koncentrácie kolchicínu na toxickú úroveň. U detí je potrebné vylúčiť makrolidové antibiotiká, ktoré interakciou s kolchicínom môžu navodiť ťažkú agranulocytózu [50]. Toxicitu kolchicínu zvyšujú aj imidazolové chemoterapeutiká (metronidazol, fluconalzol, ketokonazol). U pacientov po transplantácii obličky je nutná zvýšená opatrnosť pri liečbe cyklosporínom A. Podobne ako pacienti liečení cyklosporínom A ani pacienti liečení kolchicínom by nemali konzumovať grapefruity.
Na rozdiel od iných autoinflamačných ochorení, väčšina pacientov s FMF vďaka vysokej účinnosti kolchicínu nevyžaduje biologickú liečbu. Pri rezistencii alebo intolerancii kolchicínu bola u 15 dospelých a detí použitá blokáda IL1, ktorá bola bez výnimky účinná [51,52]. Zatiaľ nie sú dostupné údaje o účinnosti týchto preparátov na amyloidózu.
Záver
V poslednej dekáde sme boli svedkami objasnenia podstaty viacerých porúch univerzálnych mechanizmov prirodzenej imunity, ktoré vyúsťujú do jej spontánnej a nekontrolovanej aktivácie so vznikom autoinflamačných ochorení. Dostupná a vysoko účinná liečba (blokáda IL1, ev. TNFα, resp. kolchicín pri FMF) z nich robí medicínsky veľmi atraktívne nozologické jednotky.
Aj keď povedomie odbornej verejnosti o autoinflamačných ochoreniach v detskom veku (deficit mevalonátkinázy, kryopyrinopatie [53], syndróm asociovaný s deficitom receptora pre TNFα a pravdepodobne aj najčastejší PFAPA-syndróm (Periodic Fever, Aphthous Stomatitis, Pharyngitis and Cervical Adenitis, Marshall‘s syndrome) [54–56]) a v dospelom veku (Schnitzlerovej syndróm) [57] narastá aj v našom regióne, ich zriedkavý výskyt, s tým súvisiaci nedostatok skúseností a relatívna nedostupnosť genetickej analýzy sťažujú ich diagnostiku. Aj naše skúsenosti s FMF potvrdzujú, že ich nízka prevalencia v našej etnicky nedisponovanej populácii sa dodnes podpisuje na zložitom osude postihnutých pacientov [8]. Ako vždy pri zriedkavých ochoreniach, správna diagnóza stojí na tom, že sa na ňu myslí. Nepredvídateľné, krátke a spontánne ustupujúce epizódy horúčok sprevádzané bolesťami brucha či na hrudníku a vysokými zápalovými parametrami, ale aj ich nevysvetlené pretrvávanie medzi epizódami, by mali aj pri negatívnej rodinnej anamnéze viesť k zváženiu tejto diagnózy. Analýza génu MEFV môže byť veľmi nápomocná, je dostupná a v konečnom dôsledku umožní začatie celoživotnej liečby kolchicínom, ktorá má rozhodujúci význam pre kvalitu života aj prognózu pacienta. Hoci sa FMF manifestuje vždy pred 20. rokom života, v našich podmienkach je potrebné o nediagnostikovanom ochorení uvažovať aj v dospelom veku.
Liečba FMF kolchicínom je účinná a bezpečná vo všetkých vekových kategóriách aj u tehotných a dojčiacich matiek. U dospelých je možné využiť skúsenosti reumatológov získané liečbou dnavej artritídy. U detí žiaľ takéto skúsenosti úplne chýbajú. Napriek tomu je v záujme pacienta, aby sa preklenuli predsudky o podávaní tohto „bunkového jedu“ dieťaťu a aby aj detský pacient s FMF dostal adekvátnu liečbu podľa platných odporúčaní [58].
MUDr. Tomáš Dallos, PhD.
dallos@dfnsp.sk
II. detská klinika LF UK a DFNsP, Bratislava, Slovensko
www.detskaklinika.sk
Doručeno do redakce: 7. 5. 2013
Přijato po recenzi: 3. 9. 2013
Sources
1. Dinarello CA. Interleukin-1beta and the autoinflammatory diseases. N Engl J Med 2009; 360(23): 2467–2470.
2. Janeway TC, Mosenthal HO. Unusual paroxysmal syndrome, probably allied to recurrent vomiting, with a study of the nitrogen metabolism. Trans Assoc Am Physicians 1908; 23 : 504–518.
3. Siegal S. Benign paroxysmal peritonitis. Ann Intern Med 1945; 23 : 1–22.
4. Siegal S. Familial paroxysmal polyserositis: analysis of fifty cases. Am J Med 1964; 36 : 893–918.
5. French FMF Consortium. A candidate gene for familial Mediterranean fever. Nat Genet 1997; 17(1): 25–31.
6. The International FMF Consortium. Ancient missense mutations in a new member of the RoRet gene family are likely to cause familial Mediterranean fever. Cell 1997; 90(4): 797–807.
7. Toplak N, Doležalová P, Constantin T et al. Periodic fever syndromes in Eastern and Central European countries: results of a pediatric multinational survey. Pediatr Rheumatol Online J 2010; 8 : 29.
8. Dallos T, Lukáčiková Gálová L, Macejková E et al. Familiárna stredomorská horúčka – prvé skúsenosti na Slovensku. Vnitř Lék 2014; XXX: 60(1): 80–85.
9. Buskila D, Zaks N, Neumann L et al. Quality of life of patients with familial Mediterranean fever. Clin Exp Rheumatol 1997; 15(4): 355–360.
10. Booth DR, Gillmore JD, Lachmann HJ et al. The genetic basis of autosomal dominant familial Mediterranean fever. QJM 2000; 93(4): 217–221.
11. Sood R, Blake T, Aksentijevich I et al. Construction of a 1-Mb restriction-mapped cosmid contig containing the candidate region for the familial Mediterranean fever locus (MEFV) on chromosome 16p13.3. Genomics 1997; 42(1): 83–95.
12. Chae JJ, Wood G, Masters SL et al. The B30.2 domain of pyrin, the familial Mediterranean fever protein, interacts directly with caspase-1 to modulate IL-1beta production. Proc Natl Acad Sci USA 2006; 103(26): 9982–9987.
13. Aksentijevich I, Torosyan Y, Samuels J et al. Mutation and haplotype studies of familial Mediterranean fever reveal new ancestral relationships and evidence for a high carrier frequency with reduced penetrance in the Ashkenazi Jewish population. Am J Hum Genet 1999; 64(4): 949–962.
14. Sohar E, Gafni J, Pras M et al. Familial Mediterranean fever. A survey of 470 cases and review of the literature. Am J Med1967; 43(2): 227–253.
15. Padeh S, Livneh A, Pras E et al. Familial Mediterranean fever in children presenting with attacks of fever alone. J Rheumatol 2010; 37(4): 865–869.
16. Padeh S, Livneh A, Pras E et al. Familial Mediterranean Fever in the first two years of life: a unique phenotype of disease in evolution. J Pediatr 2010; 156(6): 985–989.
17. Samli H, Içduygu FM, Ozgöz A et al. Surgery for acute abdomen and MEFV mutations in patients with FMF. Acta Reumatol Port 2009; 34(3): 520–524.
18. Brik R, Shinawi M, Kasinetz L et al. The musculoskeletal manifestations of familial Mediterranean fever in children genetically diagnosed with the disease. Arthritis Rheum 2001; 44(6): 1416–1419.
19. Heller H, Gafni J, Michaeli D et al. The arthritis of familial Mediterranean fever (FMF). Arthritis Rheum 1966; 9(1): 1–17.
20. Kees S, Langevitz P, Zemer D et al. Attacks of pericarditis as a manifestation of familial Mediterranean fever (FMF). QJM 1997; 90(10): 643–647.
21. Majeed HA, Quabazard Z, Hijazi Z et al. The cutaneous manifestations in children with familial Mediterranean fever (recurrent hereditary polyserositis). A six-year study. Q J Med 1990; 75(278): 607–616.
22. Senel K, Melikoglu MA, Baykal T et al. Protracted febrile myalgia syndrome in familial Mediterranean fever. Mod Rheumatol 2010; 20(4): 410–412.
23. Lachmann HJ, Sengül B, Yavuzşen TU et al. Clinical and subclinical inflammation in patients with familial Mediterranean fever and in heterozygous carriers of MEFV mutations. Rheumatology (Oxford) 2006; 45(6): 746–750.
24. Akar S, Soyturk M, Onen F et al. The relations between attacks and menstrual periods and pregnancies of familial Mediterranean fever patients. Rheumatol Int 2006; 26(7): 676–679.
25. Lidar M, Yaqubov M, Zaks N et al. The prodrome: a prominent yet overlooked pre-attack manifestation of familial Mediterranean fever. J Rheumatol 2006; 33(6): 1089–1092.
26. Gafni J, Ravid M, Sohar E. The role of amyloidosis in familial mediterranean fever. A population study. Isr J Med Sci 1968; 4(5): 995–999.
27. Livneh A, Langevitz P, Zemer D et al. The changing face of familial Mediterranean fever. Semin Arthritis Rheum 1996; 26(3): 612–627.
28. Gershoni-Baruch R, Brik R, Lidar M et al. Male sex coupled with articular manifestations cause a 4-fold increase in susceptibility to amyloidosis in patients with familial Mediterranean fever homozygous for the M694V-MEFV mutation. J Rheumatol 2003; 30(2): 308–312.
29. Cazeneuve C, Sarkisian T, Pêcheux C et al. MEFV-Gene analysis in Armenian patients with Familial Mediterranean fever: diagnostic value and unfavorable renal prognosis of the M694V homozygous genotype-genetic and therapeutic implications. Am J Hum Genet 1999; 65(1): 88–97.
30. Cazeneuve C, Ajrapetyan H, Papin S et al. Identification of MEFV-independent modifying genetic factors for familial Mediterranean fever. Am J Hum Genet 2000; 67(5): 1136–1143.
31. Ozçakar ZB, Yalçınkaya F. Vascular comorbidities in familial Mediterranean fever. Rheumatol Int 2011; 31(10): 1275–1281.
32. Rozenbaum M, Rosner I. Severe outcome of juvenile idiopathic arthritis (JIA) associated with familial Mediterranean fever (FMF). Clin Exp Rheumatol 2004; 22(4 Suppl 34): 75–78.
33. Fidder HH, Chowers Y, Lidar M et al. Crohn disease in patients with familial Mediterranean fever. Medicine (Baltimore) 2002; 81(6): 411–416.
34. Livneh A, Langevitz P, Zemer D et al. Criteria for the diagnosis of familial Mediterranean fever. Arthritis Rheum 1997; 40(10): 1879–1885.
35. Yalçinkaya F, Ozen S, Ozçakar ZB et al. A new set of criteria for the diagnosis of familial Mediterranean fever in childhood. Rheumatology (Oxford) 2009; 48(4): 395–398.
36. Kondi A, Hentgen V, Piram M et al. Validation of the new paediatric criteria for the diagnosis of familial Mediterranean fever: data from a mixed population of 100 children from the French reference centre for auto-inflammatory disorders. Rheumatology (Oxford) 2010; 49(11): 2200–2203.
37. Touitou I. The spectrum of Familial Mediterranean Fever (FMF) mutations. Eur J Hum Genet 2001; 9(7): 473–483.
38. Marek-Yagel D, Berkun Y, Padeh S et al. Clinical disease among patients heterozygous for familial Mediterranean fever. Arthritis Rheum 2009; 60(6): 1862–1866.
39. Caglayan AO, DemiryilmazF, Ozyazgan I et al. MEFV gene compound heterozygous mutations in familial Mediterranean fever phenotype: a retrospective clinical and molecular study. Nephrol Dial Transplant 2010; 25(8): 2520–2523.
40. Ozen S, Aktay N, Lainka E et al. Disease severity in children and adolescents with familial Mediterranean fever: a comparative study to explore environmental effects on a monogenic disease. Ann Rheum Dis 2009; 68(2): 246–248.
41. Ter Haar N, Lachmann H, Ozen S et al. Treatment of autoinflammatory diseases: results from the Eurofever Registry and a literature review. Ann Rheum Dis 2013; 72(5): 678–685.
42. Goldfinger SE. Colchicine for familial Mediterranean fever. N Engl J Med 1972; 287(25): 1302.
43. Zemer D, Pras M, Sohar E et al. Colchicine in the prevention and treatment of the amyloidosis of familial Mediterranean fever. N Engl J Med 1986; 314(16): 1001–1005.
44. Ozçakar ZB, Yalçinkaya F, Yüksel S et al. Possible effect of subclinical inflammation on daily life in familial Mediterranean fever. Clin Rheumatol 2006; 25(2): 149–152.
45. Tatsuta E, Sipe JD, Shirahama T et al. Colchicine inhibition of serum amyloid protein SAA and SAP synthesis in primary mouse liver cell cultures. Arthritis Rheum 1984; 27(3): 349–352.
46. Livneh A, Zemer D, Langevitz P et al. Colchicine treatment of AA amyloidosis of familial Mediterranean fever. An analysis of factors affecting outcome. Arthritis Rheum 1994; 37(12): 1804–1811.
47. Ben-Chetrit E, Scherrmann JM, Levy M. Colchicine in breast milk of patients with familial Mediterranean fever. Arthritis Rheum 1996; 39(7): 1213–1217.
48. Savgan-Gürol E, Kasapçopur O, Hatemi S et al. Growth and IGF-1 levels of children with familial Mediterranean fever on colchicine treatment. Clin Exp Rheumatol 2001; 19: (5 Suppl 24): 72–75.
49. Ozçakar ZB, Kadioğlu G, Siklar Z et al. The effect of colchicine on physical growth in children with familial mediterranean fever. Eur J Pediatr 2010; 169(7): 825–828.
50. Rollot F, Pajot O, Chauvelot-Moachon L et al. Acute colchicine intoxication during clarithromycin administration. Ann Pharmacother 2004; 38(12): 2074–2077.
51. Meinzer U, Quartier P, Alexandra JF et al. Interleukin-1 targeting drugs in familial Mediterranean fever: a case series and a review of the literature. Semin Arthritis Rheum 2011; 41(2): 265–271.
52. Moser C, Pohl G, Haslinger I et al. Successful treatment of familial Mediterranean fever with Anakinra and outcome after renal transplantation. Nephrol Dial Transplant 2009; 24(2): 676–678.
53. Vargová, V, Macejová Ž. Naše skúsenosti s liečbou anakinrou u pacientky s CINCA/NOMID syndrómom. Rheumatologia 2011; 25(3): 121–125.
54. Kovács L, Hlavatá A, Paulovičová E et al. Syndrómy periodických horúčok – PFAPA syndróm. Pediatr prax 2009; 10(3): 141–144.
55. Kovács L, Hlavatá A, Baldovič M et al. Elevated immunoglobulin D levels in children with PFAPA syndrome. Neuro Endocrinol Lett 2010; 31(6): 743–746.
56. Król P, Katra R, Doležalová P. Syndromy periodických horeček. Čes-slov Pediat 2009; 64(10): 480–487.
57. Szturz P, Adam Z, Sedivá A et al. Schnitzler syndrome: diagnostics and treatment. Klin Onkol 2011; 24(4): 271–277.
58. Kallinich T, Haffner D, Niehues T et al. Colchicine use in children and adolescents with familial Mediterranean fever: literature review and consensus statement. Pediatrics 2007; 119(2): 474–483.
Labels
Diabetology Endocrinology Internal medicineArticle was published in
Internal Medicine

2014 Issue 1
Most read in this issue
- Effect of alcohol consumption on cardiac electrophysiology
- Familiar Mediterranean fever in Czech Republic
- Familial Mediterranean Fever – clinical picture, diagnosis and treatment
- International guidelines for management of severe sepsis and septic shock 2012 – comment