
Faktory vedoucí k poškození a destrukci B-buněk Langerhansových ostrůvků pankreatu
Factors causing damage and destruction of beta-cells of the islets of Langerhans in the pancreas
Insulin secretion in patients with manifested diabetes mellitus tends to disappear months to decades after the diagnosis, which is a clear sign of a gradual loss of pancreatic islet beta-cells. In our sample of 30 type 2 diabetic patients, whose disease manifested between 30 and 45 years of age, about a half have retained or even increased insulin secretion 30 years later, while the other half exhibit a much diminished or lost insulin secretion. Factors that can damage or destroy beta-cells can be divided into the following groups: Metabolic factors: hyperglycemia and glucotoxicity, lipotoxicity, hypoxia, reactive oxygen species; Pharmacological factors: antimicrobial medication pentamidine, SSRI antidepressants; Factors related to impaired insulin secretion: MODY type diabetes; Environmental toxic factors: rat poison Vacor, streptozotocin, polychlorinated and polybrominated hydrocarbons; Disorders of the exocrine pancreas: tumor infiltration, fibrous infiltration, chronic pancreatitis, cystic fibrosis; Infections, inflammation, autoimmunity, viral factors: Coxsackie viruses, H1N1 influenza, enteroviruses. We are currently working on finding other factors leading to beta-cell damage, studying their effect on apoptosis and necrosis and looking for possible protective factors to prevent this damage. We our increasing knowledge about the mechanisms of beta-cell damage and destruction we come ever closer to suggest measures for their prevention. In this review we offer a brief and simplified summary of some of the findings related to this area.
Key words:
pancreatic islet beta-cells of Langerhans – factors damaging or destroying beta-cells – insulin secretion
Authors:
Michal Anděl; Vlasta Němcová; Nela Pavlíková; Jana Urbanová; Marie Čecháková; Andrea Havlová; Radka Straková; Livia Večeřová; Václav Mandys; Jan Kovář; Petr Heneberg; Jan Trnka; Jan Polák
Authors‘ workplace:
Centrum výzkumu diabetu, metabolizmu a výživy 3. LF UK a FN Královské Vinohrady Praha, přednosta prof. MUDr. Michal Anděl, CSc.
Published in:
Vnitř Lék 2014; 60(9): 684-690
Category:
Overview
U pacientů, u kterých se manifestoval diabetes mellitus, dochází po měsících až desetiletích trvání choroby k vyhasnutí sekrece inzulinu, což je téměř jistým znamením úbytku B-buněk Langerhansových ostrůvků. U souboru 30 pacientů, u kterých se choroba manifestovala mezi 30–45 roky a byli diagnostikováni jako diabetici 2. typu, má po 30 letech trvání choroby polovina zachovanou nebo vyšší sekreci inzulinu, druhá polovina pak sekreci výrazně sníženou nebo vyhaslou. Faktory, které postihují B-buňky a vedou k jejich destrukci, můžeme shrnout do následujících skupin: 1. Faktory chemické: faktory metabolické: hyperglykemie a glukotoxicita, lipotoxicita, hypoxie, volné kyslíkové radikály, faktory farmakologické: anitimikrobiální prostředek pentamidin, antidepresiva typu SSRI, faktory spojené s poruchou sekrece inzulinu: MODY typy diabetu, toxické látky ze zevního prostředí: jed na krysy Vacor, streptozotocin, polychlorované či polybromované uhlovodíky 2. Onemocnění zevně sekretorické části pankreatu: nádorová infiltrace, vazivová infiltrace, chronická pankreatitida 3. Infekce, zánět a autoimunita: faktory virové: Coxsackie viry, virus chřipky H1N1, enteroviry, záněty: autoimunní faktory, představující patogenetický faktor diabetu 1. typu. V současné době pracujeme jak na další specifikaci dalších faktorů vedoucích k poškození B-buněk, tak na studiu poznání jejich účinku na buněčnou apoptózu respektive nekrózu, a konečně na definici ochranných faktorů, které by účinky působení těchto faktorů snížily. S nárůstem vědomostí o mechanizmech poškození a destrukce B-buněk se rýsují návrhy některých opatření, která by je mohla chránit. V našem přehledu podáváme zestručnělý a s ohledem na rozsah článku také notně zjednodušený přehled některých znalostí, které se poškození a destrukce B-buněk týkají.
Klíčová slova:
B-buňky Langerhansových ostrůvků pankreatu – faktory vedoucí k destrukcí B-buněk – sekrece inzulinu
Úvod
U dospělého člověka se v pankreatu vyskytuje 800 000–2 000 000 ostrůvků, které jsou lokalizovány především v jeho těle s maximem na přechodu hlavy a těla. Celková hmotnost ostrůvků tvoří 1–2 % celkové váhy pankreatu, která činí 60–90 g, pohybuje se tedy celkem okolo 1 g. Langerhansův ostrůvek je velký 0,1–0,5 mm. U člověka jsou ve fetálním období a v raném dětství B-buňky uloženy v ostrůvku centrálně. Okolo tohoto centra se na periferii ostrůvků nalézá neúplná vrstva z A, D a PP buněk. Takovéto ostrůvky byly popsány i u potkana a myši a dalších hlodavců [53]. U dospělého člověka jsou však nacházeny jednotlivé buněčné typy více mozaikovitě uspořádány. Stejné jsou nálezy např. u plášťového paviána, dospělého morčete, psa, ovce či prasete. U člověka tvoří B-buňky 50–60 % všech buněčných elementů ostrůvků, u hlodavců je to více, většinou 70–80 %. Hlodavci se od člověka zásadně liší v regeneraci B-buněk: zatímco myš denně obnoví asi 3 % všech B-buněk, tedy za měsíc se obnoví všechny, člověk má v dospělosti zcela nepatrnou míru jejich obnovy. Také proto jsou výsledky experimentů prováděných jak na izolovaných buňkách pocházejících z hlodavců, tak na hlodavcích přenositelné na člověka jen s velkou dávkou opatrnosti. Bližší nálezy o vývoji Langerhansových ostrůvků i z jejich komparativní histologie je možné nalézt v unikátní monografii Milana Tittelbacha (2001) [53].
Postupné vyhasínání sekrece inzulinu z B-buněk Langerhansových ostrůvků je u velkého procenta diabetiků 2. typu poměrně dobře známé. Dysfunkce B-buněk je prakticky vždy přítomná od počátečních stadií diabetu. Oproti tomu inzulinová rezistence je stabilní, od období zhoršené glukozové tolerance až po plně rozvinutý diabetes [32]. Pokles sekrece začal zřejmě již před diagnózou diabetu v období porušené glukozové tolerance a souvisí s faktem, že u části obézních pacientů s hyperinzulinemií a inzulinovou rezistencí dochází k poklesu sekrece inzulinu, která již není dostatečná k tomu, aby vyvážila inzulinovou rezistenci [15]. Hladina plazmatického inzulinu je však u naprosté většiny pacientů s čerstvě manifestovaným diabetem 2. typu zvýšena, může však být v době manifestace normální či již snížená [15,51].
V našem sledování 30 pacientů s manifestací choroby mezi 30. a 45. rokem a s trváním diabetu delším než 30 let jsme u dvou třetin nemocných nalezli hladinu inzulinu nalačno nižší než 300 nmol/l. Toto sledování je v souladu s etablovanými názory v této oblasti. Stále zásadnější se jeví otázka, jakými mechanizmy k poškození B-buněk Langerhansových ostrůvků dochází. V následujícím článku podáváme přehled faktorů, které byly jak v experimentu na izolovaných B-buňkách, tak experimentech zvířecích či u lidí nalezeny jako reálně či potenciálně pro B-buňky toxické. Přitom je zřejmé, že mnohé aspekty poškození B-buněk se týkají jak diabetu 1., tak 2. typu.
Přehled těchto faktorů je uvádí tab.
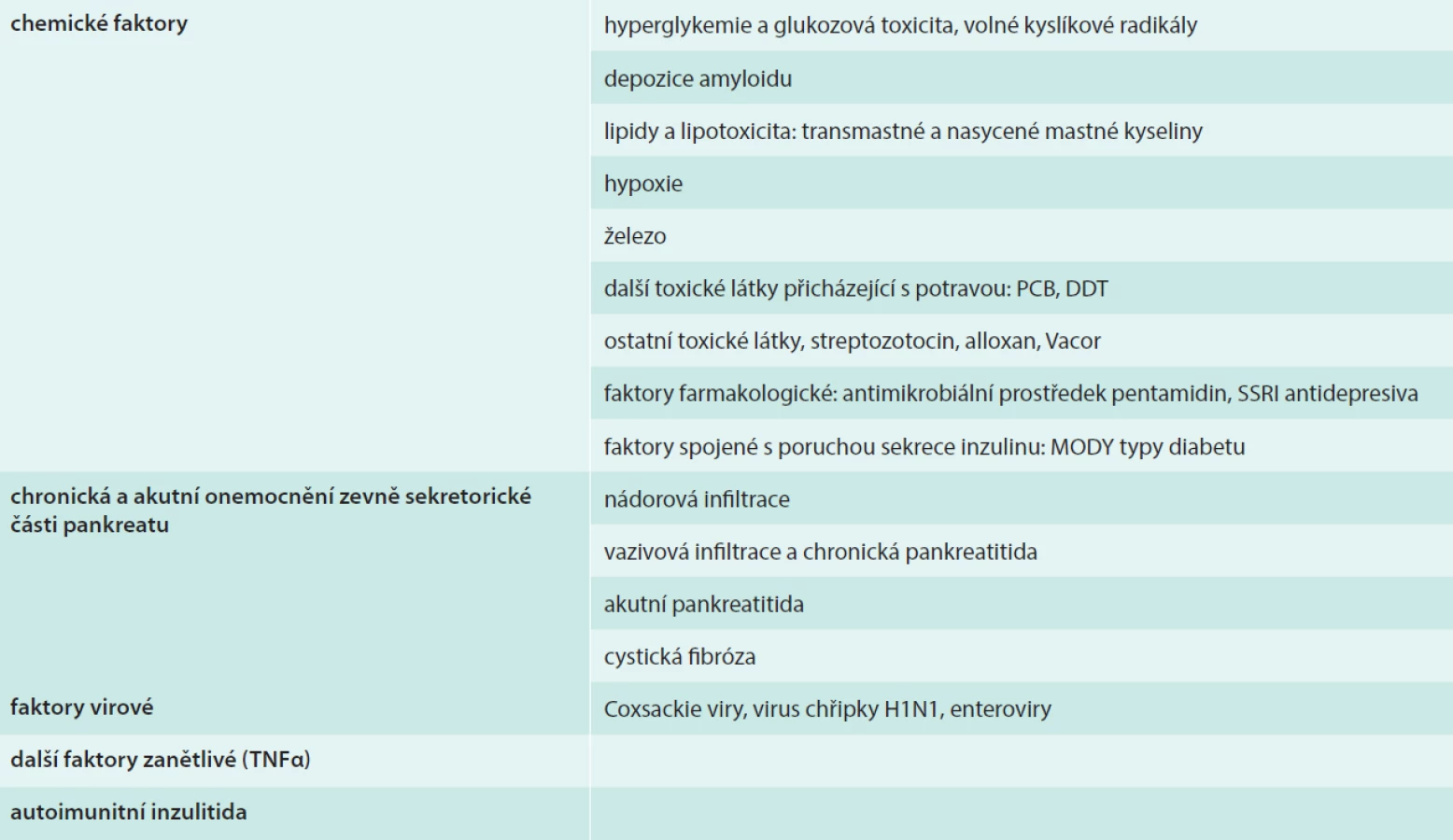
Faktory chemické
Faktory metabolické
Metabolické faktory jsou podrobně studovány celou řadu let a pojem glukózová toxicita pro B-buňky je dobře známý [47]. Mechanizmy, kterými působí hyperglykemie a hypoglykemie, jsou extenzivně zkoumány, přesto však obraz tohoto mechanizmu poškození není úplný.
Apoptózu B-buněk indukuje jak hyperglykemie, tak také hypoglykemie. Pro optimální funkci B-buněk je nezbytná pravidelná stimulace fyziologickými koncentracemi glukózy.
Mechanizmus poškození B-buněk hyperglykemií není zcela objasněn. Výraznou roli hraje zvýšený oxidativní stres, v jeho důsledku pak poškození mitochondriální DNA, aktivace stresové odpovědi endoplazmatického retikula, dlouhodobě pak zvýšená intracelulární tvorba pokročilých produktů glykace (AGEs). Hyperglykemie vede i ke zvýšení exprese IL1β, a dále ke zvýšení v důsledku dysfunkce endoplazmatického retikula k zvýšení tvorby IAPP oligomerů a k depozici amyloidu v B-buňkách.
Exogenní AGEs konzumujeme s potravou (maso, mléko, káva, sýry), zejména pokud je jejich příprava za vysokých teplot, vznikají i během dlouhodobého skladování potravin a k jejich vzniku může přispívat i obsah aditiv. V myším a krysím modelu vede vyšší příjem AGEs ke zvýšené tvorbě superoxidu v mitochondriích a ke zvýšené apoptóze B-buněk.
Depozice amyloidu
Depozice amyloidu je nacházena v B-buňkách diabetiků 2. typu poměrně často. Ve sledování Kamata et al [30] bylo u 118 pitvaných japonských diabetiků 26 případů s depozicí amyloidu. Tíže amyloidové depozice korelovala s redukovaným objemem B-buněk i A-buněk, se zvýšeným BMI, ale ne s věkem nemocných, s hladinou HbA1c ani s trváním diabetu. Ostrůvky bohaté na amyloid byly zvýšeně infiltrované makrofágy. V B-buňkách těchto ostrůvků byly známky poškození DNA ve vztahu k oxidativnímu poškození – exprese yH2AX a potlačení exprese (pro)inzulinové mRNA.
Lipidy a lipotoxicita
Existují jasné důkazy o souvislosti obezity, stavu charakteristickém mimo jiné zvýšenou hladinou mastných kyselin v krvi, s incidencí inzulinové rezistence a diabetu 2. typu [40].
Krátkodobé působení mastných kyselin na B-buňky vede ke zvýšení glukózou indukované sekrece inzulinu, avšak jejich dlouhodobé působení (hodiny a více) snižuje viabilitu B-buněk a vede k jejich apoptóze (lipotoxicita), jak bylo prokázáno v in vitro experimentech na lidských i zvířecích B-buňkách i in vivo ve zvířecích modelech [14,16,33,49]. Z hlediska vlivu na viabilitu B-buněk jsou výrazně toxičtější nasycené mastné kyseliny (např. kyselina palmitová, kyselina stearová), zatímco nenasycené mastné kyseliny (např. kyselina olejová, kyselina palmitolejová) jsou B-buňkami dobře tolerovány a jsou schopné dokonce apoptózu indukovanou nasycenými mastnými kyselinami inhibovat [36], což jsme prokázali i v naší laboratoři na linii lidských pankreatických B-buněk NES2Y [39]. Chronické působení transnenasycených mastných kyselin (kyselina elaidová) snižuje proliferaci B-buněk a též jejich antiapoptotický účinek je nižší v porovnání s jejich cis protějšky [10,16].
Palmitát je reprezentant mastných kyselin s dlouhým řetězcem, které poškozují B-buňky. V buněčném modelu odvozeném od krysího inzulinomu (INS-1D buňky) 24hodinová expozice koncentraci 0,6 mmol/l palmitátu vedla ke zvýšení aktivity oxidázy nikotinamidadenindinukleotidfosfátu (NOX) a hladin NOX2, což je patologický zdroj volných kyslíkových radikálů (ROS) v B-buňkách. Expozice palmitátu zvýšila v buněčné membráně B-buňky hladinu proteinu p47phox, což je regulační protein NOX2. Suprese proteinu p47phox vedla k poklesu palmitátem zvýšené produkce ROS i ke zlepšení palmitátem zhoršené glukózou stimulované sekrece inzulinu [46].
Mechanizmy uplatňující se v regulaci apoptózy B-buněk nasycenými a nenasycenými mastnými kyselinami nejsou uspokojivě objasněny. Jasná je úloha kaspáz (kaspáza 3, kaspáza 8, kaspáza 9, kaspáza 2) v exekuční fázi apoptózy [24,38], avšak děje předcházející aktivaci těchto klíčových enzymů apoptózy jsou méně jasné. V současnosti nejvíce skloňovaný mechanizmus proapoptického účinku nasycených mastných kyselin se týká indukce tzv. stresu endoplazmatického retikula (ER) [3], procesu, při němž je po narušení funkce ER nejprve aktivována složitá kaskáda signálních drah směřujících k obnovení funkce této pro syntézu sekretovaných proteinů včetně inzulinu esenciální organely, avšak v případě neúspěchu této odpovědi dochází k indukci apoptózy. V indukci apoptózy působením nasycených mastných kyselin se zcela jistě uplatňují i další mechanizmy, např. změna exprese propoptotických a antiapoptotických proteinů rodiny Bcl2 [21], indukce tvorby ceramidu [18] a ROS [42] a zcela jistě dochází i k ovlivnění procesu autofagie. Výše popsané mechanizmy apoptózy B-buněk indukované nasycenými mastnými kyselinami jsou inhibovány působením nenasycených mastných kyselin, a to i v případě, že jsou přítomny v několikanásobně nižší koncentraci nebo přidány až po několika hodinách působení nasycených mastných kyselin [36,37,39,57].
Hypoxie
B-buňky jsou výrazně citlivé na hypoxii. Z tohoto hlediska jsou logické pokusy studovat v experimentu vliv hypoxie na metabolizmus sacharidů a na sekreci inzulinu a citlivost k inzulinu. V experimentu bylo nalezeno, že myš exponovaná intermitentní hypoxii má jak poruchu sekrece inzulinu, tak také zhoršení citlivosti k inzulinu. Pokud dojde k normalizaci přívodu kyslíku, dojde také k částečné úpravě těchto poruch [43]. Spánková apnoe by u obézních pak mohla být jedním z faktorů, které spojují obezitu, inzulinovou rezistenci a diabetes mellitus 2. typu [35,41]. Je zajímavé, že zatímco hypoxie Langerhansových ostrůvků vede v B-buňkách k významnému nárůstu apoptózy, v A-buňkách ostrůvků apoptóza výrazně nenarůstá [4].
Železo
Při dlouhodobé expozici železu, ať již z genetických důvodů při hemochromatóze, nebo ze získaných důvodů při opakovaných transfuzích, při hepatitis C či porphyria cutanea tarda dochází k depozici železa v pankreatu, k jeho sekundární fibróze a dle literárních údajů v desítkách procent případů k diabetu [19]. B-buňky jsou extrémně citlivé k oxidačnímu poškození z volných kyslíkových radikálů derivovaných právě z účinku nadbytečného železa [50].
Další toxické látky přicházející s potravou
Perzistentní organické polutanty (tzv. POPs, mezi které patří např. polychlorované bifenyly, DDT a další) se v dnešní době běžně nacházejí v potravě, vodě i vzduchu. Poté, co se dostanou do organizmu, se primárně ukládají v tukové tkáni, odkud jsou permanentně ve velmi malém množství uvolňovány do krevního oběhu. První data, která naznačují možné spojení mezi znečištěním životního prostředí a výskytem diabetu, dodaly epidemiologické studie. Byly měřeny koncentrace různých POPs v krvi nebo v tukové tkáni a byla nalezena pozitivní korelace s výskytem diabetes mellitus [6–8,12,13,48].
Další výzkum se zaměřil na in vivo studie. Jako zdroj perzistentních organických polutantů je běžně používané maso ryb s vysokým obsahem tuku, např. maso lososa (konzumace lososa představuje běžný zdroj POPs i pro člověka) [8]. Příjem potravy s vysokým obsahem tuku kontaminované různými POPs vedl (ve srovnání se stejnou dietou, ale bez POPs) k signifikantním změnám v metabolizmu glukózy u myší a krys. Tato dieta vedla např. nárůstu obezity a inzulinové rezistence [27,45], hyperinzulinemii [20] nebo k hyperglykemii na lačno [23] v závislosti na konkrétních detailech expozice (druh a koncentrace POPs, délka expozice). Nicméně jsou i studie, které tvrdí, že vliv POPs na glukózovou toleranci nemusí být nutně negativní [26].
Prakticky neexistují informace o tom, na kterou oblast B-buňky tyto látky konkrétně působí. Důvodem je velmi specifická expozice. Lidé přijímají POPs do organizmu celý život, nepravidelně a nerovnoměrně (záleží na znečištění v místě pobytu) ve velmi malých dávkách, které se ovšem v organizmu kumulují. Je v podstatě nemožné vytvořit model, který by dostatečně napodobil tento proces. Přesto jsme v naší nedávné studii zkusili exponovat lidské pankreatické B-buňky NES2Y subletální koncentrací DDT po dobu 1 měsíce. Následně jsme analyzovali změny v proteomu buněk pomocí 2-D elektroforézy. Našli jsme změny v expresi některých cytoskeletárních proteinů a enzymu podílejícího se na glykolýze. Tyto výsledky korelují s jinými studiemi, které exponovaly B-buňky jiným stresovým vlivům, např. kyselině palmitové [22] nebo vysoké koncentraci glukózy [17]. Tyto proteiny se zdají být náchylné ke změnám v důsledku expozice negativním faktorům a mohou reprezentovat „zranitelná“ místa B-buňky.
Ostatní toxické látky
Streptozotocin a alloxan se používají jako induktory diabetu 2. typu v experimentálním krysím a myším modelu. Obecně je více preferován streptozotocin, protože vede k trvalejším hyperglykemiím a může tak být snadněji použit pro studium vzniku komplikací [25].
Vacor je jed na krysy na bazi N-3-pyridylmetyl-N-p - nitrofenylurey (PNU, pyriminil), který způsobil ať již při náhodné nebo suicidální expozici několik desítek případů těžkého inzulindependentního diabetu s ketoacidózou a pozdějším rozvojem autonomní a periferní neuropatie. PNU je látkou přímo toxickou pro B-buňky, v experimentu se tomuto účinku dá zabránit, podobně jako u streptozotocinového diabetu, nikotinamidem. V dávkách klinických však léčba nikotinamidem při otravě vacorem nerevertovala diabetes [29,31,44].
Faktory farmakologické
Farmakologické faktory souvisejí s celou řadou více nebo méně známých účinků farmak na B-buňky. Při tom je třeba odlišit dobře známé účinky farmak na inzulinovou senzitivitu respektive rezistenci. Je dobře známé, že například B-adrenolytika zhoršuji citlivost k inzulinu, zatímco ACE inhibitory působí opačně: citlivost buněk k inzulinu zlepšují. Těmito aspekty se však v našem sdělení nezabýváme. Popisujeme především přímý toxický účinek farmak na B-buňky.
Ten je dobře známý u antipararazitika pentamidinu, léku, který se používá u pneumocystové pneumonie, pokud selhala terapie biseptolem, zejména u nemocných s HIV infekcí. Jinou jeho indikací je leishmanióza. U nemocných s pneumocystovou pneumonií léčených pentamidinem se v týdnech až měsících po zahájení léčby v 70–100 % rozvine ireverzibilní inzulindependentní diabetes [1].
Fluoxetin (Prozac), lék užívaný v léčbě deprese a v dalších indikacích v psychiatrii, je selektivním inhibitorem reuptake serotoninu (SSRI). SSRI je v současnosti nejčastěji předpisovanou skupinou antidepresivních léků. Na INS-1E buňkách krysí buněčné linie bylo zjištěno, že současně s podáním fluoxetinu do media byla v buňkách detekovatelná zvýšená produkce reaktivních kyslíkových molekul (ROS) a současně hypotetizováno, že oxidativní poškození může přispět ke snížení aktivity enzymů mitochondriálního elektronového transportního řetězu (ETC). Fluoxetin vede k poklesu glukózou stimulované sekrece inzulinu (GSIS). Fluoxetinem indikovaný deficit ve funkci B-buňky byl preventabilní přidáním kyseliny listové [11]. Přestože bylo sledování provedeno v pokusech na izolovaných buňkách, mohou osvětlit vztah mezi SSRI a T2 diabetem a naznačují, že by toxický účinek SSRI na B-buňky mohl být v klinické praxi preventabilní poměrně jednoduše.
Faktory spojené s MODY typem diabetu
Ve skupině 28 pacientů s nejběžnějšími typy MODY diabetu (HNF1A-MODY, HNF4A-MODY a GCK-MODY) jsme nalezli pozitivní autoprotilátky proti B-buňkám (GADA a IA2A). Tento nález byl častější u pacientů s vyšším HbA1c a korespondoval s pozdější manifestací diabetu, nikoliv však se signifikantním poklesem B-celulární funkce [55,56]. V klinické praxi je běžné, že pacienti s MODY typy diabetu spjatých s mutací v genech pro transkripční faktory HNF jsou po mnoho let dobře léčitelní deriváty sulfonylurey, po desetiletích trvání choroby však (v souvislosti s klesající inzulinovou sekrecí) velmi často vyžadují léčbu inzulinem. Mechanizmy, kterými u nemocných s MODY diabetem dochází k poškození B-buněk, nejsou dobře známé. U HNF1A-MODY či HNF4A-MODY je pravděpodobně příčinou úbytku B-buněk přímo mutace v HNF4A nebo HNF1A genu, jenž podmiňuje defektní buněčný obrat B-buněk. Patofyziologickým podkladem je změna v expresi genů ovlivňující buněčný růst a proliferaci, které vedou k poklesu celkového obratu pankreatických B-buněk, tedy nárůstu apoptózy a snížení schopnosti proliferace, jak bylo prokázáno na zvířecích modelech [54,58]. Je však možné, že se u těchto pacientů na poškození B-buněk podílí více faktorů, včetně hyperglykemie.
Chronická onemocnění zevně sekretorické části pankreatu
Chronická pankreatitis
Chronická pankreatitida je faktorem, který je často spojen se sekundárním diabetem. Všeobecně se má za to, že jde především o inzulinopenický diabetes vznikající v důsledku vazivové přestavby pankreatu a současného zničení ostrůvků. Zda se jedná jen o důsledek mechanického postižení, nebo zda lokální zánět hraje také nějakou roli, je předmětem našich úvah.
Akutní pankreatitida
Akutní pankreatitida při prudkém průběhu a destrukci exokrinní a endokrinní tkáně může vést k inzulinopenickému diabetu. Mechanizmy zániku B-buněk souvisí zřejmě s akutní nekrózou pankreatu [19].
O tom, že další pankreatitidy mohou být příčinou diabetu, jsou zprávy spíše sporadické. Sami jsme popsali granulomatózní pankreatitidu u pacientky, která v jiné nemocnici zemřela na těžký akutní zánět s manifestací akutního diabetického syndromu [34].
Karcinom pankreatu
Mechanizmus vzniku diabetu u nemocných s karcinomem pankreatu není jasný. Zdá se, že původní představa, že by mohlo jít o mechanickou destrukci ostrůvků či o ischemii ostrůvků, není jednoznačně potvrzena. U pacientů s karcinomem pankreatu se sekundárním diabetem byla ve tkáni pankreatu nalezena vyšší exprese MIF (macrophage migration inhibitory factor) než u vzorků od nemocných s chronickou pankreatitidou či v normálních vzorcích pankreatické tkáně. MIF zhoršuje funkci B-buněk tím, že snižuje proud Ca2+, snižuje expresi proteinu α1 podjednotky L typu kalciového kanálu. Průměrné hodnoty MIC byly významně větší u pacientů s nově manifestovaným diabetem a karcinomem pankreatu než u nemocných s chronickou pankreatitis či u zdravých osob [52].
Cystická fibróza
V počátečních stadiích cystické fibrózy je funkce B-buněk dobrá, postupně však dochází ke zhoršení sekrece inzulinu jako důsledek selhání B-buněk způsobený fibrózou, tukovou infiltrací a také ukládáním amyloidu [19].
Infekce, zánět, autoimunita
Autoimunní proces
Autoimunitní proces, který vede k diabetu 1. typu, je od Botazzova popisu autoimunitní inzulitidy rozsáhle studovaný více než 25 let a není cílem tohoto přehledu zacházet do jeho detailů. Proto se zmíníme jen o některých, dle nás zajímavých aspektech tohoto procesu publikovaných recentně. Proces je zprostředkovaný autoreaktivními T-lymfocyty, které útočí na B-buňky. T-lymfocyty uvolňují cytokiny, jakými jsou např. interleukin 1, které nejen že inhibují sekreci inzulinu, ale působí i destrukci B-buněk. V odpovědi na cytokinový efekt B-buňky exprimují inducibilní syntázu oxidu dusíku a produkuji reaktivní oxid dusíku. V důsledku toho dochází k inhibici mitochondriální oxidace glukózy, což vede ke zhoršení sekrece inzulinu. Současně je reaktivní oxid dusíku také zodpovědný za poškození DNA v B-buňkách. Zatímco na jedné straně oxid dusíku zprostředkovává buněčné poškození v důsledku účinku cytokinů, současně spouští i protektivní cesty, které vedou k zotavení B-buněk [5]. Oxid dusíku má tedy v oblasti B-buněk duální efekt.
Podle hypotézy Jaberi-Douraki et al [28] z roku 2014 autoimunitní útok způsobí stres endoplazmatického retikula tím, že zbývající buňky ve zvýšené míře syntetizují a secernují inzulin. Autoři expresivně pojmenovávají útok T-lymfocytů jako vraždu B-buněk a následný stres endoplazmatického retikula jako jejich sebevraždu.
Další zánětlivé faktory
Zánět tukové tkáně je poměrně recentně diskutovaným aspektem obezity. Při tom je zajímavé, že mezi desítkami hormonálních a humorálních látek, které adipocyty produkují, se vyskytuje jak proapoptiticy působící TNFα, který snižuje v ostrůvcích Bcl2 a indukuje iNOS, tak antiapoptotický IL6, který v B-buňkách snižuje produkci NO a v myším modelu zvyšuje viabilitu v přítomnosti cytotoxických cytokinů. V této souvislosti je zajímavé, že dva z nejvýznamnějších hormonů produkovaných adipocyty, leptin a adiponektin, mají antiapoptotický účinek. Leptin snižuje obsah triglyceridů, inhibuje aktivitu iNOS a v krysím modelu zvyšuje Bcl2. Adiponektin inhibuje v krysí B-buňce apoptózu indukovanou jak palmitátem, tak cytokiny.
V minulosti byly popisovány různé infekce ve vztahu k ateroskleróze nebo diabetu 2. typu, typicky byla rozsáhle např. diskutována chronická zubní infekce nebo infekce Helicobacter pylori. Zdá se však, že jakákoliv infekce cestou aktivace TNFα a syntézy CRP může vést ke spuštění dalších mechanizmů poškozujících B-buňky [2].
Závěr
V současné době jsme schopni detekovat více než dvě desítky faktorů, které potenciálně nebo reálně vedou k dysfunkci a posléze k poškození a buněčné smrti B-buněk Langerhansových ostrůvků. Stupeň tohoto poznání dává naději na možnosti mnohem širší a současně cílenější prevence diabetu, zejména diabetu 2. typu.
Práce vznikla díky programu výzkumu Univerzity Karlovy Prvouk Iniciální stadia obezity, diabetu, aterosklerózy a dalších metabolických, endokrinních a nutričních postižení organizmu a díky podpoře programu Univerzitní centrum energetického metabolizmu Univerzity Karlovy.
prof. MUDr. Michal Anděl, CSc.
michal.andel@lf3.cuni.cz
Centrum výzkumu diabetu, metabolizmu a výživy II. interní kliniky 3. LF UK a FN Královské Vinohrady, Praha
www.fnkv.cz
Doručeno do redakce 21. 7. 2014
Přijato po recenzi 24. 7. 2014
Sources
1. Anděl M, Kraml P, Šilhová E. Hypoglykémie a sekundární diabetes po pentamidinu. DMEV 2011; 14(2): 68–70.
2. Anděl M, Polák J, Kraml P et al. Chronický mírný zánět spojuje obezitu, metabolický syndrom, aterosklerózu a diabetes. Vnitř Lék 2009; 55(7): 659–665.
3. Biden TJ, Boslem E, Chu KY et al. Lipotoxic endoplasmic reticulum stress, B cell failure, and type 2 diabetes mellitus. Trends Endocrinol Metab 2014; 25(8): 389–398.
4. Bloch K, Vennäng J, Lazard D et al. Different susceptibility of rat pancreatic alpha and beta cells to hypoxia. Histochem Cell Biol 2012; 137(6): 801–810.
5. Broniowska KA, Oleson BJ, Corbet JA. B-cell responses to nitric oxide. Vitam Horm 2014; 95 : 299–322.
6. Codru N, Schymura MJ, Negoita S et al. Diabetes and relation to serum levels of polychlorinated biphenyls and chlorinated pesticides an adult native Americans. Environ Health Perspect 2007; 115(10): 1442–1447.
7. Cox S, Niskar AS, Narayan KMV et al. Prevalence of self-reported diabetes and exposure to organochlorine pesticides among Mexican Americans: Hispanic Health and Nutrition Examination Survey, 1982–1984. Environ Health Perspect 2007; 115(12): 1747–1752.
8. Crinnion WJ. The Role of Persistent Organic Pollutants in the Worldwide Epidemic of Type 2 Diabetes Mellitus and the Possible Connection to Farmed Atlantic Salmon (Salmo salar). Altern Med Rev 2011; 16(4): 301–313.
9. Čecháková M, Kratochvíl A, Anděl M. MIkrovaskulární komplikace u pacientů s diabetes mellitus 2. typu manifestovaném mezi 30. a 45. rokem s trváním diabetu nad 30 let na základě hodnocení inzulinosekrece a kompenzace (poster 38). Abstrakta 50. Luhačovických diabetologických dnů. DMEV 2014; 17(Suppl): 54.
10. Dhayal S, Welters HJ, Morgan NG. Structural requirements for the cytoprotective actions of mono-unsaturated fatty acids in the pancreatic beta-cell line, BRIN-BD11. Br J Pharmacol 2008; 153(8): 1718–1727.
11. De Long NE, Hyslop JR, Raha S et al. Fluoxetine-induced pancreatic beta-cell dysfunction: New insight into benefits of folic acid in the treatment of depression. J Affect Disord 2014; 166 : 6–13.
12. Everett CJ, Frithsen I, Player M. Relationship of polychlorinated biphenyls with type 2 diabetes and hypertension. J Environ Monit 2011; 13(2): 241–251.
13. Everett CJ, Matheson EM. Biomarkers of pesticide exposure and diabetes in the 1999–2004 National Health and Nutrition Examination Survey. Environ Int 2010; 36(4):398–401.
14. Eitel K, Staiger H, Brendel MD et al. Different role of saturated and unsaturated fatty acids in beta-cell apoptosis. Biochem Biophys Res Commun 2002; 299(5): 853–856.
15. Fonsenca V, John-Kalarickal J. Type 2 Diabetes Mellitus: Epidemiology, Genetics, Pathogenesis and clinical Manifestations. In: Poretsky L (ed). Principles of Diabetes Mellitus. 2nd ed. Springer: New York, Dordrecht, Heidelberg, London 2010 : 203–220. ISBN 978–0-387–09840–1.
16. Fürstova V, Kopska T, James RF et al. Comparison of the effect of individual saturated and unsaturated fatty acids on cell growth and death induction in the human pancreatic beta-cell line NES2Y. Life Sci 2008; 82(13–14): 684–691.
17. Fernandez C, Fransson U, Hallgard E et al. Metabolic and proteomic analysis of a clonal insulin-producing beta-cell line (INS-1 832/13). J Proteome Res 2008; 7(1): 400–411.
18. Galadari S, Rahman A, Pallichankandy S et al. Role of ceramide in diabetes mellitus: evidence and mechanisms. Lipids Health Dis 2013; 12 : 12–98. Dostupné z DOI: <http://doi: 10.1186/1476–511X-12–98>.
19. Garger YB, Joshi PM, Pareek AS et al. Secondary Causes of Diabetes Mellitus. In: Poretsky L (ed.): Principles of Diabetes Mellitus. (2nd ed). Springer: New York, Dordrecht, Heidelberg, London 2010 : 245–258. ISBN 978–0-387–09840–1.
20. Gray SL, Shaw AC, Gagne AX et al. Chronic exposure to PCBs (Aroclor 1254) exacerbates obesity-induced insulin resistance and hyperinsulinemia in mice. J Toxicol Environ Health A 2013; 76(12): 701–715.
21. Gurzov EN, Eizirik DL. Bcl-2 proteins in diabetes: mitochondrial pathways of B-cell death and dysfunction. Trends Cell Biol 2011; 21(7): 424–431.
22. Hovsepyan M, Sargsyan E, Bergsten P. Palmitate-induced changes in protein expression of insulin secreting INS-1E cells. J Proteomics 2010; 73(6): 1148–1155.
23. Howell GE, Meek E, Kilic J et al. Exposure to p,p ‘-dichlorodiphenyldichloroethylene (DDE) induces fasting hyperglycemia without insulin resistance in male C57BL/6H mice. Toxicology 2014; 320 : 6–14.
24. Hui H, Dotta F, Di Mario U et al. Role of caspases in the regulation of apoptotic pancreatic islet beta-cells death. J Cell Physiol 2004; 200(2): 177–200.
25. Christopher RJ, Takeuchi K, Lee B. Rodent models of diabetes. In: Poretsky L (ed). Principles of Diabetes Mellitus. (2nd ed). Springer: New York, Dordrecht, Heidelberg, London 2010 : 165–178. ISBN 978–0-387–09840–1.
26. Ibrahim MM, Fjaere E, Lock EJ et al. Metabolic impacts of high dietary exposure to persistent organic pollutants in mice. Toxicol Lett 2012; 215(1): 8–15.
27. Ibrahim MM, Fjaere E, Lock EJ et al. Chronic Consumption of Farmed Salmon Containing Persistent Organic Pollutants Causes Insulin Resistance and Obesity in Mice. PloS One 2011; 6(9): e25170. Dostupné z DOI: <http://doi: 10.1371/journal.pone.0025170>.
28. Jaberi-Douraki M, Schnell S, Pietropaolo M et al. Unraveling the contribution of pancreatic betas-cell suicide in autoimmune type-1 diabetes. J Theor Biol 2014; 5193: S0022–00270. Dostupné z DOI: <http://10.1016/j.jtbi.2014.05.003>.
29. Johnson D, Kubic P, Levitt C. Accidental ingestion of Vacor rodenticide: the symptoms and sequelae in a 25-month-old child. Am J Dis Child 1980; 134(2): 161–164.
30. Kamata K, Mizukami H, Inaba W et al. Islet amyloid with macrophage migration correlates with augmented b-cell deficit in type 2 diabetic patients. Amyloid 2014; 35 : 1–11. Epub ahead of print.
31. Karam JH, Lewitt PA, Young CW et al. Insulinopenic diabetes after rodenticide (Vacor) ingestion: a unique model of acquired diabetes in man. Diabetes 1980; 29(12): 971–978.
32. Leahy JL. Beta-cell dysfunction In: Kahn CR, King GL, Moses A et al (eds). Joslin’s Diabetes Mellitus. Selected chapters from Fourteenth Edition. Lippincott Williams and Williams: Boston 2004 : 223–235. ISBN 9780781727969.
33. Maedler K, Spinas GA, Dyntar D et al. Distinct effects of saturated and monounsaturated fatty acids on beta-cell turnover and function. Diabetes 2001; 50(1): 69–76.
34. Mandys V, Kheck M, Anděl M. Granulomatous pancreatitis in a patient with acute manifested insulin-dependent diabetes mellitus. Case Rep Pathol 2014; 615426. Dostupné z DOI: <http://doi: 10.1155/2014/615426>.
35. Mesarwi O, Polak J, Jun J et al. Sleep disorders and the development of insulin resistance and obesity. Endocrinol Metab Clin North Am 2013; 42(3): 617–634.
36. Morgan NG, Dhayal S. Unsaturated fatty acids as cytoprotective agents in the pancreatic beta-cell. Prostaglandins Leukot Essent Fatty Acids 2010; 82(4–6): 231–236.
37. Němcová-Fürstová V, James RF, Kovář J. Inhibitory effect of unsaturated fatty acids on saturated fatty acid-induced apoptosis in human pancreatic B-cells: activation of caspases and ER stress induction. Cell Physiol Biochem 2011; 27(5): 525–538.
38. Nemcova-Furstova V, Balusikova K, Sramek J et al. Caspase-2 and JNK activated by saturated fatty acids are not involved in apoptosis induction but modulate ER stress in human pancreatic B-cells. Cell Physiol Biochem 2013; 31(2–3): 277–289.
39. Němcová-Fürstová V, James RF, Kovář J. Comparison of the effect of individual saturated and unsaturated fatty acids on cell growth and death induction in the human pancreatic beta-cell line NES2Y. Life Sci 2008 : 26; 82(13–14):684–691.
40. Ozcan U, Cao Q, Yilmaz E et al. Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science 2004; 306(5695): 457–461.
41. Pallayova M, Lazurova I, Donic V. Hypoxic damage to pancreatic beta cells--the hidden link between sleep apnea and diabetes. Med Hypotheses 2011; 77(5): 930–934.
42. Pi J, Collins S. Reactive oxygen species and uncoupling protein 2 in pancreatic B-cell function. Diabetes Obes Metab 2010; 12(Suppl 2): S141-S148.
43. Polak J, Shimoda LA, Drager LF et al. Intermittent hypoxia impairs glucose homeostasis in C57BL6/J mice: partial improvement with cessation of the exposure. Sleep 2013; 36(10): 1483–1490.
44. Pont A, Rubino JM, Bishop D et al. Diabetes mellitus and neuropathy following Vacor ingestion in man. Arch Intern Med 1979; 139(2): 185–187.
45. Ruzzin J, Petersen R, Meugnier E et al. Persistent Organic Pollutant Exposure Leads to Insulin Resistance Syndrome. Environ Health Perspect 2009; 118(4): 465–471.
46. Sato Y, Fujimoto S, Mukai E et al. Palmitate induces reactive oxygen species production and B‐cell dysfunction by activating nicotinamide adenine dinucleotide phosphate oxidase through Src signaling. J Diabetes Investig 2014; 5(1):19–26.
47. Smiley T. The role of declining beta cell function in the progression of type 2 diabetes: implications for outcomes and pharmacological management. Can J Diabetes 2003; 27(3): 277–286.
48. Son HK, Kim SA, Kang JH et al. Strong associations between low-dose organochlorine pesticides and type 2 diabetes in Korea. Environ Int 2010; 36(5): 410–414.
49. Sone H, Kagawa Y. Pancreatic beta cell senescence contributes to the pathogenesis of type 2 diabetes in high-fat diet-induced diabetic mice. Diabetologia 2005; 48(1): 58–67.
50. Swaminathan S, Fonseca V, Alam M et al. The role of iron in diabetes and its complications. Diabetes Care 2007; 30(7): 1926–1933.
51. Škrha J. Patogeneze diabetes mellitus 1. a 2. typu v roce 2011 – jednotící model poruchy glykoregulace. Vnitř Lék 2011; 57(11): 949–953.
52. Tan L, Ye X, Zhou Y et al. Macrophage migration inhibitory factor is overexpressed in pancreatic cancer tissues and impairs insulin secretion of b-cell. J Transl Med 2014; 12 : 92. Dostupné z DOI: <http://doi: 10.1186/1479–5876–12–92>.
53. Tittlbach M. Ostrůvky pankreatu, fylogenetický a ontogenetický vývoj, primární struktury hormonů. In: Anděl M, Saudek F, Klimeš I (eds). Horizonty diabetologie. Díl 1. Tigis: Praha 2001. ISBN 80–900130–1-7.
54. Uchizono Y, Baldwin AC, Sakuma H et al. Role of HNF-1α in regulating the expression of genes involved in cellular growth and proliferation in pancreatic beta-cells. Diabetes Res Clin Pract 2009; 84(1):19–26.
55. Urbanová J, Rypáčková B, Procházková Z et al. Positivity for islet cell autoantibodies in patients with monogenic diabetes is associated with later diabetes onset and higher HbA1c level. Diabet Med 2014; 31(4): 466–471.
56. Urbanová J, Rypáčková B, Kučera P et al. Should the negativity for islet cell autoantibodies be used in a prescreening for genetic testing in maturity-onset diabetes of the young? The case of autoimmunity-associated destruction of pancreatic B-cells in a family of HNF1A-MODY subjects. Int Arch Allergy Immunol 2013; 161(3): 279–284.
57. Welters HJ, Tadayyon M, Scarpello JH et al. Mono-unsaturated fatty acids protect against beta-cell apoptosis induced by saturated fatty acids, serum withdrawal or cytokine exposure. FEBS Lett 2004; 560(1–3): 103–108.
58. Wobser H, Düssmann H, Kögel D et al. Dominant-negative suppression of HNF-1 alpha results in mitochondrial dysfunction, INS-1 cell apoptosis, and increased sensitivity to ceramide-, but not to high glucose-induced cell death. J Biol Chem 2001; 277(8): 6413–6421.
Labels
Diabetology Endocrinology Internal medicineArticle was published in
Internal Medicine

2014 Issue 9
Most read in this issue
- Životní prognóza osob s diabetem 1. typu dříve a dnes
- Gliptiny: bezpečná a účinná léčba diabetu
- Inzulinová rezistence – příčiny a možnosti ovlivnění
- AGEs a RAGE – konečné produkty pokročilé glykace a jejich receptor v otázkách a odpovědích