
Inzulinová rezistence – příčiny a možnosti ovlivnění
Insulin resistance – its causes and therapy possibilities
Insulin resistance (IR) is defined as a condition where normal plasma free insuconcentrations induce a reduced response of the body. In the narrower sense we understand IR as the impairment of insulin action in the target structure which may arise at any level of the insulin signalling cascade. In the clinical conditions we usually define it as the impairment of insulin action in glucose metabolism, although it is true that the impairment may concern different effects of insulin and different cell structures. The characteristic feature of IR linked to the metabolic syndrome or Type 2 diabetes is defective signalling which affects PI3-kinase branch of insulin signalling cascade. Other insulin actions depending on the signalling through the Ras complex and MAP-kinase, may not be affected. Due to compensatory hyperinsulinemia they may be even increased. The article summarizes some recent findings regarding the structure and regulation of insulin signalling cascade and analyses selected primary and secondary causes of IR which include genetic and epigenetic factors, the microRNA regulation role, metabolic, humoral and immunological factors. The detailed knowledge of the causes of IR opens possibilities of its rational treatment. This is currently based on the treatment of curable causes of IR, i.e. consistent compensation of diabetes, weight reduction, regimen arrangements (diet, physical activity), re-assessment of the need to use corticosteroids in therapy, treatment of coexisting conditions and possibly administration of metformin or pioglitazone.
Key words:
cytokines – insulin resistance – insulin signalling cascade
Authors:
Terezie Pelikánová
Authors‘ workplace:
Centrum diabetologie IKEM Praha, přednostka prof. MUDr. Terezie Pelikánová, DrSc.
Published in:
Vnitř Lék 2014; 60(9): 746-755
Category:
Overview
Inzulinovou rezistenci (IR) definujeme jako stav, při němž normální koncentrace volného plazmatického inzulinu vyvolává sníženou odpověď organizmu. V užším slova smyslu IR chápeme jako poruchu účinku inzulinu v cílové struktuře, která může vzniknout na kterékoli úrovni inzulinové signalizační kaskády. V klinických podmínkách ji obvykle definujeme jako poruchu účinku inzulinu v metabolizmu glukózy, i když platí, že porucha se může se týkat různých účinků inzulinu a různých buněčných struktur. Charakteristickým rysem IR svázané s metabolickým syndromem či diabetem 2. typu je defektní signalizace, která postihuje PI3-kinázovou větev inzulinové signalizační kaskády. Další účinky inzulinu, které závisí na signalizaci vedoucí přes Ras komplex a MAP-kinázu, nemusí být postiženy. Vlivem kompenzatorní hyperinzulinemie mohou být dokonce zvýšeny. Článek shrnuje některé novější poznatky týkající struktury a regulací inzulinové signalizační kaskády a rozebírá vybrané primární a sekundární příčiny IR, které zahrnují faktory genetické a epigenetické, regulační roli mikroRNA a metabolické, humorální a imunologické faktory. Detailní poznání příčin IR nabízí možnosti její racionální léčby. Ta se v současné době opírá o léčbu odstranitelných příčin IR, tj. důslednou kompenzaci diabetu, redukci hmotnosti, režimová opatření (dieta, fyzická aktivita), přehodnocení potřeby léčby kortikoidy, léčbu přidružených onemocnění, a případně podání metforminu či pioglitazonu.
Klíčová slova:
cytokiny – inzulinová rezistence – inzulinová signalizační kaskáda
Úvod
Pojem inzulinová rezistence/senzitivita se objevil v souvislosti s možností stanovit imunoreaktivní inzulin v plazmě v 50. letech minulého století, protože jeho vysoké hladiny, nalezené u obézních nemocných s diabetem, implikovaly poruchu jeho účinku. Od té doby je zájem o tuto problematiku trvalý. Počet publikací, které se zabývají inzulinovou rezistencí (IR) na nejrůznějších úrovních, roste zásluhou zavádění nových metod biomedicínského výzkumu (např. proteomika, lipidomika, metabolomika) geometrickou řadou [1]. Počet odkazů, které se objevily na informačním portálu PubMed za 5 let k 2. 6. 2014 po zadání hesla „insulin resistance“ byl 31 971. Proto se bude následující text zabývat pouze vybranými okruhy této problematiky.
Účinek inzulinu – fyziologické poznámky
Účinek inzulinu v cílových tkáních je zprostředkován komplikovaným buněčným signálním mechanizmem, jehož komponenty jsou přítomny prakticky ve všech buňkách lidského těla. Po specifické vazbě inzulinu na inzulinový receptor následuje kaskáda fosforylačně-defosforylačních reakcí, která je zahájena autofosforylací tyrozinu na β podjednotce inzulinového receptoru, při níž je atakována celá řada intracelulárních proteinů, jejichž konečným cílem je např. syntéza a aktivace enzymů metabolických drah, aktivace transportérů glukózy či ovlivnění mitogenní aktivity buněk a apoptózy. Klasická signální cesta má 2 základní větve. První z nich vede přes aktivaci fosfatidylinozitol 3kinázy (PI3K) a druhá přes aktivaci Ras komplexu a MAP-kinázu. Přitom už zdaleka neplatí, že PI3-kinázová cesta odpovídá pouze za metabolické účinky inzulinu a MAP-kinázová cesta za účinky inzulinu na buněčný cyklus a proliferaci. Obě signální dráhy se úzce prolínají a vzájemně ovlivňují. Detailní signální mechanizmus stále není objasněn. Molekul, které vstupují do hry a které jsou citlivě regulovány, jsou stovky. Fosforylaci zajišťují serin-treoninkinázy a defosforylaci fosfatázy. Roli hrají cAMP, G-proteiny, fosfatidylinozitol, proteinkináza C, fosfolipáza C, specifické fosfatázy – PTP 1B, SHIP2, PTEN, Foxa2 (forkhead transcription factor), ubikvitinový systém a řada dalších molekul, z nichž některé znázorňuje schéma 1. Schéma 1 ukazuje také zapojení vybraných molekul v inzulinové signalizační kaskádě z hlediska jejich výsledného metabolického efektu.
![Schéma 1. Inzulinová signalizační kaskáda a její interakce s humorálními (cytokiny) a nutričními (FFA, aminokyseliny a glukóza) podněty s vybranými účinky inzulinu v metabolizmu cukrů, tuků a bílkovin a dalšími biologickými účinky (autofagie, apoptóza, mitochondriální biogeneze, příjem potravy, metabolizmus vápníku, růst kostí a vazodilatace). Upraveno podle [2].](https://pl-master.mdcdn.cz/media/image/fbe270f4d19164e293601d33d39c1f2c.png?version=1537797771)
Inzulinová signalizační kaskáda podléhá velmi sofistikované regulaci. Odpovídá na nejrůznější metabolické (glukózy lipidy, aminokyseliny) a humorální podněty (cytokiny). Je ovlivněna oxidačním stresem a funkcí mitochondriálního dýchacího řetězce. Uplatňují se i další regulátory (např. SOCS, Sirt1, ARNT/HIF1, mikroRNA), z nichž každý by zasloužil pozornost [2,3].
Specifická regulace složek inzulinové signalizační kaskády a její poznání nabízí možnosti racionální intervence a léčby inzulinové rezistence.
Inzulinová rezistence a její příčiny
Definice pojmu IR
IR obecně rozumíme poruchu účinku inzulinu. Jde o stav, kdy normální koncentrace volného plazmatického inzulinu vyvolává sníženou odpověď organizmu. V užším slova smyslu chápeme IR jako poruchu účinku inzulinu v cílové struktuře, která může vzniknout kdekoli v kaskádě dějů, které zajišťují normální účinek. V klinických podmínkách ji obvykle definujeme jako poruchu účinku inzulinu v metabolizmu glukózy, i když je jasné, že porucha se může se týkat různých účinků inzulinu a různých buněčných struktur. Charakteristickým rysem IR svázané s metabolickým syndromem či diabetem 2. typu (DM2T) je defektní signalizace, která postihuje PI3-kinázovou větev inzulinové signalizační kaskády. Další účinky inzulinu, které závisí na signalizaci vedoucí přes Ras komplex a MAP-kinázu nemusí být postiženy. Vlivem kompenzatorní hyperinzulinemie mohou být dokonce zvýšeny.
Příčiny IR
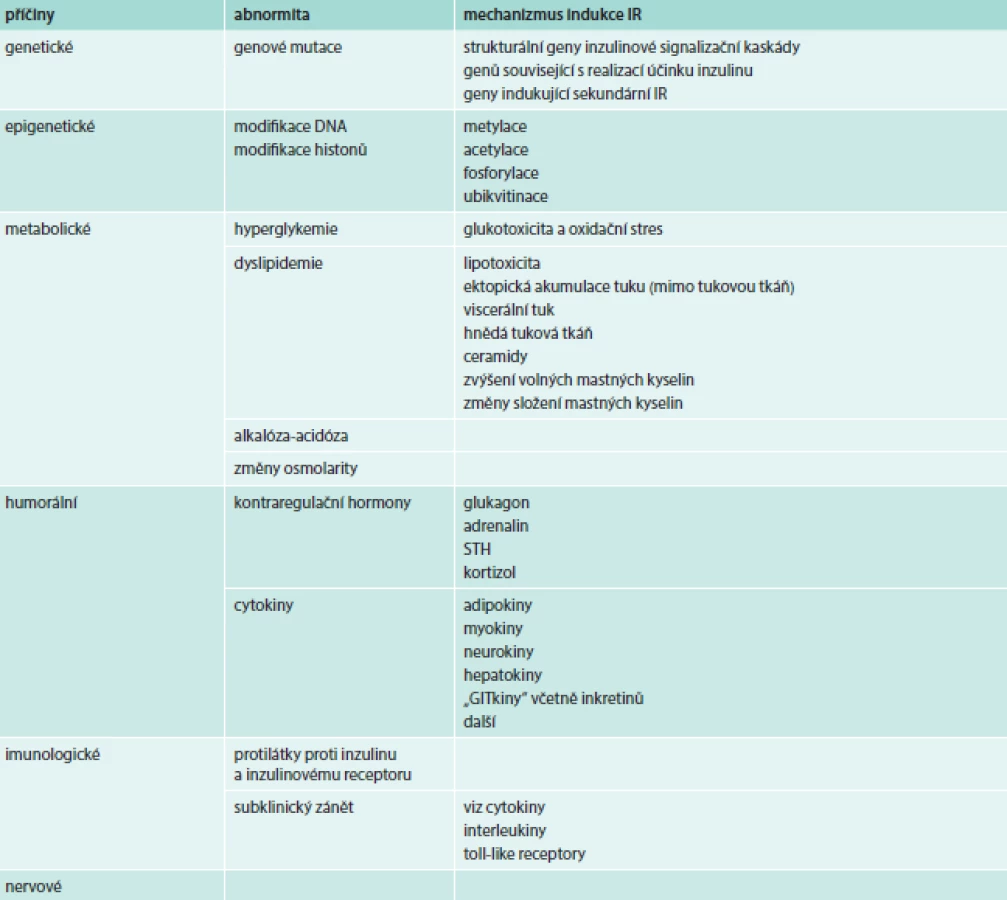
Podle příčiny vzniku můžeme rozlišovat IR geneticky podmíněné (primární) a sekundární. Obecně se při rozvoji IR mohou uplatňovat mechanizmy, které zasahují cestou vegetativního nervstva, humorální cestou či změnami metabolického charakteru. Někdy se pod příčiny vedoucí k IR zahrnují i abnormity v sekreci inzulinu. Při nich nacházíme v plazmě vysoké aktivity imunoreaktivního inzulinu a imponují jako IR, ačkoli nejde o poruchu účinku inzulinu v pravém slova smyslu.
Klinické stavy spojené s poruchou účinku inzulinu uvádí tab. 1. IR se prohubuje se fyziologicky při stárnutí, v pubertě, v těhotenství, při stresu a hladovění a inzulinová senzitivita se naopak zvyšuje při fyzickém tréninku. IR je asociována s řadou onemocnění. Objevuje se nejen při onemocněních, která s IR tradičně spojujeme, jako je metabolický syndrom, obezita a DM2T, ale prakticky každá patologie, ať již chronická nebo akutně vzniklá, je sledována rozvojem určitého stupně IR (renální selhání, cirhóza, febrilie, infekční onemocnění atd).

V klinickém měřítku hraje při rozvoji IR nejvýznamnější roli přejídání a obezita, nevhodné složení stravy, malá fyzická aktivita, stres, kouření a některá farmaka.
Genetické příčiny IR
Raketový nárůst počtu genetických studií, ať už typu asociačních studií kandidátních genů, či studií, které využívají analýzy celého genomu pomocí mikročipových metod (genome-wide association studies – GWAS) vedly k objevení již desítek genových mutací, které jsou asociované s IR. U některých z nich je známy i mechanizmy, které k IR vedou, zatímco u většiny z popisovaných genů zůstává jejich význam a funkce záhadou. Přes uvedené pokroky však i v roce 2014 platí, že mutace, které by hrály roli u většiny nemocných s metabolickým syndromem/DM2T, zatím nebyly objeveny. Složitost problematiky je dána množstvím signálních a regulačních proteinů, jejichž strukturální nebo funkční změna by se mohla uplatňovat. Pouze u malé části nemocných (1–2 %) je etiologie IR uspokojivě vysvětlena průkazem genové mutace [4,5].
Epigenetika
Žhavým tématem v etiopatogenze řady onemocnění je možnost modifikace genové exprese. Vlivy, které se uplatňují jinou cestou, než jsou klasické mutace v DNA, označujeme jako epigenetické faktory. I v oblasti IR se ukazuje, že epigenetické mechanizmy hrají důležitou roli. Modifikace DNA a histonů vlivem metylace, acetylace, fosforylace či ubikvitinace, což jsou klasické epigenetické mechanizmy, vedou ke změnám genové exprese a uplatňují se při rozvoji IR [6].
MikroRNA (miR)
MiR jsou krátké nekódující molekuly RNA, které obsahují asi 22 nukleotidů. Nekódující znamená, že nekódují žádný protein a mají pouze roli regulační. Kontrolují genovou expresi na posttranskripční úrovni cestou suprese již nasyntetizované mRNA. Po vazbě na mRNA buď zabrání její translaci (tj. blokují tvorbu bílkoviny) nebo způsobí její degradaci.
MiR je možné prokázat nejen ve tkáních, ale i v cirkulaci. Změny hladin různých typů miR byly nalezeny při DM2T, DM1T i při gestačním diabetu. Ve vazbě s diabetem byl opakovaně dokladován pokles např. miR-126, -29, -107, -143. Změny miR byly nalezeny u osob s metabolickým syndromem. Ve finské práci, která analyzovala 71 osob s metabolickým syndromem pomocí stanovení vybraných miR v krvi, celogenomového skríningu a analyzovala metabolom v séru, bylo 9 různých miR asociováno s projevy inzulinové rezistence [7]. Zdá se, že miR by bylo použitelné jako případný biomarker rizika diabetu a dokonce jako biomarker jeho komplikací.
Vybrané miR jsou ve vazbě k sekreci inzulinu i účinku inzulinu. Schéma 2 shrnuje dosavadní nálezy změn hladin miR ve vybraných tkáních s vazbou na inzulinovou rezistenci [8], a to nejen z hlediska nově objevených miR, ale zejména z hlediska nových potenciálních mechanizmů, kterými jsou miR v indukci IR zapojeny. Příkladem může být zcela recentní práce autorů Mohamed et al [9], která ukazuje na dosud nediskutovanou miR-149. Její hladina v kosterním svalu se zvyšuje při cvičení, a naopak klesá při vysokotukové dietě. Zdá se, že miR-149 rozhodujícím způsobem ovlivňuje, jak bude spotřebováván NAD+ v kosterním svalu [10], zda povede ke zvýšení biologické aktivity mitochondrií cestou aktivace SIRT-1/PGC-1a, nebo se naopak biologická aktivita mitochondrií utlumí a zvýší se PARP-2, a tudíž bude akcentován zánět a apoptóza.
![Schéma 2. MiRNA uplatňující se v účinku inzulinu (IR/IS). Upraveno podle [8].](https://pl-master.mdcdn.cz/media/image/7075b304d2daf8e2b2107b5f88a8f9e6.png?version=1537794564)
Hyperglykemie a oxidační stres
Hyperglykemie je klasickou metabolickou příčinou IR, která se významně uplatňuje v klinické praxi. Hyperglykemie aktivuje glukózové transportéry, včetně GLUT4, a glukóza se hromadí v buňkách, jejichž morfologii a funkce může poškozovat různými mechanizmy. Po vstupu glukózy do buněk je glukóza fosforylována na glukózo-6-fosfát. Za fyziologických podmínek probíhá oxidace glukózy, glykolýza, která je provázena tvorbou glycerol-3-fosfátu a diacyl-glycerolu (DAG) a je syntetizován zásobní glykogen. Při hyperglykemii a nadbytku glukózy intracelulárně jsou patologicky zvýšené náhradní metabolické cesty, které se za fyziologických podmínek uplatňují pouze minimálně. Jsou to:
- neenzymatická glykace a tvorau pokročilých produktů glykace (AGE)
- tvorba hexosaminu
- akcentace polyolové cesty a zvýšení osmoticky aktivního sorbitolu
- indukce oxidačního stresu
Mechanizmy, kterými hyperglykemie zvyšuje oxidační stres, je několik. Sama glukóza podléhá autooxidaci (glykooxidace) a je zdrojem reaktivních forem kyslíku, které mohou indukovat řetězovou reakci vzniku dalších kyslíkových radikálů (oxidační stres) provázenou poškozením řady struktur. Zároveň se takto mění v reaktivní dikarbonyly (karbonylový stres), které reagují s aminoskupinami bílkovin ještě rychleji než intaktní glukóza a ještě účinněji podporují vznik síťových vazeb a AGE. Reaktivní dikarbonyly mohou vznikat nejen jako produkty glykooxidace, ale polyolovou cestou zpracování glukózy při přeměně fruktózy, či při oxidaci lipidů. Zdrojem reaktivních forem kyslíku (ROS) je i zvýšená „fyziologická“ oxidace glukózy. Dalším samostatným mechanizmem je zvýšená aktivace klasických izoforem proteinkinázy C (cPKC-α, -βI, -βII, -γ), které jsou jasně ve vztahu k rozvoji IR a které jsou při hyperglykemii aktivované v nadbytku se tvořícím DAG. Výsledné produkty mohou působit toxicky a přímo interferovat s inzulinovou signalizační kaskádou nebo se mohou uplatnit nepřímo, tak že indukují tvorbu prozánětlivých cytosinů [11,12]. Výsledkem je inzulinová rezistence.
Poruchy metabolizmu lipidů
Rozvoj IR při vzestupu volných mastných kyselin (MK) je znám řadu desetiletí. Tradičním mechanizmem její indukce, který se nepochybně uplatňuje, je kompetice energetických substrátů (Randlův cyklus) [13]. V posledních letech je však větší pozornost věnována jiným mechanizmům působení poruch metabolizmu lipidů, než je pouhá kompetice substrátů. Faktorem, který nepochybně vede k IR, je ektopická akumulace lipidů. Ektopickou akumulací tuku rozumíme ukládání lipidů mimo tukovou tkáň, v nichž nadbytečný obsah tuků vede k funkčnímu a morfologickému poškození. Mechanizmus, kterým se tak děje, označujeme jako „lipotoxicitu“. Ektopická akumulace tuku byla prokázána např. v kosterním svalu, kardiomyocytech či v jaterní tkáni, v nichž je asociována s inzulinovou rezistencí, a řada autorů se domnívá, že je dokonce její hlavní příčinou [14]. Kromě zvýšené hladiny volných MK se na ektopické akumulaci tuku může podílet alterace transportních systémů, které facilitují vstup masných kyselin do buněk (fatty acid binding proteins – FABP) [15], či porucha v intracelulární homeostáze lipidů. Poměr mezi oxidací a ukládáním lipidů je regulován aktivitou AMP-aktivované protein kinázy (AMPK) a podléhá řadě regulačních vlivů, např. miR, které byly zmíněny výše. Rovnováhu mezi oxidací tuků a lipogenezou ovlivňují působky produkované tukovou tkání (adipokiny), z nichž některé, jako je rezistin, retinyl vážící protein 4 (RBP4) či TNFα jsou asociované s rozvojem IR, a naopak adiponektin, visfatin či leptin jsou spojovány s opačnými účinky. Leptin a adiponektin zvyšují oxidaci MK v játrech, svalech, Langerhansových ostrůvcích (LO) cestou aktivace AMP-aktivované protein kinázy (AMPK), která zároveň snižuje aktivitu lipogeních enzymů. Deficience leptinu či leptinová rezistence stejně jako deficience adiponektinu tak vede k ektopickému hromadění tuku ve svalech, játrech, B-buňkách a k rozvoji IR.
AMPK je enzym aktivovaný zvýšením poměru AMP/ATP, tj. reaguje na pokles ATP např. při fyzické aktivitě nebo hladovění. Zvyšuje produkci ATP, tak že zvyšuje oxidaci tuků a potlačuje jejich syntézu. Je logické, že její dysregulace může stát v pozadí ektopického hromadění tuků a IR. AMPK má však řadu dalších účinků, např. účinky protizánětlivé potlačuje oxidační stres a stres endoplazmatického retikula, aktivuje autofagii, což jsou všechno děje, které hrají roli v indukci IR. Proto je dysregulace AMPK považována v současné době nejen za důležitý patogenetický faktor, ale mohla by být i terapeutickým cílem v léčbě IR [16].
Zvýšená nabídka MK interferuje s inzulinovou signalizační kaskádou a může navodit IR řadou mechanizmů. Experimentální studie dokládají, že důležitější než prostá kompetice substrátů při oxidadaci (glukóza vs MK) [13] je toxický efekt nahromaděných lipidových metabolitů v cytozolu. Jedním z takových metabolitů s lipotoxickými účinky jsou ceramidy. Zvýšená nabídka MK a intracelulární akumulace lipidů je doprovázena zvýšením de novo syntézy ceramidů z palmitátu a buněčnou apoptózou. Ve studiích in vitro bylo prokázáno, že ceramidy interferují s inzulinovou signalizační kaskádou na úrovni proteinkinázy B (PKB/Akt) [14]. Vyšší koncentrace ceramidů v plazmě i tkáních byly nalezeny u osob s diabetem či poruchou glukózové tolerance. Nicméně jasný důkaz potvrzující kauzální vztah mezi hladinou ceramidů a mírou IR u člověka chybí [17].
Dalším z mechanizmů toxického působení MK je produkce reaktivních forem kyslíku (ROS) a dusíku (RNS) a indukce oxidačního stresu. ROS a RNS přímo poškozují DNA, strukturální bílkoviny a lipidy. Navíc ROS a RNS působí i nepřímo přes řadu stresem indukovaných systémů, např. aktivují NF-κB, p38 MAPK, zvyšují tvorbu hexosaminů atd [12].
MK a jejich acyl-CoA mohou také přímo měnit genovou expresi stovek genů. Slouží jako specifické ligandy řady nukleárních receptorů a kontrolují expresi řady transkripčních faktorů, jako je např. sterol regulační element vázající protein (sterol response element-binding proteins – SREBP) nebo receptory aktivované proliferátory peroxizomů (peroxisome proliferator-activated receptors – PPARs) [18]. Kromě toho mají funkci strukturálních lipidů, jsou součástí buněčných membrán, a ovlivňují fluiditu buněčných membrán, funkci receptorů a enzymů vázaných na membrány, mají funkce transportní a jsou prekurzorem prostanoidů (prostaglandiny, tromboxany, interleukiny).
Posledním faktorem, který bych ráda v souvislosti s rolí lipidů v patogenezi IR zmínila, je složení MK v sérových a tkáňových lipidech. MK dělíme podle stupně elongace a desaturace a podle umístění dvojných vazeb na nasycené, monoenové, a polynenasycené MK řady n-6 a n-3. V současné době je již mimo jakoukoli pochybnost, že výše uvedené účinky MK závisí na typu MK. Naše pracovní skupina jako první poukázala na významnou závislost mezi mírou inzulinové rezistence, testovanou in vivo pomocí hyperinzulinového euglykemického clampu a složením MK ve fosfolipidech séra [19]. Účinek inzulinu negativně koreloval se zastoupením nasycených MK a pozitivně závisel na obsahu esenciálních MK řady n-6. Nejvýznamnější byly vztahy k obsahu kyseliny linolové (C18 : 2 n6). Tyto závislosti byly patrné i v izolovaných erytrocytech mezi složením fosfolipidů erytrocytárních membrán a specifickou vazbou inzulinu na receptory [20]. V 90. letech 20. století byly potvrzeny závislosti mezi účinkem inzulinu a složením MK v kosterním svalu u experimentálních zvířat [21] a u člověka [22] také jinými výzkumnými skupinami. Rizikový profil MK snížení obsahu kyseliny linolové a zvýšení polynenasycených elongovaných forem n-6 MK jsme nalezli u DM2T, u pacientů s metabolickým syndromem a zdravých prvostupňových příbuzných hypertoniků [23,24]. A potěšující je, že i v intervenčních studiích, které provádíme v současné době, se ukazuje, že zlepšení inzulinové senzitivity u nemocných s DM2T vlivem vegetariánské diety [25] i vlivem změny počtu jídel za den [26] je vázáno na zvýšení obsahu kyseliny linolové ve fosfolipidové frakci.
Humorální příčiny IR
Zvýšení hladin kontraregulačních hormonů je klasickým příkladem příčin IR.
Pozornost se však v posledních letech soustřeďuje na působky, které mohou být produkovány adipocyty, hepatocyty, endotelem, buňkami střevní sliznice, imunokompetentními buňkami či CNS. Tyto působky mohou účinkovat lokálně i systémově a mohou zprostředkovávat komunikaci mezi buňkami i tkáněmi. Cytokiny, které hrají roli v indukci inzulinové rezistence, mají obvykle i různou měrou vyjádřené vlastnosti prozánětlivé, vazokonstrikční, trofické, prokoagulační a protrombotické vlastnosti. Za fyziologických podmínek jsou v rovnováze s působky, které mají účinky opačné. Protože jednotlivé mediátory často působí prostřednictvím vazby na své receptory, je jejich biologický účinek navíc negativně ovlivněn cirkulujícími solubilními receptory či receptorovými antagonisty a protilátkami.
Adipokiny jsou působky produkované adipocyty. V současné době se jejich počet rozšířil na více než 600. Z nich některé modulují imunitní odpověď (adipsin, ASP, SAA3,CSFs,IL-17D), mají prozánětlivé účinky (IL1b, IL6, IL8, IL10, CRP, MPC1, osteopontin, progranulin, chemerin), ovlivňují metabolizmus glukózy (leptin, adiponektin, DPP4, rezistin, vaspin), inzulinovou senzitivitu (leptin, adiponektin, chemerin, rezistin RBP4), krevní tlak (angiotenzinogen), adhezi buněk (PAI1), angiogenezi a vazokonstrikci/vazodilataci (VEGF), adipogenezi a tvorbu kostní hmoty (BMP7), růstové faktory (IGF1,TGFβ, fibronectin), metabolizmus lipidů (CD36), regulace chuti k jídlu (leptin, vaspin) a další biologické funkce [27].
Myokiny jsou působky produkované buňkami kosterního svalu. Proteomickými přístupy jich bylo odhaleno několik stovek, byť pouze u zlomku je známa jejich funkce. Jsou indukované svalovou kontrakcí (fyzickou aktivitou). Mají parakrinní a endokrinní účinky. Ovlivňují citlivost kosterního svalu k inzulinu, subklinický zánět a další. Mezi myokiny, které by mohly být předmětem farmakologického využití, patří fibroblastový růstový faktor 21 (fibroblast growth factor 21 – FGF21) a foilistatinu podobný faktor 21 (follistatin-like 1 – FSTL1). FGF21 stimuluje sekreci adiponektinu, a tak zvyšuje inzulinovou senzitivitu. Zároveň tento působek indukuje změnu bílé tukové tkáně v hnědou. Syntetický FGF21 analog LY2405319 je v současné době v prvních fázích klinického zkoušení.
FSTL1 je označovaný jako kardioprotektivní cytokin. Jde o glykoprotein, který má vliv na endotelové buňky cévní stěny. Je produkován nejen kosterním svalem, ale i myokardem či adipocyty. Má antiapoptotické a protizánětlivé účinky. Zdá se, že brání ischemicko-reperfuznímu poškození myokardu [28].
Podobně mohou důležitou roli zprostředkovávat neurokiny produkované v CNS, hepatokiny secernované jaterní buňkou, „GITkiny“ tvořené buňkami střevní sliznice, které zahrnují také inkretiny, endoteliny produkované buňkami cévního endotelu a celou řadu dalších cytokinů, které jsou produkovány buňkami imunitního systému (interleukiny, TNFα a další).
Imunologické příčiny
IR je asociována se subklinickým zánětem. Imunologické abnormity mohou být nejen projevem IR, ale i její bezprostřední příčinou. Velká pozornost je věnována bílé tukové tkáni, a zejména viscerálnímu tuku, byť infiltrace makrofágy a dalšími imunokompetentními buňkami je popsána i v dalších tkáních souvisejících s inzulinovou rezistencí (např. játra, střevo a další). Makrofágy a další buňky pak produkují proinflamatorní cytokiny (interleukin 1), které interferují s inzulinovou signalizační kaskádou. Kromě makrofágů se stále více pozornosti věnuje také ostatním imunokompetentním buňkám, jako jsou T - a B-lymfocyty [29]. Mimo rámec tohoto přehledu stojí řada mechanizmů, které odpovídají za indukci imunitních reakcí (např. toll-like receptory), či cesty indukce tvorby prozánětlivých cytokinů, jako je IKKb a aktivace NFκB a molekulární mechanizmy a jejich interference s inzulinovou signalizační kaskádou [2]. Klinicky důležité by mohlo být, že potlačení subklinického zánětu by mohlo být jednou z cest léčby IR a DM2T.
Úloha vybraných orgánů a tkání v patogenezi IR
Vybrané tkáně, které hrají roli v projevech a patogenezi IR, ukazuje schéma 3.

Kosterní sval
Z hlediska regulace glykemie je nejdůležitějším místem, v němž se projeví IR v metabolizmu glukózy, kosterní sval. Kromě defektů v inzulinové signalizační kaskádě, snížené aktivaci GLUT4 a vybraných enzymů metabolizmu glukózy, které mohou mít primární i sekundární charakter, hraje roli také morfologie svalových vláken. Ve vztahu k účinku inzulinu je denzita kapilár v kosterním svalstvu a převládající typ svalových vláken. Kosterní sval je producentem stovek myokinů popsaných výše a experimenty s geneticky modifikovanými zvířaty (MIRKO – knock out myši s odstraněným genem pro inzulinový receptor v kosterním svalu) prokázaly, že izolovaný defekt v účinku inzulinu vázaný na kosterní sval indukuje rozvoj IR i v dalších tkáních (v tukové tkáni a játrech). Přesto v současné době stále převládá názor, že IR v kosterním svalu je spíše důsledkem poruchy funkce jiných tkání. Na prvním místě je za zvýšení hladin volných mastných kyselin a produkci látek s endokrinní aktivitou, které také patrně odpovídají za ektopickou akumulaci tuku v kosterním svalu, zodpovědná tuková tkáň. Zmnožení intramyocelulárních triacylglycerolů (TG) se opakovaně nalézá u osob s inzulinovou rezistencí a DM2T. Dalším faktorem, který ovlivňuje odsun glukózy do kosterního svalu, je krevní průtok. Při IR byla potvrzena porucha vazodilatace a další primární strukturou, která odpovídá za snížený odsun glukózy do svalu, může být endotel.
Játra
IR je v blokádě produkce glukózy v játrech jedním z mechanizmů, které hrají roli při rozvoji hyperglykemie nalačno, a jedním z projevů IR jsou metabolická játra (nealkoholová steatóza, steatohepatitida a steatofibróza). Experimenty s geneticky modifikovanými zvířaty (LIRKO – knock out myši s odstraněným genem pro inzulinový receptor v játrech nebo s defektem PDK1, mTORC2, či Akt1 a Akt2) však prokázaly, že izolovaný defekt v účinku inzulinu vázaný na hepatocyt indukuje rozvoj IR v dalších tkáních (v tukové tkáni a kosterním svalu), stejně jako izolované defekty v inzulinové signalizační kaskádě [2]. Játra proto mohou být primární lokalizací IR a hrát podstatnou roli v indukci IR a rozvoji hyperglykemie. Navíc se játra uplatňují v regulaci koncentrace inzulinu. Asi jedna třetina inzulinu, který je uvolňován do portální krve, se vychytá v játrech při první pasáži.
Tuková tkáň, viscerální tuk a hnědá tuková tkáň
Experimenty s geneticky modifikovanými zvířaty (ADIPOIRKO – knock out myši s odstraněným genem pro inzulinový receptor v adipocytech) prokázaly, že izolovaný defekt v účinku inzulinu vázaný na tukovou tkáň indukuje rozvoj inzulinové rezistence v dalších tkáních (v játrech, kosterním svalu i B-buňkách). Diskutují se 2 možné mechanizmy, a to zvýšené uvolňování volných mastných kyselin při nedostatečné blokádě lipolýzy inzulinem a endokrinní aktivita tukové tkáně, ať již cestou adipokinů či cytotinů tvořených vcestovanými imunokompetentními buňkami (viz výše). Vyšší metabolická a endokrinní aktivita je spojena zejména s viscerálním tukem, a proto je IR asociována s centrální obezitou. Nově se v této oblasti diskutuje možná pozitivní úloha hnědé a „béžové“ tukové tkáně, jejíž indukce u člověka by mohla být cestou ovlivnění IR [30].
CNS
Nervová tkáň je dalším příkladem tkáně, která z hlediska metabolizmu glukózy není závislá na inzulinu, ale buňky CNS mají na svém povrchu inzulinové receptory a obsahují inzulinovou signalizační kaskádu. Experimenty s geneticky modifikovanými zvířaty (NIRKO – knock out myši s odstraněným genem pro inzulinový receptor v neuronech) prokázaly, že izolovaný defekt v účinku inzulinu vázaný na CNS indukuje rozvoj inzulinové rezistence v dalších tkáních (v játrech, adipocytech, kosterním svalu i B-buňkách). V hypotalamu je centrum hladu a sytosti a k rozvoji IR a diabetu může dojít cestou indukce obezity [31].
Střevo
Buňky střevní sliznice produkují celou řadu látek, které ovlivňují sekreci i účinek inzulinu. Střevo je místem produkce hormonů enteroinzulární osy, z nichž nejdůležitější z hlediska regulace sekrece inzulinu a glukagonu jsou inkretiny. Navíc střevo produkuje řadu prozánětlivých cytokinů (TNFα, IL6 a další), které mohou přímo indukovat IR, a látek, které působí v CNS a ovlivňují chuť k jídlu (oxyntomodulin, enterostatin, NPY, grelin a další). Buňky střevní sliznice také vychytávají glukózu a podílejí se na inzulinem stimulovaném odsunu glukózy po přísunu potravy. Je zajímavé, že pokud je v experimentu navozena izolovaná IR, která postihuje pouze střevo (GIRKO – knock out myši s odstraněným genem pro inzulinový receptor ve střevním epitelu), dojde opět k rozvoji IR i v dalších tkáních. Funkci enterocytů ovlivňuje střevní mikroflóra a v posledních letech se pozornost soustřeďuje na možnosti léčby IR prostřednictvím modifikace střevní mikroflóry [32].
Endotel
Endoteliální dysfunkce je jedním z projevů IR. Ale endotelové buňky jsou také mohutným producentem látek humorální povahy, které mají vazoaktivní účinky, ovlivňují trombogenezi a mohly být i mediátorem IR v dalších cílových tkáních. Buňky endotelu obsahují inzulinovou signalizační kaskádu, přestože transport glukózy do nich není závislý na inzulinu. Aktivace inzulinové signalizační kaskády v endotelových buňkách vede cestou proteinkinázy B (Akt) k tvorbě kysličníku dusnatého (NO) a zprostředkovává vazodilataci. Porucha vazodilatace může být při IR jednou z příčin sníženého odsunu glukózy do kosterního svalu.
Úloha imunitního systému a imunokompetentních buněk v indukci IR byla zmíněna v předchozím textu. Mezi další producenty vazoaktivních látek, které by mohly hrát roli při rozvoji IR, patří také ledviny. Ledviny by se mohly v indukci IR uplatňovat i nepřímo, cestou ovlivnění hladin glykemie. V ledvinách totiž probíhá glukoneogeneza a filtrace a reabsorpce glukózy. V indukci IR může hrát roli i pankreas a hormony, které produkuje. Konečně poslední tkání, která doplňuje konečný počet na magickou desítku, je kostní dřeň.
Úloha vybraných buněčných struktur v patogenezi IR
Příčiny rozvoje se hledají také na úrovni buněčných organel. Řada in vitro studií prokazuje roli dysfunkce mitochondrií. Mitochondrie jsou buněčnými organelami, v nichž probíhá oxidace lipidů a cukrů v Krebsově cyklu a tvorba ATP. Jejich dysfunkce je asociována s inzulinovou rezistencí. Navíc je mitochondriální dysfunkce a porucha energetického metabolizmu spojena se zvýšenou tvorbou redukovaného NADPH, a tudíž se zvýšeným oxidačním stresem, který indukuje IR. Podobně existují důkazy o vazbě IR na poškození endoplazmatického retikula (ER), pro které se v literatuře používá termín stres ER. Snad nejvíce diskutovaným okruhem v patogenezi IR jsou poruchy funkce nukleárních receptorů a transkripčních faktorů [33], které jsou zapojené do regulací energetického metabolizmu včetně kontroly cirkadiálních rytmů. Právě poruchy v expresi genů, které kontrolují cirkadiální rytmy (clock genes), jsou další oblastí studovanou ve vazbě na IR [34].
Léčba IR
Léčba IR se v současné době opírá o odstranění „ovlivnitelných“ příčin IR. Mezi ně patří dekompenzace diabetu (hyperglykemie, dyslipidemie), obezita, fyzická inaktivita, interkurentní akutní onemocnění, chronické onemocnění (jater, ledvin a další) a některá farmaka (např. glukokortikoidy). Vlastní léčebné přístupy zahrnují okruhy, které uvádí tab. 3.
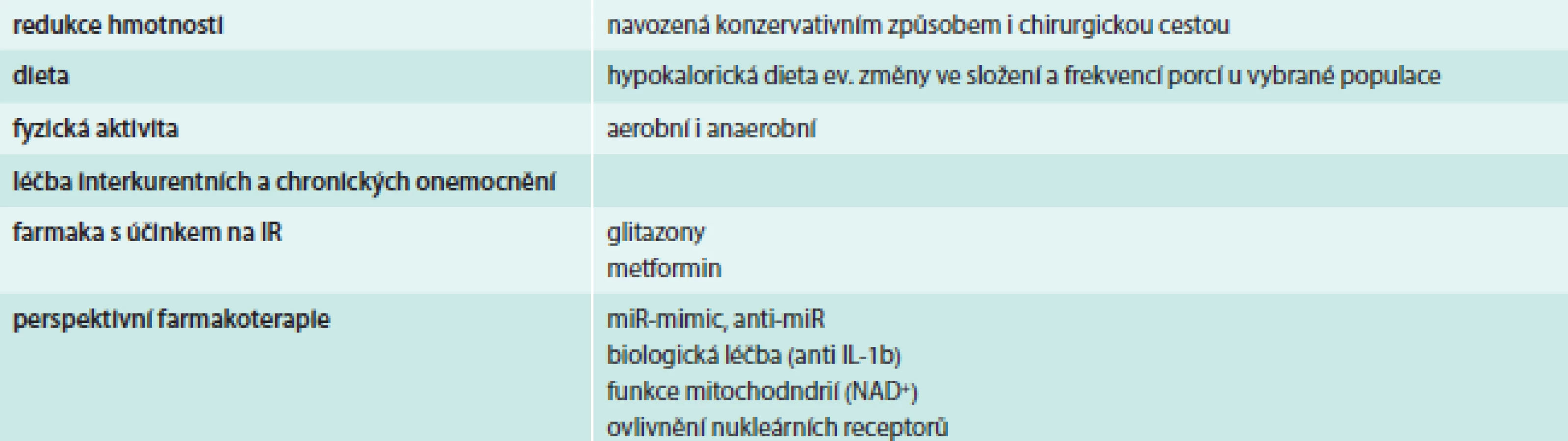
Přístupy, které vycházejí z poznání patogenetických mechanizmů a možností regulace inzulinové signalizační kaskády, jsou velmi nadějné, byť zatím vzdálené reálnému klinickému použití. Příkladem mohou být pokusy o ovlivnění regulačních miR, které byly zmíněny výše, a to buď se snahou navodit jejich zvýšení pomocí tzv. miR mimik nebo jejich potlačení pomocí tzv. anti-miR. Vývoj léčebných postupů založených na ovlivnění miR je patrný zejména v onkologii, ale klinická hodnocení začínají probíhat i na poli metabolických chorob. Jednou z prvních vlaštovek v pokusu touto cestou snížit kardiovaskulární riziko, IR a obezitu je zahájení klinického zkoušení anti-miR-208 (informace dostupné z: http://miragentherapeutics.com/piperine) [35].
Další nadějnou oblastí je biologická léčba IR, která je zaměřená na ovlivnění funkce cytokinů. Reálné klinické výsledky jsou zatím k dispozici pouze u látek blokujících IL1b. Z nich v klinickém zkoušení zatím zůstává kanakinumab – monoklonální protilátka proti IL1b. Má za sebou nadějná pilotní data a v běhu je multicentrická randomizovaná studie Cardiovascular Risk Reduction Study (CANTOS), která testuje efekt 2letého podávání kanakinumabu na kardiovaskulární příhody u nemocných po infarktu myokardu. V této studii budou jako vedlejší cíle hodnoceny u nemocných s DM2T také inzulinová senzitivita a sekrece inzulinu [36].
Protože jedním ze základních projevů IR je dysfunkce mitochondrií, vyvíjejí se nové molekuly, jejichž cílem je ovlivnit metabolizmus mitochondrií. Nadějná je cesta pomocí zvýšení hladin intracelulárního NAD+. Manipulace vedoucí ke zvýšení NAD+ jsou buď nepřímé prostřednictvím ovlivnění PARP2, SIRT1, zkoušejí se látky jako PJ34, resveratrol, SRT1720, nikotinamid ribosid, nikotinamid mononukleotid, event. přímým dodáním substrátu. V úvahu připadá i použití miR149 [10].
Zcela nové obzory v léčbě IR nabízí cílené ovlivnění nukleárních receptorů [33].
Podpořeno grantem IGA MZČR č. NT14250-3/2013
prof. MUDr. Terezie Pelikánová, DrSc.
terezie.pelikanova@medicon.cz
Centrum diabetologie IKEM, Praha
www.ikem.cz
Doručeno do redakce 22. 7. 2014
Přijato po recenzi 28. 7. 2014
Sources
1. Kurland IJ, Accili D, Burant C et al. Application of combined omics platforms to accelerate biomedical discovery in diabesity. Ann NY Acad Sci 2013; 1287 : 1–16.
2. Guo S. Insulin signaling, resistance, and the metabolic syndrome: insights from mouse models into disease mechanisms. J Endocrinol 2014; 220(2): T1-T23.
3. Saltiel AR. Putting the brakes on insulin signaling. N Engl J Med 2003; 349(26): 2560–2562.
4. Grarup N, Sandholt CH, Hansen T et al. Genetic susceptibility to type 2 diabetes and obesity: from genome-wide association studies to rare variants and beyond. Diabetologia 20147; 57 : 1528–1541.
5. Ahlqvist E, Ahluwalia TS, Groop L. Genetics of type 2 diabetes. Clin Chem 2011; 57(2): 241–254.
6. Sookoian S, Pirola CJ. Epigenetics of insulin resistance: an emerging field in translational medicine. Curr Diab Rep 2013; 13(2): 229–237.
7. Raitoharju E, Seppala I, Oksala N et al. Blood microRNA profile associates with the levels of serum lipids and metabolites associated with glucose metabolism and insulin resistance and pinpoints pathways underlying metabolic syndrome: the cardiovascular risk in Young Finns Study. Mol Cell Endocrinol 2014; 391(1–2): 41–49.
8. Chen H, Lan HY, Roukos DH et al. Application of microRNAs in diabetes mellitus. J Endocrinol 2014; 222(1): R1-R10.
9. Mohamed JS, Hajira A, Pardo PS et al.MicroRNA-149 inhibits PARP-2 and promotes mitochondrial biogenesis via SIRT-1/PGC-1alpha network in skeletal muscle. Diabetes 2014; 63(5): 1546–1559.
10. Svensson K, Handschin C. MicroRNAs emerge as modulators of NAD+-dependent energy metabolism in skeletal muscle. Diabetes 2014; 63(5): 1451–1453.
11. Škrha J. Biochemické důsledky dlouhodobé hyperglykémie. In: Škrha J (ed). Diabetologie. Galén: Praha 2009 : 66–75. ISBN 978–80–7262–607–6.
12. Evans JL, Goldfine ID, Maddux BA et al. Are oxidative stress-activated signaling pathways mediators of insulin resistance and beta-cell dysfunction? Diabetes 2003; 52(1): 1–8.
13. Randle PJ, Garland PB, Hales CN et al. The glucose fatty-acid cycle. Its role in insulin sensitivity and the metabolic disturbances of diabetes mellitus. Lancet 1963; 1(7285): 785–789.
14. Unger RH, Clark GO, Scherer PE et al. Lipid homeostasis, lipotoxicity and the metabolic syndrome. Biochim Biophys Acta 2010; 1801(3): 209–214.
15. Krusinova E, Pelikanova T. Fatty acid binding proteins in adipose tissue: a promising link between metabolic syndrome and atherosclerosis? Diabetes Res Clin Pract 2008; 82(Suppl 2): S127-S134.
16. Ruderman NB, Carling D, Prentki M et al. AMPK, insulin resistance, and the metabolic syndrome. J Clin Invest 2013; 123(7): 2764–2772.
17. Larsen PJ, Tennagels N. On ceramides, other sphingolipids and impaired glucose homeostasis. Mol Metab 2014; 3(3): 252–260.
18. Masi LN, Rodrigues AC, Curi R. Fatty acids regulation of inflammatory and metabolic genes. Curr Opin Clin Nutr Metab Care 2013; 16(4): 418–424.
19. Pelikanova T, Kohout M, Valek J et al. Insulin secretion and insulin action related to the serum phospholipid fatty acid pattern in healthy men. Metabolism 1989; 38(2): 188–192.
20. Pelikanova T, Kohout M, Hilgertova J et al. Erythrocyte insulin receptor characteristics and erythrocyte membrane lipid composition in healthy men. Physiol Bohemoslov 1989; 38(5): 419–425.
21. Storlien LH, Jenkins AB, Chisholm DJ et al. Influence of dietary fat composition on development of insulin resistance in rats. Relationship to muscle triglyceride and omega-3 fatty acids in muscle phospholipid. Diabetes 1991; 40(2): 280–289.
22. Borkman M, Storlien LH, Pan DA et al. The relation between insulin sensitivity and the fatty-acid composition of skeletal-muscle phospholipids. N Engl J Med 1993; 328(4): 238–244.
23. Pelikanova T, Kazdova L, Chvojkova S et al. Serum phospholipid fatty acid composition and insulin action in type 2 diabetic patients. Metabolism 2001; 50(12): 1472–1478.
24. Suchankova G, Vlasakova Z, Zicha J et al. Effect of acute hyperglycemia on erythrocyte membrane ion transport in offspring of hypertensive parents. J Hypertens 2003; 21(7): 1325–1330.
25. Kahleova H, Matoulek M, Bratova M et al. Vegetarian diet-induced increase in linoleic acid in serum phospholipids is associated with improved insulin sensitivity in subjects with type 2 diabetes. Nutr Diabetes 2007; 3: e75. Dostupné w DOI: <http://doi: 10.1038/nutd.2013.12>.
26. Kahleova H, Belinova L, Malinska H et al. Eating two larger meals a day (breakfast and lunch) is more effective than six smaller meals in a reduced-energy regimen for patients with type 2 diabetes: a randomised crossover study. Diabetologia 2014; 57(8): 1552–1560.
27. Bluher M. Adipokines – removing road blocks to obesity and diabetes therapy. Mol Metab 2014; 3(3): 230–240.
28. Eckardt K, Gorgens SW, Raschke S et al. Myokines in insulin resistance and type 2 diabetes. Diabetologia 2014; 57 (6): 1087–1099.
29. Chatzigeorgiou A, Karalis KP, Bornstein SR et al. Lymphocytes in obesity-related adipose tissue inflammation. Diabetologia 2012; 55(10): 2583–2592.
30. Hocking S, Samocha-Bonet D, Milner KL et al. Adiposity and insulin resistance in humans: the role of the different tissue and cellular lipid depots. Endocr Rev 2013; 34(4): 463–500.
31. Duarte AI, Candeias E, Correia SC et al. Crosstalk between diabetes and brain: glucagon-like peptide-1 mimetics as a promising therapy against neurodegeneration. Biochim Biophys Acta 2013; 1832(4): 527–541.
32. Cani PD, Everard A, Duparc T. Gut microbiota, enteroendocrine functions and metabolism. Curr Opin Pharmacol 2013; 13(6): 935–940.
33. Hong SH, Ahmadian M, Yu RT et al. Nuclear receptors and metabolism: from feast to famine. Diabetologia 2014; 57(5): 860–867.
34. Bass J. Circadian topology of metabolism. Nature 2012; 491 (7424): 348–356.
35. Novák J, Bienertová-Vašků J, Slabý O. MikroRNA a diabetes mellitus. In: Kvapil M (ed). Diabetologie 2014. Triton: Praha 2014 : 225–233. ISBN 978–80–7387–755–2.
36. Ridker PM, Howard CP, Walter V et al. Effects of interleukin-1beta inhibition with canakinumab on hemoglobin A1c, lipids, C-reactive protein, interleukin-6, and fibrinogen: a phase IIb randomized, placebo-controlled trial. Circulation 2012; 126(23): 2739–2748.
Labels
Diabetology Endocrinology Internal medicineArticle was published in
Internal Medicine

2014 Issue 9
Most read in this issue
- Životní prognóza osob s diabetem 1. typu dříve a dnes
- Gliptiny: bezpečná a účinná léčba diabetu
- Inzulinová rezistence – příčiny a možnosti ovlivnění
- AGEs a RAGE – konečné produkty pokročilé glykace a jejich receptor v otázkách a odpovědích