
Játra, ledviny a diabetes: tři tváře deficitu genu HNF1B
Liver, kidneys and diabetes: three faces of HNF1B gene deficit
The renal cysts and diabetes syndrome (RCAD), also known as HNF1B-MODYor MODY5, is caused by the deletion or point mutation of HNF1B gene which leads to the depletion of HNF1B transcription factor. The main clinical components of RCAD include cystic kidney disease or other developmental anomalies of the kidneys and diabetes mellitus which typically manifests in the second decade of life or later. Renal disorders may lead to the development of chronic renal insufficiency already in childhood or young adulthood. The other symptoms include hepatic impairment - cholestatic jaundice in middle-aged patients, sometimes even neonatal cholestasis, atrophy of the pancreas with the impairment of exocrine pancreatic secretion and some congenital anomalies of the genital tract. As opposed to the other forms of MODY diabetes, the family history may not be positive because most of the deviations of HNF1B appear de novo. We associate RCAD in particular with adults suffering from diabetes and cystic kidney disease and/or cholestatic jaundice and children with cystic kidney disease of unclear etiology, even without the presence of diabetes. A supportive finding may be hypomagnesemia which occurs in up to 70 % of patients diagnosed with HNF1B related disease and hyperuricemia.
Key words:
HNF1B – MODY – RCAD – diabetes mellitus – cholestatic jaundice
Authors:
Jan Lebl; Stanislava Koloušková; Petra Dušátková; Ondřej Cinek; Lenka Dušátková; Tomáš Dědič; Radana Kotalová; Zdeněk Šumník; Tomáš Seeman; Štěpánka Průhová
Authors‘ workplace:
Pediatrická klinika 2. LF UK a FN Motol Praha, přednosta prof. MUDr. Jan Lebl, CSc.
Published in:
Vnitř Lék 2014; 60(9): 725-729
Category:
Overview
Syndrom renálních cyst a diabetu (RCAD), zvaný také HNF1B-MODY či MODY5, je způsoben delecí nebo bodovou mutací genu HNF1B, která vede k depleci transkripčního faktoru HNF1B. Hlavními klinickými součástmi RCAD jsou cystické onemocnění ledvin nebo jiné vývojové anomálie ledvin a diabetes mellitus, který se projeví nejčastěji ve 2. dekádě života nebo později. Poruchy ledvin mohou vést k rozvoji chronické renální insuficience už v dětství nebo mladé dospělosti. K dalším příznakům patří poruchy funkce jater – cholestatický ikterus ve středním věku, ale někdy už novorozenecká cholestáza, atrofie pankreatu s poruchou exokrinní sekrece a některé vrozené anomálie pohlavního ústrojí. Na rozdíl od ostatních forem MODY diabetu rodinná anamnéza nemusí být pozitivní, protože většina odchylek HNF1B vzniká de novo. Na RCAD pomýšlíme zejména u dospělých osob s diabetem a cystickým onemocněním ledvin nebo cholestatickým ikterem a u dětí s cystickým onemocněním ledvin nejasné etiologie, i bez přítomnosti diabetu. Podpůrným nálezem může být hypomagneziemie, která se vyskytuje až u 70 % nositelů poruchy genu HNF1B, a hyperurikemie.
Klíčová slova:
diabetes mellitus – cholestatický ikterus – HNF1B – MODY – RCAD
Úvod
Hepatocytární nukleární faktory (HNF) jsou transkripční faktory, které řídí embryonální a fetální vývoj jater a diferenciaci intrahepatálních i extrahepatálních žlučových cest. Jejich celoživotní exprese v hepatocytech (HNF4A a HNF6) i v buňkách epitelu žlučových cest (HNF1B, HNF3A a HNF3B) nasvědčuje tomu, že regulují také postnatální funkci buněk, jejich přežívání a regeneraci [1–3]. Jeden z nich, hepatocytární nukleární faktor 1-β (HNF1B) kódovaný genem HNF1B, se podílí na transkripční regulaci a řízení funkce nejen jater a žlučových cest, ale také ledvin a urogenitálního traktu a v neposlední řadě B-buněk pankreatu. Do transkripční regulační sítě B-buněk patří i další dva HNF-HNF1A a HNF4A, jejichž poruchy vedou k častým formám diabetu MODY [4,5], ale poruchy funkce jater do jejich klinického obrazu nepatří nebo se projevují jen okrajově [6]. Chybění transkripčního faktoru HNF1B v důsledku mutace nebo delece genu HNF1B bylo nejdříve zjištěno u malé podskupiny pacientů s diabetem MODY a označeno jako MODY5, resp. HNF1B-MODY [7–9]. Protože prakticky všichni tito pacienti mají také cysty ledvin nebo jinou vývojovou odchylku urogenitálního systému, nazýváme dnes tuto poruchu RCAD (renal cysts and diabetes syndrome – syndrom renálních cyst a diabetu), tab. 1. Většina odchylek HNF1B jsou de novo vzniklé delece celého genu; proto se RCAD (na rozdíl od všech ostatních forem MODY) jen vzácně vyskytuje ve více generacích [10–13]. V posledních letech se objevují také zprávy o vztahu HNF1B k funkci jater a žlučových cest, zpravidla ve spojení s diabetem a cystami ledvin. Tím se otevírá nový pohled na vzájemnou souvislost diabetu a poruchy funkce ledvin a jater: klasické pojetí nefropatie a hepatopatie jako komplikací diabetu je v některých případech vystřídáno poznáním, že jde o paralelně probíhající klinické příznaky poruchy tohoto jednoho genu. Zatímco u ostatních forem diabetu MODY ze skupiny tzv. diabetu transkripčních faktorů (schéma, tab. 2) je diabetes prvním a hlavním příznakem, u RCAD se diabetes může přidružit až později jako nová součást nemoci u pacienta sledovaného pro poruchu ledvin či jater.

![Spektrum klinických příznaků při defektu genu HNF1B Procenta ukazují, u jakého podílu nositelů poruchy byl daný projev přítomen. Tučně jsou zvýrazněny nejvýznamnější příznaky. Upraveno dle [19,25].](https://www.prelekara.sk/media/cache/resolve/media_object_image_large/media/image/f20757ae5b99e4156fccbac8d2b035a6.png)
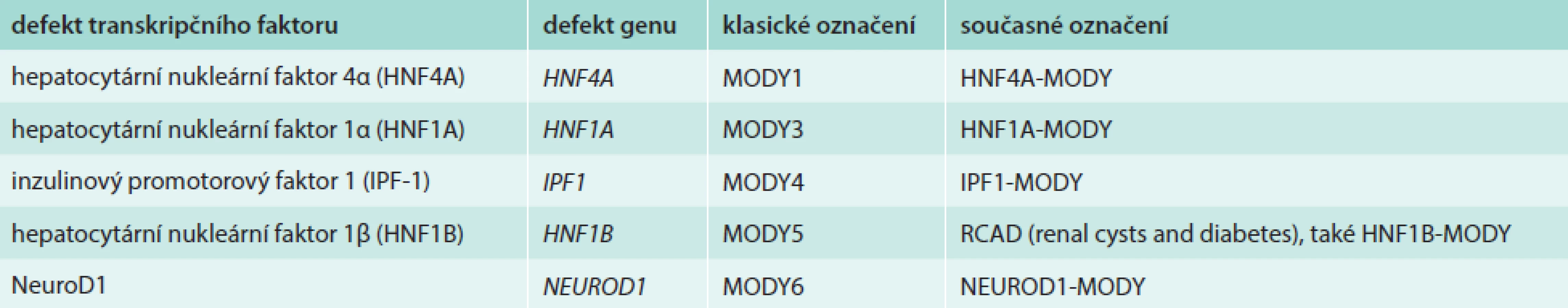
HNF1B a játra
Roelandt et al [14] popsali kazuistiky 3 dospělých pacientů (2 ženy, 1 muž), u kterých diabetes vznikl ve věku 14–51 let. Všichni měli příznaky cholestatického ikteru, a to od věku 29, 30 a 53 let. Laboratorní známky cholestázy postupně progredovaly. U jednoho z nich byla za příčinu biliární obstrukce pokládána chronická pankreatitida, v té době trvající 9 let, u druhého alkoholem indukovaná hepatopatie. Biopsie jater byla v jednom případě zhodnocena jako nespecifické změny s mírnou steatózou, u dvou pacientů byla zjištěna lehká dilatace sinusů, z toho jednou ve spojení se ztluštěním bazální membrány kolem žlučových vývodů. Všichni měli počínající renální insuficienci připisovanou diabetické nefropatii. Další vyšetření ukázala hypomagnezemii a ledvinné cysty. U všech byla ultrazvukem a magnetickou rezonancí také prokázána zmenšená velikost („atrofie“) pankreatu.
Díky souboru podobných příznaků bylo všem provedeno vyšetření genu HNF1B jak metodou MLPA, která detekuje deleci celého genu, tak přímou sekvenací. U 33leté ženy byla příčinou diabetu, biliární cholestázy, ledvinných cyst a hypoplazie pankreatu delece v rozsahu 1423 kB v kritické oblasti 17. chromozomu (17q12) se ztrátou celého genu HNF1B; u 53letého muže se stejným spektrem příznaků a mylnou diagnózou 9 let trvající chronické pankreatitidy delece stejné oblasti v rozsahu 1427 kB. U 30leté ženy s cholestatickým ikterem, diabetem, hypoplazií pankreatu a cystami ledvin byla zjištěna bodová mutace HNF1B c.544C>T.
I když výsledek molekulárně-genetického vyšetření v těchto případech zpravidla nezmění léčbu, přinese racionální pohled na pacienta a jednotlivé součásti jeho nemoci. Navíc lze konstatovat 50% riziko přenosu nemoci na potomky probanda.
U 3 nepříbuzných novorozenců a kojenců vedl defekt genu HNF1B ke vzniku cholestatického ikteru od prvních dnů či týdnů života [15–17]. Všichni prodělali intrauterinní růstovou retardaci a rodili se malí vzhledem ke gestačnímu věku (SDS porodní hmotnosti ve vztahu ke gestačnímu věku v rozmezí -2,08 až -3,70). Měli mnohočetné kortikální ledvinné cysty velikosti od 10 do 20 mm, u jednoho ve spojení s jednostrannou agenezí ledviny a u jednoho se stenózou uretry. Cholestatický ikterus během 2. poloviny prvního roku života postupně ustoupil, známky hepatopatie ale přetrvávaly. Během prvních dvou desetiletí života se u všech rozvinula chronická renální insuficience. U 2 pacientů se projevila postupná atrofizace pankreatu a úbytek zevní pankreatické sekrece s nutností pankreatické substituce u jednoho z nich. Klinické příznaky diabetu nastoupily mezi 5 a 13 lety věku. Diabetes je kompenzován při denní dávce inzulinu 0,40–1,34 IU/kg. Příčinou nemoci byla v jednom případě záměna skupiny bazí uvnitř HNF1B genu c.499_504delGCTCTGinsCCCCT, ve druhém bodová mutace c.457C>A a ve třetím delece celého genu a přilehlé oblasti v 17q12 rozsahu 1590 kB.
Mírnější jaterní fenotyp (chronicky zvýšené hodnoty AST a ALT bez progrese) byl pozorován u několika dalších nositelů mutací HNF1B s RCAD [12,17,18].
HNF1B a ledviny
Poruchy vývoje ledvin mají téměř všichni pacienti (90–100 %) se změnou v HNF1B [19]. Nejčastější jsou renální cysty (70–80 %), tíže postižení ale kolísá od prostých renálních cyst přes cystickou dysplazii ledvin, multicystickou dysplazii až po glomerulocystickou chorobu ledvin.
Prvním projevem ledvinné léze může být prenatálně zjištěná hyperechogenita ledvin (60–96 % případů) [19,20]. V nitroděložním období nebývají ledviny zvětšené a cysty nejsou viditelné, ale po narození je lze ultrasonograficky rozpoznat. Anomálie HNF1B jsou nejčastější příčinou prenatálně zjištěných oboustranně hyperechogenních ledvin (29 %). Vzácně vede anomálie HNF1B k jednostranné multicystické dysplazii ledviny (multicystic dysplastic kidney – MCDK), přičemž je téměř vždy strukturálně postižena i druhá, funkčně solitární ledvina [20,21]. Nejvzácnějším ledvinným projevem poruchy HNF1B je familiární hypoplastická forma glomerulocystické choroby ledvin (hypoplastic familial glomerulocystic kidney disease – FGCKD). Tu lze prokázat pouze biopsií ledviny.
U malé části pacientů vede porucha HNF1B k hypo-dysplazii ledvin bez cyst (3–10 % pacientů) [19,22,23], k agenezi nebo hypoplazii jedné ledviny, ke zdvojené či podkovovité ledvině, případně k vrozené anomálii vývodných močových cest, např. vezikoureterálnímu refluxu [24]. Téměř vždy je patologicky změněná i kontralaterální ledvina, postižení ledvin je typicky oboustranné.
Funkce ledvin může být při RCAD normální, ale většinou je snížená a může vést již v dětském věku k terminálnímu chronickému selhání ledvin, které vyžaduje dialýzu a transplantaci ledviny (13–20 %) [19].
Pacienti s poruchou HNF1B mají často hypomagneziemii (44–67 %) [24] v důsledku snížené tubulární resorpce magnezia. Příčinou je porušená transkripce genu FXYD2, jehož produkt ovlivňuje tubulární resorpci magnezia. Hypomagneziemie je empirickým skríningovým testem pro diferenciální diagnostiku ledvinných cyst.
HNF1B, diabetes a pankreas
Součástí syndromu RCAD je diabetes, který patří do skupiny „diabetu transkripčních faktorů“ (tab. 2, obr. 1). Na rozdíl od dalších forem diabetu MODY se diabetes při RCAD často objevuje u pacienta v rodině poprvé (až v 50 % případů je příčinou RCAD de novo vzniklá delece celého genu), poměrně pozdě (typicky od 2. poloviny druhého decennia) a je často zjištěn náhodně jako glykosurie při vyšetření moči z jiného důvodu. Pankreas je strukturálně menší, je redukována jeho endokrinní i exokrinní tkáň a většina pacientů má subklinickou exokrinní pankreatickou insuficienci, která jen vzácně vyžaduje léčbu [7–9]. Autoprotilátky jsou při diabetu negativní.
Lékem volby diabetu při RCAD je obecně inzulin. Nicméně úbytek vlastní sekrece inzulinu se rozvíjí zvolna a může být dlouho kompenzován dietou; u mladých osob následně volíme deriváty sulfonylurey, které mohou mít po jistou dobu dobrý efekt. V další fázi je k udržení dobré kompenzace diabetu nutné přejít na léčbu inzulinem. V dětství a dospívání může být provokujícím momentem, kdy se projeví diabetes, léčba růstovým hormonem nebo kortikoidy.
HNF1B a další orgány
Postižení dalších orgánů je při RCAD vzácné (1–10 %). Mohou se vyskytnout vývojové odchylky pohlavního ústrojí jak u dívek a žen (dělohy, vaginy, ovarií), tak u chlapců a mužů (kryptorchizmus, testikulární hypoplazie, epididymální cysty, hypospadie). Mentální retardace, opoždění neuropsychického vývoje či autizmus se objevují spíše u osob s rozsáhlejší delecí v kritické oblasti chromozomu 17q12, které zahrnují také jiné geny v sousedství HNF1B [17,19].
Celkově lze říci, že fenotyp pacientů s poruchou genu HNF1B může být velmi pestrý, jsou možné nejrůznější kombinace nebo jen izolované projevy. Z prvních výsledků analýzy českých pacientů s defektem HNF1B vyplývá (zatím nepublikované výsledky), že se nijak výrazně neliší tíže projevů u pacientů s bodovou mutací či rozsáhlou delecí zahrnující vedle HNF1B i řadu sousedících genů. HNF1B tedy hraje nejvýraznější roli. Nicméně lze vypozorovat, že ve fenotypu vždy dominuje jedna složka výrazněji nad jinou. Např. pacienti s multicystickým onemocněním ledvin a tendencí k renálnímu selhání nemají zatím velký výskyt diabetu. Oproti tomu pacienti s manifestovaným diabetem (medián věku manifestace diabetu u českých pacientů je 17 let) mají často jen diabetes či kombinaci s nevýznamnou změnou ledvin, např. charakteru solitární cysty či podkovovité ledviny.
Kdy pomýšlet na RCAD?
RCAD je pravděpodobný u pacienta léčeného pro diabetes od pozdního dětství, adolescence či mladé dospělosti, který má současně renálními cysty, případně jiné anomálie ledvin. Podpůrným nálezem je nízká hladina magnezia v séru, případně hyperurikemie. Funkce ledvin může být normální nebo snížená.
RCAD může vést u dospělých s diabetem a cystami ledvin k biliární obstrukci či hepatopatii zdánlivě nejasné etiologie.
U dětí na RCAD pomýšlíme i před vznikem diabetu při prenatálním nálezu hyperechogenních ledvin, při cystickém onemocněními ledvin nejasné etiologie (cystické dysplazii ledvin, polycystickém onemocnění ledvin, prostých cystách ledvin), při multicystické dysplazii ledviny s jakýmkoliv poškozením kontralaterální ledviny, při agenezi ledviny s jakýmkoliv poškozením kontralaterální ledviny, při hypomagneziemii spojené se strukturálním poškozením ledvin (cystickým i necystickým) nebo při cystickém onemocnění ledvin, které je provázeno také anomálií pankreatu, jater nebo pohlavních orgánů.
Stejné nebo podobné postižení ledvin nebo pankreatu u jednoho z rodičů pacienta zvyšuje pravděpodobnost RCAD, avšak většina abnormit HNF1B vzniká de novo. Negativní rodinná anamnéza tedy RCAD nevylučuje.
Diagnózu RCAD potvrdí nález delece nebo mutace HNF1B genu. Tato diagnostika je po předcházející konzultaci příslušné indikace k dispozici na pracovišti autorů.
Studie HNF1B genu jsou podpořeny granty IGA MZ ČR NT11457 a NT11402 a výzkumným projektem MZ ČR koncepčního vývoje výzkumné organizace 00064203 (FN Motol).
prof. MUDr. Jan Lebl, CSc.
jan.lebl@lfmotol.cuni.cz
Pediatrická klinika 2. LF UK a FN Motol, Praha
www.fnmotol.cz
Doručeno do redakce 1. 6. 2014
Přijato po recenzi 24. 7. 2014
Sources
1. Limaye PB, Alarcón G, Walls AL et al. Expression of specific hepatocyte and cholangiocyte transcription factors in human liver disease and embryonic development. Lab Invest 2008; 88(8): 865–872.
2. Raynaud P, Carpentier R, Antoniou A et al. Biliary differentiation and bile duct morphogenesis in development and disease. Int J Biochem Cell Biol 2011; 43(2): 245–256.
3. Strazzabosco M, Fabris L. Development of the bile ducts: Essentials for the clinical hepatologist. J Hepatol 2012; 56(5): 1159–1170.
4. Pruhova S, Ek J, Lebl J et al. Genetic epidemiology of MODY in the Czech Republic: Novel mutations in the MODY genes HNF-4a, GCK and HNF-1a. Diabetologia 2003; 46(2): 291–295.
5. Hansen SK, Parrizas M, Jensen ML et al. Genetic evidence that HNF-1alpha-dependent transcriptional control of HNF-4alpha is essential for human pancreatic beta cell function. J Clin Invest 2002; 110(6): 827–833.
6. Pearson ER, Pruhova S, Tack CJ et al. Molecular genetics and phenotypic characteristics of MODY caused by hepatocyte nuclear factor 4alpha mutations in a large European collection. Diabetologia 2005; 48(5): 878–885.
7. Horikawa Y, Iwasaki N, Hara M et al. Mutation in hepatocyte nuclear factor-1 beta gene (TCF2) associated with MODY. Nat Genet 1997; 17(4): 384–385.
8. Nishigori H, Yamada S, Kohama T et al. Frameshift mutation, A263fsinsGG, in the hepatocyte nuclear factor-1beta gene associated with diabetes and renal dysfunction. Diabetes 1998; 47(8): 1354–1355.
9. Beards F, Frayling T, Bulman M et al. Mutations in hepatocyte nuclear factor 1beta are not a common cause of maturity-onset diabetes of the young in the U.K. Diabetes 1998; 47(7): 1152–1154.
10. Lindner TH, Njolstad PR, Horikawa Y et al. A novel syndrome of diabetes mellitus, renal dysfunction and genital malformation associated with a partial deletion of the pseudo-POU domain of hepatocyte nuclear factor-1beta. Hum Mol Genet 1999; 8(11): 2001–2008.
11. Kolatsi-Joannou M, Bingham C, Ellard S et al. Hepatocyte nuclear factor-1beta: a new kindred with renal cysts and diabetes and gene expression in normal human development. J Am Soc Nephrol 2001; 12(10): 2175–2180.
12. Montoli A, Colussi G, Massa O et al. Renal cysts and diabetes syndrome linked to mutations of the hepatocyte nuclear factor-1 beta gene: description of a new family with associated liver involvement. Am J Kidney Dis 2002; 40(2): 397–402.
13. Bingham C, Hattersley AT Renal cysts and diabetes syndrome resulting from mutations in hepatocyte nuclear factor-1beta. Nephrol Dial Transplant 2004; 19(11): 2703–2708.
14. Roelandt P, Antoniou A, Libbrecht L et al. HNF1B deficiency causes ciliary defects in human cholangiocytes. Hepatology 2012; 56(3): 1178–1181.
15. Kitanaka S, Miki Y, Hayashi Y et al. Promoter-specific repression of hepatocyte nuclear factor (HNF)-1 beta and HNF-1 alpha transcriptional activity by an HNF-1 beta missense mutant associated with Type 5 maturity-onset diabetes of the young with hepatic and biliary manifestations. J Clin Endocrinol Metab 2004; 89(3): 1369–1378.
16. Beckers D, Bellanné-Chantelot C, Maes M. Neonatal cholestatic jaundice as the first symptom of a mutation in the hepatocyte nuclear factor-1beta gene (HNF-1beta). J Pediatr 2007; 150(3): 313–314.
17. Raile K, Klopocki E, Holder M et al. Expanded clinical spectrum in hepatocyte nuclear factor 1b-maturity-onset diabetes of the young. J Clin Endocrinol Metab 2009; 94(7): 2658–2664.
18. Gonc EN, Ozturk BB, Haldorsen IS et al. HNF1B mutation in a Turkish child with renal and exocrine pancreas insufficiency, diabetes and liver disease. Pediatr Diabetes 2012; 13(2): e1-e5.
19. Chen YZ, Gao Q, Zhao XZ et al. Systematic review of TCF2 anomalies in renal cysts and diabetes syndrome/maturity onset diabetes of the young type 5. Chin Med J (Engl) 2010; 123(22): 3326–3333.
20. Heidet L, Decramer S, Pawtowski A et al. Spectrum of HNF1B mutations in a large cohort of patients who harbor renal diseases. Clin J Am Soc Nephrol 2010; 5(6): 1079–1090.
21. Ulinski T, Lescure S, Beaufils S et al. Renal phenotypes related to hepatocyte nuclear factor-1beta (TCF2) mutations in a pediatric cohort. J Am Soc Nephrol 2006; 17(2): 497–503.
22. Weber S, Moriniere V, Knüppel T et al. Prevalence of mutations in renal developmental genes in children with renal hypodysplasia: results of the ESCAPE study. J Am Soc Nephrol 2006; 17(10): 2864–2870.
23. Thomas R, Sanna-Cherchi S, Warady BA et al. HNF1B and PAX2 mutations are a common cause of renal hypodysplasia in the CKiD cohort. Pediatr Nephrol 2011; 26(6): 897–903.
24. Adalat S, Woolf AS, Johnstone KA et al. HNF1B mutations associate with hypomagnesemia and renal magnesium wasting. J Am Soc Nephrol 2009; 20(5): 1123–1131.
25. Faguer S, Decramer S, Chassaing N et al. Diagnosis, management, and prognosis of HNF1B nephropathy in adulthood. Kidney Int 2011; 80(7): 768–776.
Labels
Diabetology Endocrinology Internal medicineArticle was published in
Internal Medicine

2014 Issue 9
Most read in this issue
- Životní prognóza osob s diabetem 1. typu dříve a dnes
- Gliptiny: bezpečná a účinná léčba diabetu
- Inzulinová rezistence – příčiny a možnosti ovlivnění
- AGEs a RAGE – konečné produkty pokročilé glykace a jejich receptor v otázkách a odpovědích