
Hodnocení pětileté léčby Erdheimovy-Chesterovy nemoci anakinrou – kazuistika a přehled literatury
Evaluation of five years of treatment of Erdheim-Chester disease with anakinra: case report and overview of literature
Erdheim-Chester disease is a histiocytic neoplasm of diseases from the group of non-Langerhans-cell histiocytoses, formed by infiltrates of foamy histiocytes. These pathological histiocytes produce pro-inflammatory cytokines. Therefore Erdheim-Chester disease is called inflammatory histiocytary neoplasm. The disease is accompanied by clinical symptoms of systemic inflammatory response, i.e. B symptoms. Imaging examinations detect typical osteosclerotic changes affecting diaphyses and metaphyses of the lower long bones and fibrotic changes which affect the aorta wall and the vessels leading from it. Also characteristic are perirenal fibrotic changes spreading in the retroperitoneum. They can cause serious complications – hydronephrosis with all its consequences. The therapy for this disease was not satisfactory in the previous years. Conventional chemotherapy or glucocorticoids do not bring any substantial and long-term improvement. Considering cytostatic drugs, only 2-chlorodeoxyadenosine (cladribine) is effective, though not in all patients. We have only reached complete remission through 2-chlorodeoxyadenosine in one of our two patients, which now lasts more than 5 years, while cladribine in the same patient did effect the reduction of infiltrates into the CNS, but it did not achieve abatement of the disease activity in other locations as shown by PET/CT with the application of the radio-pharmaceutical fluorodeoxyglucose (FDG). Another effective medicine for patients with Erdheim-Chester disease is interferon α. However its long-term administration is associated with multiple adverse effects and so we did not test it in the described patient. The introduction of anakinra, the interleukin-1 receptor blocker, to therapy brought a new hope for these patients. We are describing the patient who has been treated with anakinra for more than 5 years. The patient applies 1 ampoule of 100 mg subcutaneously per day. This treatment completely removed systemic B symptoms, relieved bone pains and attained normalization of all findings that signalled systemic inflammatory response. The treatment effect is regularly checked by CT imaging of the abdomen and by FDG-PET/CT examinations. The retroperitoneal fibrotic changes gradually regressed during the 5 years of anakinra treatment, as documented by the pictures in the text. Low-dose CT imaging which was part of the PET/CT examination, identified many osteosclerotic lesions in the skeleton, mainly in the legs, with an increased accumulation of 18F-fluorodeoxyglucose (FDG). Osteosclerotic lesions remain well visible at repeated examinations. Still during the course of the 5-year period the FDG accumulation in them decreased, as shown by the pictures in the text. Anakinra treatment has a character of maintenance therapy. The BRAFV600E mutation was not proven in the described patient, therefore we did not test vemurafenib treatment.
Conclusion:
anakinra effected regression of fibrotic changes in the retroperitoneum and disappearance of B symptoms as well as decrease in FDG accumulation at FDG-PET/CT examination.
Key words:
anakinra – Erdheim-Chester disease – cladribine – retroperitoneal fibrosis – vemurafenib
Authors:
Zdeněk Adam 1; Hana Petrášová 2; Zdeněk Řehák 3; Renata Koukalová 3; Marta Krejčí 1; Luděk Pour 1; Eva Vetešníková 1; Aleš Čermák 4; Sabina Ševčíková 5; Petr Szturz 1; Zdeněk Král 1; Jiří Mayer 1
Authors‘ workplace:
Interní hematologická a onkologická klinika LF MU a FN Brno, pracoviště Bohunice
1; Radiologická klinika LF MU a FN Brno, pracoviště Bohunice
2; Oddělení nukleární medicíny, centrum PET, RECAMO, Masarykův onkologický ústav, Brno
3; Urologická klinika LF MU a FN Brno, pracoviště Bohunice
4; Ústav patologické fyziologie LF MU, Brno
5
Published in:
Vnitř Lék 2016; 62(10): 820-832
Category:
Case Reports
Overview
Erdheimova-Chesterova nemoc je histiocytární neoplazie ze skupiny non-Langerhans-cell histiocytóz, je tvořena infiltráty pěnitých histiocytů. Tyto patologické histiocyty produkují prozánětlivé cytokiny. Proto je Erdheimova-Chesterova choroba nazývána inflamatorní histiocytární neoplazií. Nemoc je provázena klinickými projevy systémové zánětlivé odpovědi neboli B symptomy. Při zobrazovacích vyšetřeních jsou typické osteosklerotické změny postihující diafýzy a metafýzy dlouhých kostí dolních končetin a dále fibrózní změny postihující stěnu aorty a cévy z ní odstupující. Dále jsou typické perirenální fibrotické změny, které se šíří v retroperitoneu. Mohou způsobit závažné komplikace – hydronefrózu se všemi jejími důsledky. Léčba této nemoci nebyla v přechozích letech uspokojivá. Klasická chemoterapie ani glukokortikoidy nepřinášejí zásadní a dlouhodobé zlepšení. Z cytostatik je účinný pouze 2-chlorodeodyadenozin (kladribin), ale ne u všech pacientů. Pouze u jednoho z našich 2 pacientů jsme dosáhli 2-chlorodeoxyadenozinem kompletní remise, která trvá více než 5 let, zatímco u popisovaného pacienta vedl kladribin sice ke zmenšení infiltrátů v CNS, ale nevedl k ústupu aktivity nemoci v jiných lokalizacích dle PET/CT s aplikací radiofarmaka fluorodeoxyglukózy (FDG). Dalším účinným lékem pro pacienty s Erdheimovou-Chesterovou nemocí je interferon α. Jeho dlouhodobé podávání je však spojeno s četnými nežádoucími účinky a u popisovaného pacienta jsme jej proto nezkoušeli. Zavedení blokátoru receptoru interleukinu 1, anakinry, do léčby přineslo novou naději pro tyto nemocné. Popisujeme pacienta, u něhož probíhá léčba anakinrou již více než 5 let. Pacient si aplikuje 1 ampulku, 100 mg podkožně denně. Tato léčba úplně odstranila systémové B symptomy a odstranila bolesti kostí a vedla k normalizaci všech nálezů, které signalizovaly systémovou zánětlivou reakci. Efekt léčby je pravidelně sledován CT zobrazením břišní dutiny a FDG-PET/CT vyšetřením. V průběhu 5 let léčby anakinrou postupně ustupovaly fibrotické změny v retroperitoneu, jak dokumentují obrázky v textu. Při low-dose CT zobrazení, které bylo součástí PET/CT vyšetření, byla patrná četná osteosklerotická ložiska ve skeletu, zvláště v dolních končetinách, se zvýšenou akumulací 18F-fluorodeoxyglukózy (FDG). Osteosklerotická ložiska zůstávají při opakovaných vyšetřeních stále dobře zřetelná. V průběhu 5 let se ale akumulace FDG v nich snížila, jak dokumentují obrázky v textu. Léčba anakinrou má charakter udržovací léčby. U popsaného pacienta se nepodařilo prokázat mutaci BRAFV600E, takže léčbu vemurafenibem jsme netestovali.
Závěr:
Anakinra vedla k ústupu fibrózních změn v retroperitonea a vymizení B symptomů a k poklesu akumulace FDG při FDG-PET/CT vyšetření.
Klíčová slova:
anakinra – Erdheimova-Chesterova choroba – kladribin – retroperitoneální fibróza – vemurafenib
Úvod
Erdheimova-Chesterova nemoc je vzácnou nozologickou jednotkou ze skupiny non-Langerhans-cell histiocytárních chorob. První popis této choroby zveřejnili Jakob Erdheim a William Chester v roce 1930 [1].
Erdheimova-Chesterova choroba (Erdheim-Chester Disease – ECD) je definována typickým histologickým obrazem xantogranulomového ložiska s infiltrací pěnitými histiocyty CD68 pozitivními a CD1a negativními, s fibrózou a xantogranulomatózou a typickou kortikální sklerózou či hyperostózou dlouhých kostí dolních končetin. Kostní změny jsou dobře viditelné na scintigrafii skeletu nebo na CT zobrazení. Absence těchto kostních změn je zcela výjimečná, ale je možná. Pak je nutno pátrat po dalších projevech nemoci, tak jak jsou v textu popsány.
Klinické symptomy této nemoci se mohou měnit případ od případu a snad jediným společným příznakem, který provází všechny nemocné s touto nemocí, je intenzivní systémová zánětlivá reakce. Proto je tato choroba též popisována jako inflamatorní myeloidní neoplazie, či přesněji inflamatorní histiocytární neoplazie. Systémová zánětlivá reakce se projevuje jako B symptomy (subfebrilie či febrilie, noční poty, úbytek hmotnosti a patologická únava).
Na diagnózu je možné pojmout podezření, pokud u nemocného s dlouhodobě zvýšenou hodnotou CRP a s dalšími projevy systémové zánětlivé reakce provedeme PET/CT vyšetření s radiofarmakem fluorodeoxyglukózou (FDG-PET/CT), anebo scintigrafii skeletu Tc-pyrofosfátem a zjistíme typickou aktivitu radiofarmaka v dlouhých kostech dolních končetin anebo pokud CT zobrazení odhalí fibrotické změny v retroperitoneu kolem ledvin. Druhým nutným krokem je pak odebrání vzorku k histologickému vyšetření, nejlépe z ložiska, které nejvíce akumuluje FDG.
Samotná histologie xantogranulomového ložiska není pro stanovení diagnózy dostačující, protože podobný histologický obraz má také juvenilní xantogranulom, který se manifestuje dominantně v dětství a tvoří nejčastěji solitární, chirurgicky odstranitelná ložiska. Vícečetné či generizované formy juvenilního xantogranulomu ohrožující život jsou u pediatrických pacientů velmi vzácné.
WHO klasifikace krevních chorob obsahuje morfologickou jednotku xantogranulom, která má různé klinické podoby. Erdheimova-Chesterova choroba je jednou z nich, s typickým orgánovým poškozením a s typickými zánětlivými projevy [2,3].
Léčba těchto nemocných se postupně vyvíjí, četné příznaky lze utlumit či odstranit preparátem anakinra, což je hlavním tématem našeho sdělení. ECD je velmi vzácně diagnostikovaná choroba, a proto se v české a slovenské medicínské literatuře této nemoci věnuje jen několik publikací [4–10].
V následujícím sdělení popisujeme dlouhodobé (5leté) zkušenosti s léčbou této nemoci inhibitorem receptoru pro interleukin 1, anakinrou.
Popis případu
Muž, narozený roku 1965, byl až do roku 2004 (do svých 39 let) zcela zdráv. Prvním příznakem nemoci byla polydipsie a polyurie. Diagnóza centrálního diabetes insipidus byla potvrzena koncentračním testem v září roku 2004. Správná diagnóza této nemoci byla však stanovena až koncem roku 2008. Podrobný popis rozvoje této nemoci a postupy i omyly při stanovaní diagnózy jsme opakovaně zveřejnili [11,12], a proto se v tomto popisu nemocného soustředíme na hodnocení 5 let, kdy byl kontinuálně léčen preparátem anakinra (Kineret) v dávce 100 mg podkožně denně. Většina doposud publikovaných prací totiž hodnotila jen krátké období léčby anakinrou.
Po prostudování publikovaných informací o terapii této nemoci jsme v rámci léčby první linie podali 2-chlorodeoxyadenozin (kladribin). Efektem léčby bylo zmenšení infiltrátů v CNS. Dle FDG-PET/CT zobrazení však zůstávala akumulace fluorodeoxyglukózy v dlouhých kostech a v dalších patologických ložiscích stejná. V rámci druhé linie léčby jsme v rámci programu CUP (Compassionate Use Program společnosti CELGENE určený pro pacienty se vzácnými hematoonkologickymi onemocněními) použili léčbu lenalidomidem. V průběhu léčby lenalidomidem došlo k dalšímu zmenšení infiltrátů v CNS. Zásadní pro hodnocení aktivity nemocni je však celotělové FDG-PET/CT zobrazení. A dle PET/CT zobrazení nevedla u tohoto pacienta léčba 2-chlorodeoxyadenozinem ani lenalidomidem ke snížení akumulace fluorodeoxyglukózy v extrakraniálních patologických ložiscích. Léčbu jak 2-chlorodeoxyadenozinem, tak později lenalidomidem snášel pacient bez vedlejších nežádoucích účinků. Po celou dobu však pacient trpěl závažnou patologickou únavou a byla zřetelná fibróza v oblasti retroperitonea a velkých cév.
Dne 21. 3. 2011 jsme u pacienta po podepsání informovaného souhlasu s touto léčbou zahájili denní podávání podkožních injekcí anakinry (preparátu KineretTM, 100 mg denně). Tato léčba probíhá s určitými pauzami, způsobenými administrativními příčinami při zajišťování léku, nyní již 5 let.
Již 2. den po zahájení léčby anakinrou udával pacient výrazné zlepšení celkového stavu a zmenšení patologické únavy. Značně se prodloužil čas, který pacient trávil mimo lože. Toto zlepšení trvá stále, pokud je podávána anakinra. Po přerušení aplikace anakinry (z administrativních důvodů) se navrací velká patologická únava.
Laboratorní hodnocení
Prozánětlivá aktivita cytokinů produkovaných patologickými fagocytujícími histiocyty Erdheimovy-Chesterovy nemoci zvyšuje hodnotu C-reaktivního proteinu (CRP), sedimentaci erytrocytů a jsou zřetelné i změny dalších parametrů, které se mění při systémové zánětlivé odpovědi organizmu. Před zahájením léčby byla opakovaně prokázána vyšší hodnota CRP mezi 20–30 mg/l (norma 0–5 mg/l) a zvýšené byly i další laboratorní parametry, které se zvyšují při systémové zánětlivé odpovědi. Před podáním anakinry byla zvýšena hodnota fibrinogenu na 5,10 g/l (norma 1,8–4,2 g/l) a mírně vyšší byla i hodnota trombocytů 343 × 109/l (norma 150–350 × 109/l), zatímco hodnota hemoglobinu byla mírně snížená kolem 122–130 g/l (norma 135–175 g/l).
Ihned po podání anakinry došlo k normalizaci hodnot CRP, které od té doby zůstávají v normě. Upravily se i ostatní parametry provázející systémovou zánětlivou odpověď, koncentrace hemoglobinu se postupně zvyšovala a v posledních letech se udržuje kolem hodnoty 150 g/l, zatímco počet trombocytů mírně poklesl na hodnoty 194 × 109/l. Poklesla i původně zvýšená hodnota fibrinogenu na normální hodnotu 2,33 g/l.
Při léčbě anakinrou došlo tedy k normalizaci všech původně zvýšených hodnot signalizujících probíhající systémovou zánětlivou reakci.
Vývoj retroperitoneální fibrózy při léčbě anakinrou
V lednu roku 2009, před nasazením léčby anakinrou, podstoupil pacient multidetektorové CT vyšetření (MDCT) hrudníku a břicha s nálezem nepravidelně zesílené stěny sestupné hrudní aorty, šíře stěny aorty dosahuje až 8,4 mm. Zesílení je podmíněno hypodenzním, hypovaskularizovaným infiltrátem patrným zejména v dorzální porci aorty, lemovitě obkružuje lumen do tří čtvrtin obvodu. Rovněž břišní aorta má obdobně nepravidelně zesílenou stěnu šíře do 6 mm (kraniálně pouze zadní stěna, kaudálněji zesílení cirkulárně), s přestupem i na odstupy renálních tepen, dolní mezenterické tepny a společných ilických tepen. Dále byly patrné zvýšené denzity tuku v předním i zadním pararenálním prostoru, hypodenzní neostře ohraničený infiltrát byl lokalizován oboustranně v oblasti ledvinných hilů, bez dilatace vývodného systému ledvin. Infiltrát okrajově zasahoval i do okolí nadledvin. Změny odpovídají extraoseálnímu postižení při známé diagnóze Erdheimovy-Chesterovy choroby. Rozsah fibrotického postižení velkých cév hrudníku i břicha a perirenální fibrotické změny před zahájením léčby anakinrou dokumentují obr. 1–5.
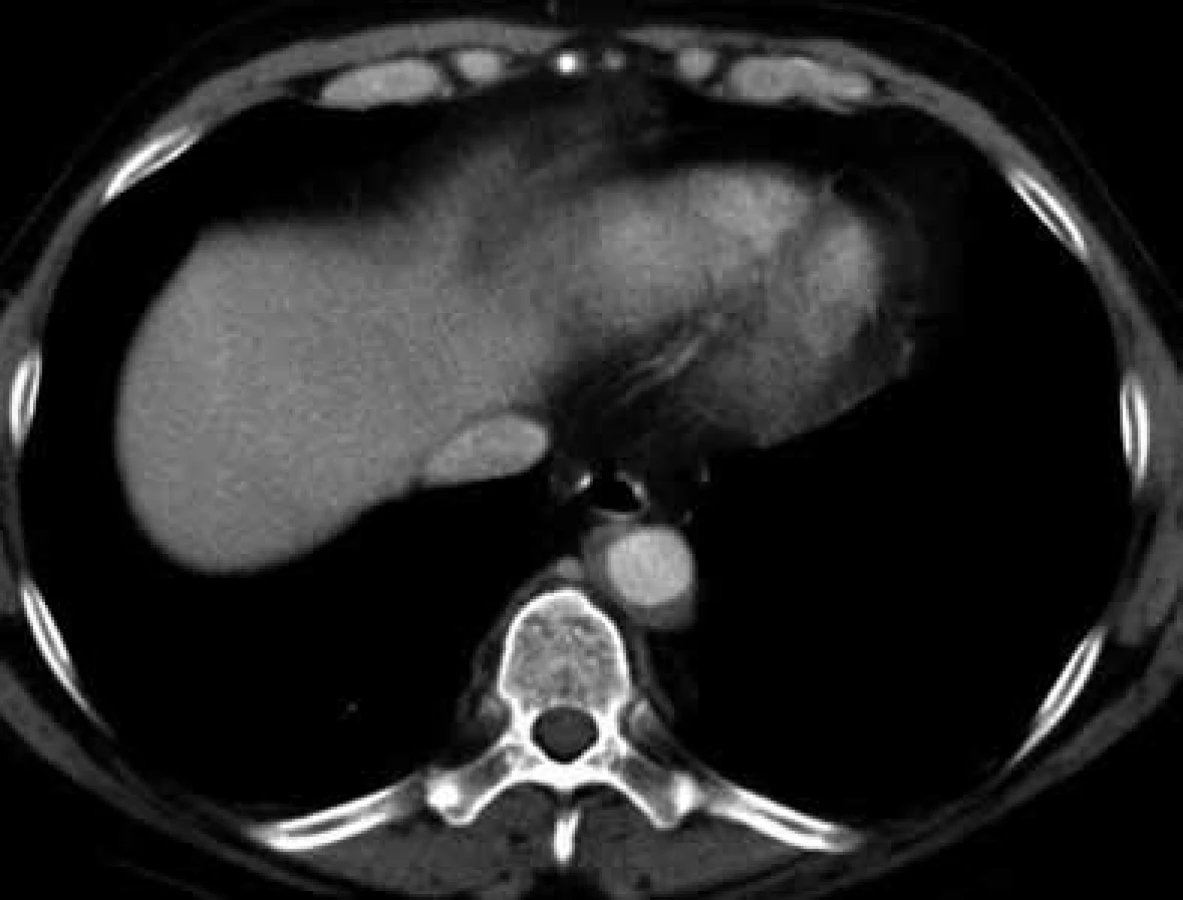


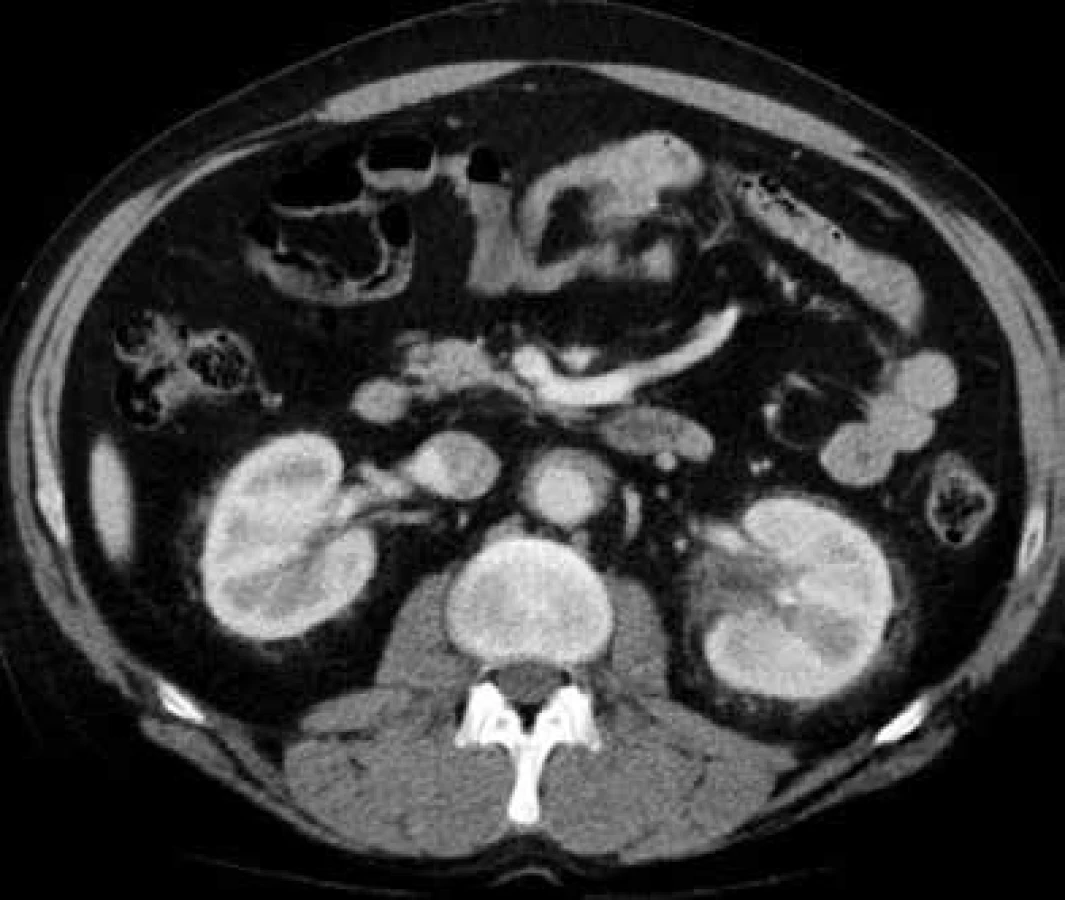
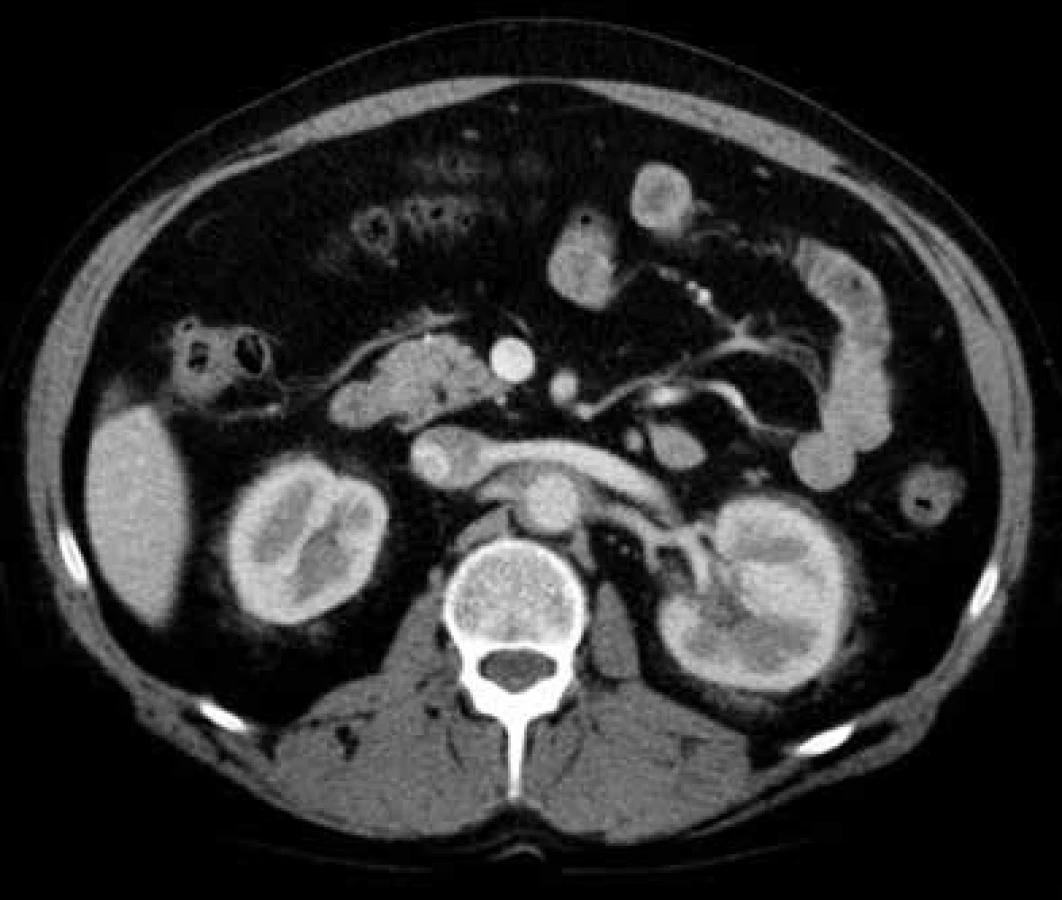
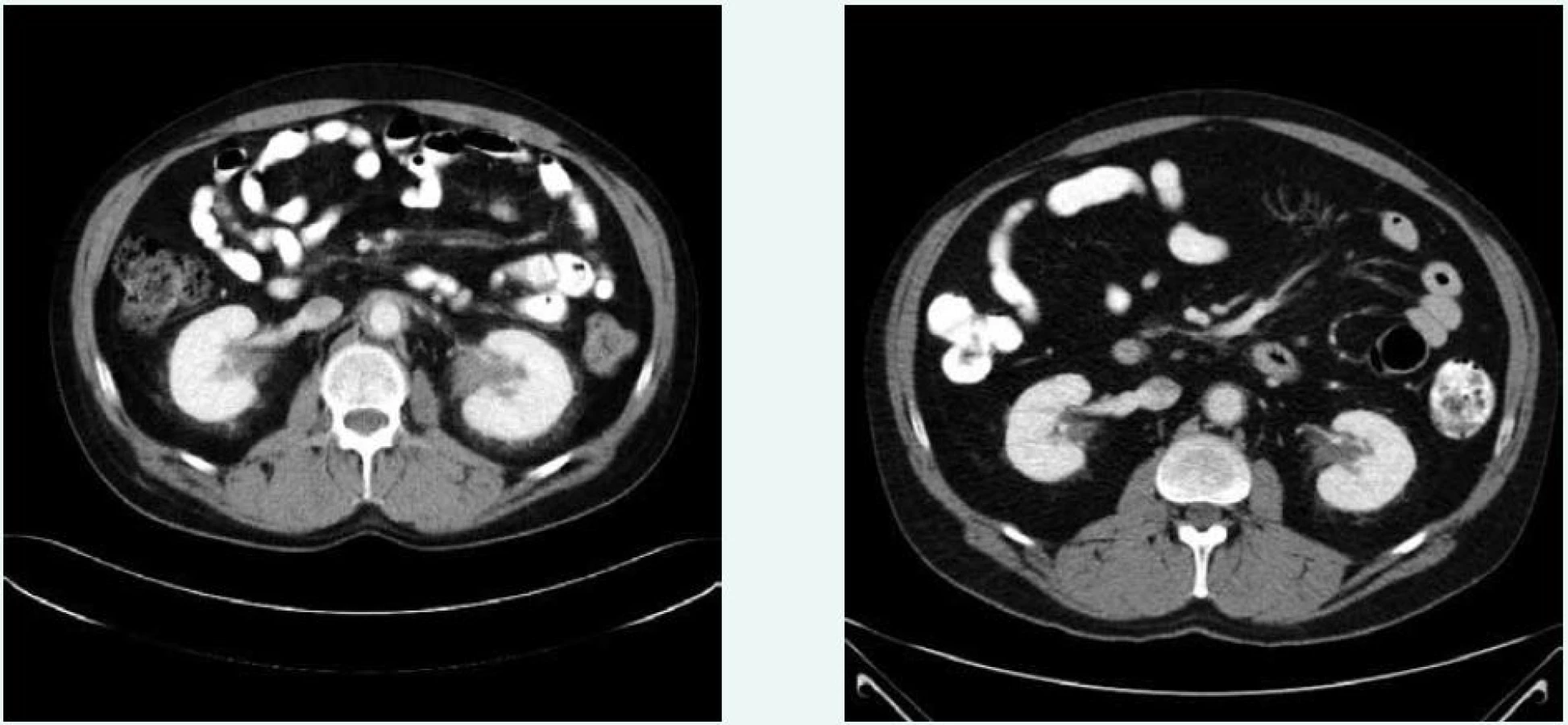
V březnu roku 2011 byla zahájena léčba anakinrou. Kontrolní CT vyšetření, které bylo provedeno 8 měsíců po zahájení léčby (listopad roku 2011), ukazuje diskrétní regresi infiltrativních změn perirenálně a v oblasti ledvinných hilů. Na následujícím kontrolním CT vyšetření, které bylo provedeno 17 měsíců po zahájení léčby anakinrou (srpen roku 2012), je patrná další mírná regrese změn perirenálně a v hilech ledvin. Minimální regrese již byla patrná i v okolí hrudní a břišní aorty, v okolí levé nadledviny. Na dalších kontrolních CT vyšetřeních provedených v květnu roku 2014 a v září roku 2015 je možné sledovat další regresi infiltrátu v udávaných lokalitách postižení, patrná je zejména regrese šíře stěny aorty, vymizení patologické tkáně perirenálně, ostřejší vykreslení struktur ledvinných hilů. Další kontrola v dubnu roku 2016 uvádí, že obraz „fibrotizujícího“ procesu v retroperitoneu (stěna aorty, perirenálně) je stabilizovaný.
Z uvedeného 5letého CT sledování je zřetelný léčebný efekt anakinry kontinuální regrese všech fibrotických změn detekovatelných při CT zobrazení, jak dokumentují obr. 6 a 7.

Hodnocení pomocí FDG-PET/CT
Při low-dose CT zobrazení, které bylo součástí PET/CT vyšetření, byla patrná četná osteosklerotická ložiska ve skeletu zvláště v dolních končetinách se zvýšenou akumulací 18F-fluorodeoxyglukózy (FDG), a tato zůstávají při opakovaných vyšetřeních stále dobře zřetelná. V průběhu 5 let se ale akumulace FDG v nich snížila, jak dokumentují obr. 8 a 9. U pacienta byla v průběhu 5 let provedena PET/CT vyšetření i s dalšími pozitronovými radiofarmaky 18F-fluorotymidinem (FLT) a 18F-natrium fluoridem (NaF), přičemž NaF-PET/CT vyšetření detekovalo kostní rozsah postižení větší, než byl detekován při PET/CT vyšetření za použití radiofarmak FLT a FDG.
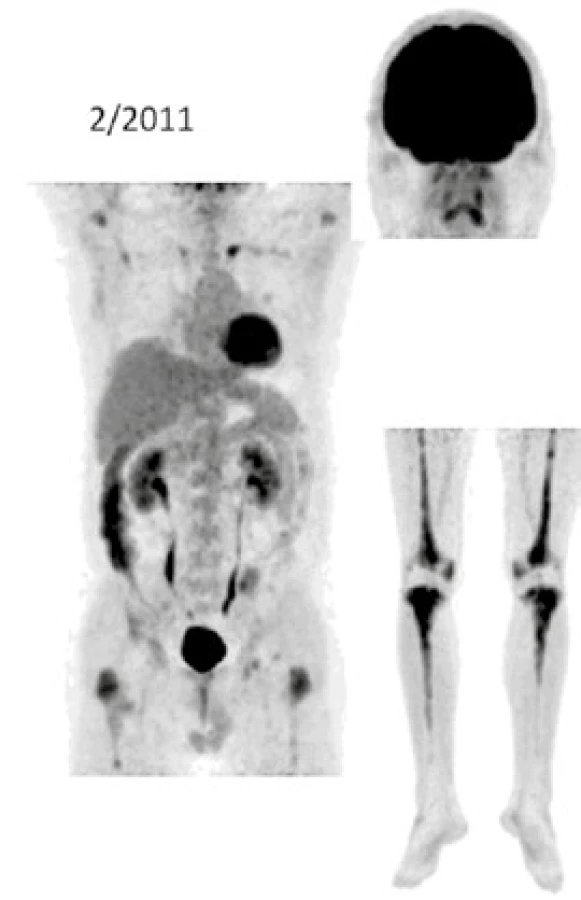
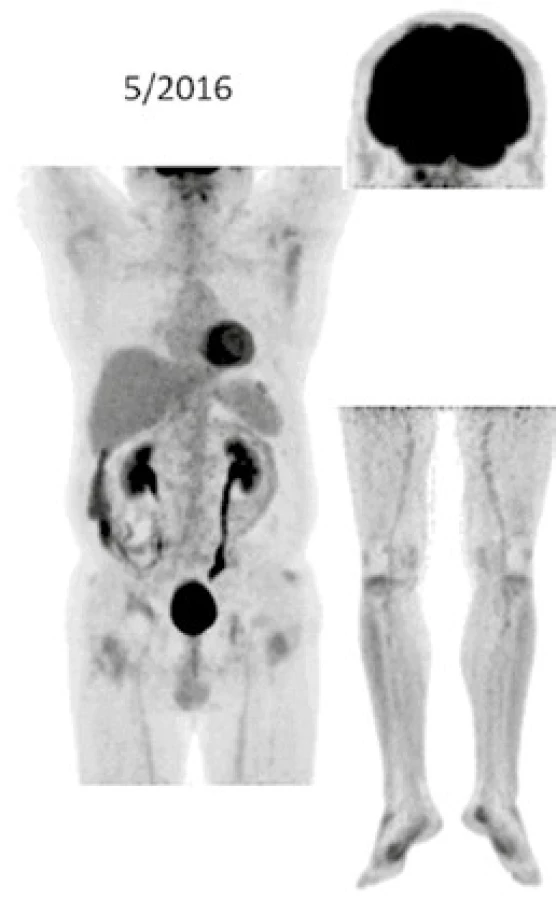
Výše uvedené sledování nemoci popisuje vývoj nemoci z pohledu různých zobrazovacích metod, dochází k regresi fibrotických změn dle klasického CT zobrazení i snížení akumulace FDG v kostních ložiscích. Osteosklerotické změny jsou na low dose CT stacionární.
Diskuse
Definici této nemoci jsme uvedli v úvodu, v rámci diskuse připomeneme základní znaky této nemoci a předložíme přehled informací o léčbě, protože tyto informace jsou pokladem pro výběr léčby, a slouží tedy i jako podklad pro žádost o úhradu této léčby revizním lékařům.
Patogeneze
Erdheimova-Chesterova choroba (ECD) je neoplastický nádorový proces s projevy systémové zánětlivé reakce. V předchozích desetiletích byla diskutována otázka, zda se jedná o reaktivní nebo o nádorové onemocnění. Teprve v posledním desetiletí přinesla molekulární biologie na tuto otázku odpověď. Prokázané klonální cytogenetické abnormality svědčí pro maligní etiologii této nemoci [13,14]. Přesvědčivým průkazem je přítomnost mutace BRAFV600E. Tato mutace byla zprvu prokázána týmem Badaliena u histiocytózy z Langerhansových buněk [15]. Posléze byla mutace BRAFV600E vyšetřována nejen u histiocytózy z Langerhansových buněk, ale také u vzácnějších non-Langerhans-cell histiocytóz, a také u ECD. Frekvence těchto mutací kolísá mezi 38 – 68 % [16,17].
Pouze v jediné práci byla tato mutace prokázána u všech 18 pacientů s ECD. Autoři tvrdí, že tuto mutaci lze u této nemoci prokázat vždy, pokud se použije dostatečně senzitivní metoda [18]. Ve vzorku ECD byly prokázány i další abnormality – onkogenní NRASQ61R mutace [19].
Patologické pěnité histiocyty ECD mají tedy klonální původ. Další jejich důležitou vlastností je skutečnost, že tyto buňky tvoří masivně proinflamatorní cytokiny a chemokiny, které přispívají k lokální aktivaci (recruitment) dalších histiocytů [20].
Analýza séra pacientů s ECD identifikovala zvýšené hladiny cytokinů, zvýšenou koncentraci interferonu α, interleukinu 12, monocytového chemotaktickému proteinu 1 a snížené hladiny interleukinu 4 a interleukinu 7 [21].
ECD tedy lze definovat jako klonální chorobu provázenou silnou systémovou zánětlivou reakcí s nadměrnou tvorbou prozánětlivých cytokinů. Proto je u většiny těchto pacientů zvýšená hodnota CRP a jsou detekovány další projevy systémové zánětlivé odpovědi, jako je např. „anémie chronických chorob“.
V patogenezi zůstává stále ještě hodně neobjasněného. Je však prokázána příbuznost ECD s histiocytózou z Langerhansových buněk (Langerhans cell histiocytosis – LCH). Příznaky ECD a LCH se částečně překrývají [22,23].
Epidemiologie
Choroba bývá diagnostikována u osob ve věku od 40 do 70 let a je častější u mužů (73 %). Jde o chorobu velmi vzácnou, takže její výskyt je charakterizován pouze počtem popsaných případů v literatuře, pohybuje se ve stovkách, ale nejsou známé údaje typu prevalence či incidence ECD [2,3,24]. O vzácnosti této nemoci svědčí i to, že naše pracoviště za 25 let registruje více než 40 případů histiocytózy z Langerhansových buněk, ale pouze 2 případy ECD.
Histologický obraz
Stanovení diagnózy ECD je založeno na histopatologickém nálezu u pacientů s typickými zobrazovacími a klinickými nálezy. Morfologicky je nutno prokázat pro tuto nemoc charakteristická ložiska pěnitých (čili lipidové inkluze obsahujících) histiocytů s fibrózou v okolí ložiska. Často jsou přítomny Toutonovy obrovské buňky. Pří imunohistochemickém barvení vykazují patologické pěnité histiocyty pozitivitu pro CD68, CD163 a negativitu pro CD1a negativitu pro langerin (CD207). Pozitivita S-100 byla pozorována jen výjimečně. To odlišuje ECD od LCH, u níž jsou Langerhansovy buňky pozitivní pro CD1a, S-100 a langerin. Histiocyty u ECD jsou morfologicky a imunohistochemicky identické s histiocyty u juvenilního xantogranulomu (JXG), a proto je ECD považována za variantu JXG s převážně mimokožními projevy [25,26].
Klinické a zobrazovací znaky
ECD může způsobovat klasické B symptomy, jak je vídáme u jiných krevních nemocí, tedy subfebrilie či febrilie, noční poty, úbytek hmotnosti a patologickou únavu. ECD může infiltrovat téměř každý orgán, ale nejčastěji postihuje skelet, retroperitoneum, orbity a také kardiovaskulární systém, plíce, CNS a endokrinní systém. Pak se klinické příznaky pojí s odpovídajícím nálezem na zobrazovacích vyšetřeních. Přehled klinických příznaků uvádí tab. 1. ECD je obvykle symptomatické onemocnění, asymptomatická forma je zcela výjimečná. Pro ECD nebyla přijata žádná klinická stadia, pouze se doporučuje za název nemoci připojit dominující prezentaci nemoci, např. ECD s dominující CNS, kardiální, vaskulární, endokrinní, retroperitoneální manifestací či multisystémová forma. V našem případě jde o pacienta s dominující CNS a retroperitonální manifestací.
![Hlavní klinické příznaky a charakteristické zobrazovací nálezy u pacientů s Erdheimovou a Chesterovou nemocí [2]](https://www.prelekara.sk/media/cache/resolve/media_object_image_large/media/image/63a1efc550c537e93cd02f2d6243ce16.jpg)
Kostní změny
Nejčastější radiologický obraz představuje postižení skeletálního systému ve formě bilaterálních, symetrických metadiafyzárních sklerotických změn dlouhých kostí. Ačkoliv osteosklerotické změny dolních končetin jsou u této nemoci téměř vždy přítomné (v 96 %), pouze asi 39–50 % pacientů udává bolest v těchto lokalizacích. Typickými známkami jsou oboustranná skleróza a zesílení kortikalis v oblasti metafýz a diafýz, které se však neprojevují v epifyzární části. Scintigrafie skeletu Tc-pyrofosfátem je klasickou základní metodou, která má možnost na tuto nemoc upozornit zvýšenou akumulací radiofarmaka v dlouhých kostech. Novější alternativa zobrazení je metoda PET/CT s fluorodeoxyglukózou (FDG) nebo natriumfluoridem (NaF). Kostní změny lze také znázornit pomocí CT nebo MR zobrazení kostí, ale na klasických snímcích skeletu bývají často přehlédnutelné [27].
Kostní novotvorba v dlouhých kostech DK se zesílením kortikalis je sice typická pro ECD, ale provází také některé další choroby, jako je Gaucherova choroba a Fabryho choroba a také Schnitzlerův syndrom.
Někteří autoři popsali také osteosklerózu v oblasti maxilárních a sfenoidálních sinů [28], což se vyskytuje i u našeho pacienta.
Retroperitoneální fibróza
Dalším poměrně častým projevem nemoci je postižení ledvin a retroperitonea pod obrazem infiltrátů s afinitou k pojivové, tukové a perivaskulární tkáni. Postižení ledvin a perirenálního tuku probíhá často asymptomaticky, je odhaleno při CT vyšetření, typicky ve formě hypodenzních homogenních infiltrátů s chabým postkontrastním sycením v oblasti renálních hilů. Perirenální postižení se projevuje šířením infiltrátu do předního, zadního, nebo obou pararenalních prostorů. Popisuje se obraz tzv. vlasatých ledvin (hairy kidney), kvůli symetrickému, bilaterálnímu perirenálnímu postižení, které je vysoce sugestivní pro danou diagnózu. Infiltrace může přecházet z ledvinných hilů na proximální uretery, což může potenciálně vést k obstrukci vývodných cest močových. Infiltrace perinefrické tkáně typu „hairy kidney” byla detekován u 68 % vyšetřených [29], klinické známky retroperitoneální fibrózy s hydronefrózou jsou však vzácnější. Tito nemocní se tedy dostávající do péče urologů, kteří by měli zahájit diagnostický proces. Postižení jiných abdominálních orgánů (játra, slinivka) je extrémně vzácné.
Diferenciální diagnostika retroperitoneální fibrózy
V diferenciální diagnostice můžeme uvažovat o postižení při idiopatické retroperitoneální fibróze (m. Ormond), existuje však několik pomocných rozlišovacích znaků [30,31]. Periaortální infiltrace při m. Ormond je často lokalizovaná v anterolaterálních porcích, s ušetřením posteriorní stěny. Časté je spolupostižení dolní duté žíly ve formě stenózy či uzávěru žíly, dále pánevních segmentů močovodů, naopak jen vzácně dochází k fibrotizaci perirenálního prostoru. U retroperitoneální fibrózy ECD (na rozdíl od idiopatické retroperitoneální fibrózy – m. Ormond) je pelvická část ureterů a v. cava inferior ušetřena fibrotických projevů. Pokud dojde k hydronefróze, je zapotřebí zavedení stentů nebo dokonce i provedení nefrostomie [32]. Infiltrace při ECD často obkružuje celý obvod cévy, typicky bývá ušetřena dolní dutá žíla a pánevní segmenty močovodů. Obraz „vlasatých“ ledvin je poměrně typický pro ECD. Při diferenciální diagnostice nesmíme zapomenout, že fibrózní změny v retroperitoneu provázejí někdy maligní lymfomy v oblasti retroperitonea.
Kardiovaskulární projevy
Kardiovaskulární postižení je běžné, při podrobném vyšetření je prokazatelné u 70 % pacientů, ale obvykle asymptomatické. Patologické změny jsou detekovatelné pomocí CT či MR zobrazení. Nejčastějšími abnormalitami je zesílení stěny hrudní a břišní aorty a jejich větví jako tzv. coated aorta. Tento znak se popisuje u dvou třetin pacientů [33]. V CT obraze typicky odhalíme periaortální (přesněji periadventiciální) infiltrát šířící se kraniokaudálním směrem podél aorty. Postižení může být omezeno na abdominální nebo hrudní aortu, může se však šířit až k srdci s potenciálně život ohrožujícími následky. Denzita infiltrátu je podobná denzitám svalové tkáně, postkontrastně vykazuje chabé homogenní sycení.
Periarteriální infiltrace může přesahovat na cévy odstupující z aorty, ale vyjma postižení renálních a koronárních tepen bývá klinicky němá. Periarteriální infiltrace renální arterie může způsobit renovaskulární hypertenzi s nutnosti stentování. Stejně může dojít k postižení koronárních tepen, které způsobí infarkt myokardu. Postižení perikardu bývá diagnostikováno u 40–45 % nemocných a může mít formu perikarditidy s výpotkem, či dokonce i s tamponádou. Asi u třetiny pacientů může být přítomen tumorózní infiltrát pravé předsíně, který je zobrazitelný pomocí MR srdce. ECD může způsobit chlopenní poruchy či poruchy vodivosti. Zcela výjimečně byla popsána difuzní infiltrace myokardu nebo interatriálního septa vedoucí k srdečnímu selhání.
Postižení intrahepatických arterií může způsobit portální hypertenzi, postižení mezenterické arterie může způsobit ischemii [34–39].
U popisovaného pacienta je zřetelné masivní postižení aorty, jak dokumentují obr. 1–5.
Plicní manifestace
Postižení plic je detekovatelné u poloviny případů, postižen bývá plicní parenchym nebo pleura. Běžné snímky plic obvykle nevykazují patologický nález, ale zobrazení plic pomocí CT s vysokým rozlišením (high resolution CT – HRCT) může odhalit interlobulární zesílení sept, nebo centrilobulární či jiné opacity, podobně jako to bývá u LCH. Metodou bronchoalveolární laváže je možné prokázat makrofágy a pěnité histiocyty. Plicní postižení je obvykle asymptomatické, jen vzácně se manifestuje jako kašel či dušnost. Spirometrie může prokázat restrikci a sníženou difuzní kapacitu [40].
Postižení CNS
Postižení centrálního nervového systému (CNS) je poměrně časté, pohybuje se od 25 % do 50 %. Nejčastějšími neurologickými projevy jsou cerebelární a pyramidální syndromy. Z dalších neurologických projevů se popisují bolesti hlavy, neuropsychiatrické poruchy, poruchy kognitivních funkcí, poruchy senzorických funkcí, a poruchy funkce hlavových nervů [41,42]. Parenchymová ložiska v CNS jsou závažnou příčinou nestability a jsou považována za nepříznivý prognostický faktor. Infiltráty se mohou vyskytnout v celém nervovém systému, nejčastější lokalizace je ale v hypotalamo-hypofyzární ose. Diabetes insipidus je typickými příznakem mozkového postižení při ECD, ale i při jiných histiocytárních chorobách.
Expanzivní, gadoliniem zvýrazněná ložiska, bývají detekována nejčastěji inftratentoriální v cerebelu, ale mohou být detekována i v cerebrálních hemisférách. Ložiska ECD mají podobný charakter jako meningeomy, granulomatozní choroby či jako meningeální infiltrace při Rosaiově-Dorfmanově nemoci. Menší ložiska způsobují fokální příznaky dle své lokalizace, velká či difuzní ložiska způsobují celkovou deterioraci kognitivních funkcí. Ačkoliv byla popsána ložiska v kterékoliv lokalizaci v rámci CNS, nejčastější jsou ložiska postihující jádra mozečku nebo mostu. Nejčastějšími projevy jsou tedy progresivní cerebelární symptomy, tedy ataxie, dysartrie a syndrom mozkového kmene. Uvedené abnormality vykazují gadoliniový enhancement, tedy podobnou vlastnost jako jiné maligní expanze, a proto mohou být zaměněny za primární či metastatické tumory, demyelinizační onemocnění, zánětlivé procesy či leukodystrofii. Podobná intrakraniální ložiska byla popsána u juvenilního xantogranulomu [42].
Infiltrativní změny CNS u LCH mají podobnou distribuci jako u ECD, s tou výjimkou, že ložiska na meningách jsou u LCH výjimečné, zato u ECD častější. Ložiska v meningách může mít i další histiocytární choroba, nemoc Rosaiova-Dorfmanova. Postižení míchy je u ECD, možné, ale u LCH se nevyskytuje. Někteří autoři uvádějí také u ECD neurodegenerativní chorobu cerebela, podobně jak občas vídáme po mnohaletém průběhu LCH, jiní uvádějí, že neurodegenerativní choroba cerebela je typická pouze pro LCH [43–46]. Obraz postižení CNS tohoto pacienta jsme zveřejnili v předchozích publikacích.
Postižení orbit
Unilaterální či bilaterální infiltrace orbit se vyskytuje asi u 25 % pacientů a může se projevit jako exoftalmus, retroorbitální bolesti, paréza n. oculomotorius, či dokonce slepota. V rámci diferenciální diagnózy je nutno myslet na Gravesovu chorobu, granulomatózní choroby, lymfom či obrovskobuněčnou arteriitis. Xantelazmata v oblasti víček se vyskytují asi u 25 % nemocných. V očnici a kolem oka však může být přítomen xantogranulom či nekrobiotický xantogranulom izolovaný pouze na očnici a může způsobovat ty stejné problémy [47], jak jsme je již dříve popsali. Diabetes insipidus byl prvním příznakem nemoci, který popisovaného pacienta přivedl k lékaři.
Endokrinologické příznaky
Diabetes insipidus může být jak příznak LCH, tak i ECD a objevuje se u osob s ECD ve 25 % jako nejčastější endokrinopatie. Vzácněji se objevuje hyperprolaktinemie, gonadální insuficience s hypotestosteronizmem. Při zobrazení stopky hypofýzy a hypotalamu může být obraz zcela normální, či může dojít k zesílení a k abnormálnímu enhancementu těchto struktur [48].
Kožní příznaky
Nejčastějším příznakem ECD jsou masivní xantelazmata víček, vyskytují se asi u třetiny pacientů. Méně často se xantelazmata vyskytují na tváři, krku, kolem axil, trupu a třísel jako žluté či nahnědlé papuly splývající do plaků. Imunohistochemicky není možné odlišit, zda jsou tyto plaky projevem ECD nebo juvenilního xantogranulomu dospělých, ale juvenilní xantogranulom nemívá obvykle tolik systémových projevů jako ECD [2,25,29]. Podobné, ale izolované projevy mají klinické jednotky zvané xantogranulom a nekrobiotický xantogranulom. V našem případě jsme byli svědky masivních xantelazmat, která při léčbě anakinrou vymizela.
Doporučená vyšetření
Při diagnóze nového pacienta se vždy doporučuje provést CT hrudníku, břicha a pánve, případně FDG-PET/CT vyšetření. U těchto pacientů by měl být vyšetřen mozek a srdece pomocí MR zobrazení. Klasickým zobrazením hyperostotických změn končetin byla scintigrafie skeletu, ale dnes může být nahrazena zobrazení metodou FDG-PET/CT nebo NaF-PET/CT. Cílem laboratorních vyšetření je monitorovat funkci ledvin, laboratorní parametry zánětu a endokrinopatie, konkrétně deficit hypotalamických a hypofyzárních hormonů. Cílem vstupního vyšetření je tedy zmapovat rozsah nemoci a použít tyto informace pro vyhodnocování léčebné odpovědi [49–51]. Pro stanovení diagnózy je však zásadní získání vhodného vzorku tkáně. Pokud jsou infiltrovány měkké tkáně, je výhodnější odebrat vzorek z měkkotkáňové infiltrace, například z oblasti víček či orbit. Odběr vzorku kostní dřeně pro analýzu musí být cílený, z místa, v němž jsou lokalizovány pěnité buňky, a váleček musí být odvápněn, což znehodnotí materiál pro genetické vyšetření. Pro potřeby DNA analýzy je třeba provést extrakci DNA z čerstvě odebraného vzorku. Vzhledem k tomu, že průkaz BRAFV600E mutace má léčebné důsledky, je vhodné metodu odběrů a transport projednat předem s pracovištěm, které má potenciál vyšetřit uvedenou mutaci. Výsledek testování této mutace samozřejmě také závisí na množství patologických histiocytů ve vzorku.
Léčba
Informace o léčbě této nemoci lze získat z několika prospektivních klinických studií a hlavně z popisů případů. Zatím nebyla zveřejněna ani jedna randomizovaná klinická studie testující různé způsoby léčby.
Cytotoxická chemoterapie a kortikoidy
Před poznáním účinku interferonu α a posléze anakinry byly testovány různé chemoterapeutické režimy, včetně vinka-alkaloidů, antracyklinů a cyklofosfamidu. Byly sice popsány určité úspěchy, ale léčba alkylačními cytostatiky a antracykliny se přesto neukázala zásadně přínosnou a není dnes doporučována [52–57]. Byla testována i vysokodávkovaná chemoterapie, ale ani ta se nestala průlomovou terapií [58,59]. Kortikosteroidy mohou zmenšit edém při závažném exoftalmu, ale nejsou účinnou monoterapií.
Radioterapie a chirurgie
Radioterapie byla také testována, ale bylo dosaženo jen krátkodobé paliace a nemoc po několika měsících dále progredovala. Chirurgie má své místo pouze ve zmenšení patologických hmot při orbitálním postižení a dále pro léčbu intrakraniálního postižení [60,61].
Kladribin
Kladribin (2-chlorodeoxyadenozin) je toxický pro nejen pro klidové lymfocyty, ale také pro buňky odvozené od monocytů, tedy také pro Langerhansovy buňky a fagocytující histiocytární buňky [62]. Purinové analogum kladribin bylo použito v léčbě systémové LCH a u této diagnózy byl účinný. Proto byl kladribin testován také u ECD a JXG. Na základě publikovaných i vlastních zkušeností můžeme konstatovat, že kladribin je v některých případech účinným lékem, který může navodit dloudobou remisi nemoci, ale není účinný u všech nemocných [63–68]. Proč u našeho popisovaného pacienta navodil kladribin jen mírné dočasné zlepšení, nikoli však kompletní remisi, zatímco náš druhý pacient s ECD je již 5 let po léčbě kladribinem v kompletní remisi, neumíme vysvětlit [69].
Interferon α2a
Jedním z nejčastěji zmiňovaných léčebných postupů je léčba interferonem α. Nejdříve byl používán klasický interferon α (INFα), později pegylovaný interferon α (PEG-IFNα). INFα byl testován od dávky 3 mil. jednotek denně či 3krát týdně do dávky 9 mil. jednotek denně. Někteří autoři uváděli, že navýšení dávky z 3 mil. jednotek na 9 mil. jednotek výrazně zvýšilo efekt této léčby. PEG-IFNα byl použit v dávce 180 µg/týdně a tyto dávky v souboru 24 pacientů stabilizovalo či zlepšilo 64 % pacientů s postižením CNS a 79 % pacientů s postižením srdce [24,70–74].
Nicméně jak víme z našich dřívějších bohatých zkušeností s udržovací léčbou IFNα podávanou pacientům s mnohočetným myelomem, ale i z vlastní zkušenosti pacienta s jeho aplikací, podávání IFNα je spojeno často s intenzivními nežádoucími účinky a tolerance této léčby se u mnoha pacientů v čase snižuje. Takže léčba IFNα je sice podpořena četnými publikacemi s pozitivními závěry, ale protože víme, že aplikace IFNα je spojena s nezanedbatelnými nežádoucími účinky, IFNα jsme v tomto případě nepoužili.
Anakinra – antagonista receptoru IL1
Jeden z důležitých účinků IFNα je stimulace tvorby antagonisty receptoru pro interleukin 1 (IL1), a tedy potlačení účinku interleukinu 1. Proto Aouba testoval léčbu pomocí rekombinantního antagonisty receptoru IL1, jménem anakinra. Léčil anakinrou 2 pacienty a pozoroval vymizení teploty a dalších B symptomů a také vymizení bolestí kostí a vymizení i xantelazmat na víčku. Aouba popsal parciální regresi retroperitoneální fibrózy. Laboratorními důsledky léčby bylo snížení hladiny CRP a IL6 [75].
V roce 2010 to byla první publikace popisující efekt této léčby. Následovaly další publikace o této léčbě. Redukce nádorové populace a snížení hladin proinflamatorních cytokinů bylo pozorováno u pacientů léčených anakinrou v dávce 1–2 mg/kg/den. Léčba byla velmi dobře tolerována a byla účinná ve stejném rozsahu, jak popsal Aouba. Vymizely bolesti kostí a B symptomy. Bylo popsáno také zlepšení kardiální formy této nemoci. Zlepšení mozkového postižení však v průběhu léčby anakinrou zaznamenáno nebylo [76–81]. Z citované literatury je zřejmé, že tato léčba se velmi rychle rozšířila a získala oblibu v klinické praxi, a to dokonce i u pediatrických pacientů [82], a stala se tak na čas zlatým standardem léčby.
Dle současných znalostí lze jednoznačně upřednostnit anakinru před INFα u pacientů bez postižení CNS , protože anakinra je podstatně lépe tolerována. Nicméně přínos anakinry není v cytotoxickém působení na maligní histiocytární buňky, ale v blokádě důsledků nadměrné aktivity IL1. Proto při analýze relativně velkého souboru 12 pacientů s ECD s mediánem aplikace 22 měsíců bylo konstatováno, že pozitivní účinek léčby anakinrou se projevuje dominantně zmírněním či odstraněním symptomů nemoci, které jsou způsobeny zřejmě IL1, zatímco vliv anakinry na maligní infiltraci je velmi variabilní a obvykle nedochází vlivem léčby k vymizení aktivity ložisek při FDG-PET/CT zobrazení. Proto Cohen v roce 2016 v případě infiltrace srdce či CNS infiltrace doporučuje zahájit léčbu INFα nebo BRAF inhibitory [83]. A jak je vidět z popisu našeho pacienta, v průběhu léčby anakinrou skutečně ustupují fibrotické změny, vymizely B symptomy, dle opakovaných FDG-PET/CT kontrol zůstávaly osteosklerotické změny skeletu, hlavně v dolních končetinách, ale v průběbu 5leté léčby se i signifikantně snížila míra akumulace FDG. Anakinra tedy inhibuje projevy nemoci, které jsou způsobené nadprodukcí prozánětlivých cytokinů, ale v našem případě vedla také ke snížení akumulace FDG v patologických ložiscích.
Ústup retroperitonálních fibrotických změn u našeho pacienta dokumentují obr. 6 a 7. Snížení akumulace FDG v patologických ložiscích ilustruje obr. 8 a 9.
Infliximab
Celkem 4 pacienti s kardiální formou ECD byli léčeni infliximabem, anti-TNF protilátkou, a byl popsán pozitivní efekt. Zatím je s touto léčbou málo zkušeností [84,85].
Imatinib
Imatinib-mesylát byl použit s úspěchem u jiných histiocytóz, a tak byl testován i u ECD. První publikace na toto téma popsala příznivý účinek imatinibu, ale pozitivní účinek nebyl potvrzen v dalších publikacích. Proto se domníváme, že imatinib lze otestovat v léčbě další linie, pokud předchozí léčba selže [86–89].
Vemurafenib
První zkušenosti s léčbou 3 pacientů s ECD vemurafenibem, inhibitorem BRAFV600E, prokázalo dramatické zlepšení [90]. Dle současných znalostí je léčba vemurafenibem považována za léčbu první volby pro všechny nemocné s ECD, kteří mají mutaci BRAFV600E. Tato léčba ale není bez nežádoucích účinků, popisují se artralgie, bolesti hlavy, kožní komplikace a patologická únava. Zatím není jasné, jak dlouho by měla být aplikována [91–96].
Sledování a délka léčby
Základní informaci o aktivitě nemoci přináší FDG-PET/CT vyšetření, proto se zpočátku doporučuje jeho provádění v 3–6měsíčních intervalech a podle výsledků v léčbě buď pokračovat či změnit léčebný postup. Toto základní vyšetření se pak doplňuje zobrazením cíleným na nejvíce postižené orgány, ložiska v myokardu odhalí speciální MR vyšetření myokardu [97]. FDG-PET/CT zobrazovací vyšetření je zcela zásadní, protože nemoc nemá žádný laboratorní marker aktivity, vyjma zvýšené hodnoty CRP, která se při úspěšné léčbě normalizuje. Vývoj fibrózy v oblasti retroperinea a velkých cév dokumentuje klasické CT zobrazení.
Dálka léčby – pokud se považuje za udržovací, je dlouhodobá. Pokud se podaří nemoc eradikovat několika cykly 2-chlorodeoxyadenozinu, jak se podařilo u našeho druhého pacienta, je léčba jen krátkodobá.
Informací o prognóze moc není, většina popisů případů se věnuje diagnostice, klinické manifestaci či léčebné odpovědi. Našli jsme pouze jednu publikaci, která uvádí 43% přežití při mediánu sledování 32 měsíců [98]. Z toho lze usoudit, že prognóza je velmi individuální a je dána mírou poškození vitálně důležitých orgánů.
Závěry pro praxi
Dle naší limitované zkušenosti doporučujeme jako léčbu první volby u Erdheimovy-Chesterovy choroby otestovat kladribin, protože tato léčba v některých případech navodí dlouhodobou kompletní remisi nemoci.
Pokud není léčba kladribinem úspěšná, je vhodné použít anakinru – rekombinantní inhibitor receptoru pro IL1. Tento lék spolehlivě odstraní známky systémové zánětlivé reakce a s ní spojené B symptomy způsobené Erdheimovou-Chesterovou nemocí. Léčba anakinrou vede k regresi fibrotických projevů nemoci, které jinak výrazně zhoršují kvalitu života vznikem hydronefrózy a renální insuficience. Léčba anakinrou je pro pacienty přínosem, i když nemusí vždy vést k regresi ložisek viditelných při FDG-PET/CT zobrazení. V popsaném případně však došlo i k ústupu akumulace FDG v ložiscích choroby v průběhu 5leté léčby.
Výraznou léčebnou odpověď přináší vemurafenib v případě průkazu mutace BRAF v patologických histiocytech. V případě pacientů s patologickými ložisky v CNS doporučují někteří autoři INFα. V našem případně jsme již dříve popsali zmenšování ložisek v CNS při léčbě kladribinem a lenalidomidem.
Za vstřícnou a bezproblémovou spolupráci bych rád poděkoval revizním lékařům ZP MV ČR včetně MUDr. Jarmily Hruškové.
Práce byla vytvořena na podporu následujících grantových aktivit: AZV 15–29508A – MZ ČR – RVO (FNBr, 65269705) a 23/14/NAP MZ ČR – RVO (MOÚ, 00209805) a projektem LO 1413.
prof. MUDr. Zdeněk Adam, CSc.
z.adam@fnbrno.cz
Interní hematologická a onkologická klinika LF MU a FN Brno, pracoviště Bohunice
www.fnbrno.cz
Doručeno do redakce 3. 5. 2016
Přijato po recenzi 6. 6. 2016
Sources
1. Chester W. Lipoidgranulomatose. Virschow Arch Pathol Anat 1930; 279(2): 561–602.
2. Haroche J, Cohen-Aubart F, Charlotte F et al. The histiocytosis Erdheim-Chester disease is an inflammatory myeloid neoplasm. Expert Rev Clin Immunol 2015; 11(9): 1033–1142. Dostupné z DOI: <http://dx.doi.org/10.1586/1744666X.2015.1060857>.
3. Haroche J, Abla O. Uncommon histiocytic disorders: Rosai-Dorfman, juvenile xanthogranuloma, and Erdheim-Chester disease. Hematology Am Soc Hematol Educ Program 2015; 2015 : 571–578. Dostupné z DOI: <http://dx.doi.org/10.1182/asheducation-2015.1.571>.
4. Kolář J, Kučera V, Povýšil C et al. Erdheim Chester disease. Rofo 1984; 141(6): 698–701.
5. Mergancová J, Kubeš L, Elleder M. Xanthogranulomatous processes in the area of the large vessels. Česk Patol 1986; 22(3): 145–50.
6. Mergancová J, Kubeš L Elleder M. A xantogranulomatous process encircling large blood vessels (Erheim-Chester disease). Czech Med 1988; 11(1): 57–64.
7. Kučera, V, Čáp V, Kužel J et al. Vzácná příčina osteosklerózy: Erheimův Chesterův syndrome. ČS Radiol 1984; 38(6): 393–402.
8. Janková H, Říhová E. Juvenilní xantogranulom. In: Oftalmologie v kasuistikách. Triton: Praha 2007 : 214–218. ISBN 978–80–7387–025–6.
9. Vašáková M. Co je to Erdheim-Chesterova nemoc? Kazuist Alergol Pneumol ORL 2006; 3(4): 22.
10. Plank L. Diagnostická patológia non-Langerhans cell histiocytóz. Vnitř Lék 2010; 56(Suppl 2): 2S27–2S38.
11. Adam Z, Balšíková K, Pour L at al. Diabetes insipidus, následovaný po 4 letech dysartrií a lehkou pravostrannou hemiparézou – první klinické příznaky Erdheimovy-Chesterovy nemoci. Popis a zobrazení případu s přehledem informací o této nemoci. Vnitř Lék 2009; 55(12): 1173–1188.
12. Szturz P, Adam Z, Koukalová R et al. Erdheimova-Chesterova nemoc v obrazech. Vnitř Lék 2010; 56(Suppl 2): 2S170–2S178.
13. Gong L, He XL, Li YH et al. Clonal status and clinicopathological feature of Erdheim-Chester disease. Pathol Res Pract 2009; 205(9): 601–607. Dostupné z DOI: <http://dx.doi.org/10.1016/j.prp.2009.02.004>.
14. Vencio EF, Jenkins RB, Schiller JL et al. Clonal cytogenetic abnormalities in Erdheim-Chester disease. Am J Surg Pathol 2007; 31(2): 319–321.
15. Badalian-Very G, Vergilio JA, Degar BA et al. Recurrent BRAF mutations in Langerhans cell histiocytosis. Blood 2010; 116(11): 1919–1923. Dostupné z DOI: <http://dx.doi.org/10.1182/blood-2010–04–279083>.
16. Satoh T, Smith A, Sarde A et al. B-RAF mutant alleles associated with Langerhans cell histiocytosis, a granulomatous pediatric disease. PLoS ONE 2012; 7(4): e33891. Dostupné z DOI: <http://dx.doi.org/10.1371/journal.pone.003389>. Erratum in PLoS One 2012; 7(6). Dostupné z DOI: <http://dx.doi.org/10.1371/annotation/74a67f4e-a536–4b3f-a350–9a4c1e6bebbd>.
17. Sahm F, Capper D, Preusser M et al. BRAFV600E mutant protein is expressed in cells of variable maturation in Langerhans cell histiocytosis. Blood 2012; 120: e28-e34. Dostupné z DOI: <http://dx.doi.org/10.1182/blood-2012–06–429597>.
18. Cangi MG, Biavasco R, Cavalli G et al. BRAFV600E-mutation is invariably present and associated to oncogene-induced senescence in Erdheim-Chester disease. Ann Rheum Dis 2015; 74(8): 1596–602. Dostupné z DOI: <http://dx.doi.org/10.1136/annrheumdis-2013–204924>.
19. Diamond EL, Abdel-Wahab O, Pentsova E et al. Detection of an NRAS mutation in Erdheim-Chester disease. Blood 2013; 122(6): 1089–1091. Dostupné z DOI: <http://dx.doi.org/10.1182/blood-2013–02–482984>.
20. Stoppacciaro A, Ferrarini M, Salmaggi C et al. Immunohistochemical evidence of a cytokine and chemokine network in three patients with Erdheim-Chester disease: implications for pathogenesis. Arthritis Rheum 2006; 54(12): 4018–4022.
21. Arnaud L, Gorochov G, Charlotte F et al. Systemic perturbation of cytokine and chemokine networks in Erdheim-Chester disease: a single-center series of 37 patients. Blood 2011; 117(10): 2783–2790. Dostupné z DOI: <http://dx.doi.org/10.1182/blood-2010–10–313510>.
22. Naruse H, Shoda H, Okamoto A et al. A case of osteoarthropathy due to Erdheim-Chester disease with overlapping Langerhans’ cell infiltration. Intern Med 2010; 49(12): 1225–1228.
23. Caoduro C, Ungureanu CM, Rudenko B et al. 18F-fluoride PET/CT aspect of an unusual case of Erdheim-Chester disease with histologic features of Langerhans cell histiocytosis. Clin Nucl Med 2013; 38(7): 541–542. Dostupné z DOI: <http://dx.doi.org/10.1097/RLU.0b013e318270830f>.
24. Arnaud L, Hervier B, Néel A et al. CNS involvement and treatment with interferon-α are independent prognostic factors in Erdheim-Chester disease: a multicenter survival analysis of 53 patients. Blood 2011; 117(10): 2778–2782. Dostupné z DOI: <http://dx.doi.org/10.1182/blood-2010–06–294108>.
25. Haroche J, Arnaud L, Amoura Z. Erdheim-Chester disease. Curr Opin Rheumatol 2012; 24(1): 53–59. Dostupné z DOI: <http://dx.doi.org/10.1097/BOR.0b013e32834d861d>.
26. Mazor RD, Manevich-Mazor M, Shoenfeld Y. Erdheim-Chester Disease: a comprehensive review of the literature. Orphanet J Rare Dis 2013; 8 : 137. Dostupné z DOI: <http://dx.doi.org/10.1186/1750–1172–8-137>.
27. Breuil V, Brocq O, Pellegrino C et al. Erdheim-Chester disease: typical radiological bone features for a rare xanthogranulomatosis. Ann Rheum Dis 2002; 61(3): 199–200.
28. Wilejto M, Abla O. Langerhans cell histiocytosis and Erdheim-Chester disease. Curr Opin Rheumatol 2012; 24(1): 90–96. Dostupné z DOI: <http://dx.doi.org/10.1097/BOR.0b013e32834db53e>.
29. Haroche J, Arnaud L, Cohen-Aubart F et al. Erdheim-Chester disease. Rheum Dis Clin North Am 2013; 39(2): 299–311. Dostupné z DOI: <http://dx.doi.org/10.1016/j.rdc.2013.02.011>.
30. Urban ML, Palmisano A, Nicastro M. Idiopathic and and secondary forms of retroperitoneal fibrosis: a diagnostic approach. Rev Med Interne 2015; 36(1): 15–21. Dostupné z DOI: <http://dx.doi.org/10.1016/j.revmed.2014.10.008>.
31. Beyer G, Schwaiger T, Lerch MM et al. IgG4-related disease: a new kid on the block or an old aquaintance? United European Gastroenterol J 2014; 2(3): 165–172. Dostupné z DOI: <http://dx.doi.org/10.1177/2050640614532457>.
32. van Bommel EF, de Mol M, Langerak AW et al. Idiopathic retroperitoneal fibrosis mimicking malignant lymphoma. Pathol Int 2011; 61(11): 672–676. Dostupné z DOI: <http://dx.doi.org/10.1111/j.1440–1827.2011.02718.x>.
33. Serratrice J, Granel B, De Roux C et al. “Coated aorta”: a new sign of Erdheim-Chester disease. J Rheumatol 2000; 27(6): 1550–1553.
34. Nicolazzi MA, Carnicelli A, Fuorlo M et al. Cardiovascular Involvement in Erdheim-Chester Disease: A Case Report and Review of the Literature. Medicine (Baltimore) 2015; 94(43): e1365. Dostupné z DOI: <http://dx.doi.org/10.1097/MD.0000000000001365>.
35. Cavalli G, Guglielmi B, Berti A et al. The multifaceted clinical presentations and manifestations of Erdheim-Chester disease: comprehensive review of the literature and of 10 new cases. Ann Rheum Dis 2013; 72(10): 1691–1695. Dostupné z DOI: <http://dx.doi.org/10.1136/annrheumdis-2012–202542>.
36. Haroche J, Cluzel P, Toledano D et al. Images in cardiovascular medicine. Cardiac involvement in Erdheim-Chester disease: magnetic resonance and computed tomographic scan imaging in a monocentric series of 37 patients. Circulation 2009; 119(25): e597-e598. Dostupné z DOI: <http://dx.doi.org/10.1161/CIRCULATIONAHA.108.825075>.
37. Haroche J, Amoura Z, Dion E et al. Cardiovascular involvement, an overlooked feature of Erdheim-Chester disease: report of 6 new cases and a literature review. Medicine (Baltimore) 2004; 83(6): 371–392.
38. Fink MG, Levinson DJ, Brown NL et al. Erdheim-Chester disease. Case report with autopsy findings. Arch Pathol Lab Med 1991; 115(6): 619–623.
39. Loeffler AG, Memoli VA. Myocardial involvement in Erdheim-Chester disease. Arch Pathol Lab Med 2004; 128(6): 682–685.
40. Arnaud L, Pierre I, Beigelman-Aubry C et al. Pulmonary involvement in Erdheim-Chester disease: a single-center study of thirty-four patients and a review of the literature. Arthritis Rheum 2010; 62(11): 3504–3512. Dostupné z DOI: <http://dx.doi.org/10.1002/art.27672>.
41. Drier A, Haroche J, Savatovsky J et al. Cerebral, facial, and orbital involvement in Erdheim-Chester disease: CT and MR imaging findings. Radiology 2010; 255(2): 586–594. Dostupné z DOI: <http://dx.doi.org/10.1148/radiol.10090320>.
42. Lachenal F, Cotton F, Desmurs-Clavel H et al. Neurological manifestations and neuroradiological presentation of Erdheim-Chester disease: report of 6 cases and systematic review of the literature. J Neurol 2006; 253(10): 1267–1277.
43. Pautas E, Chérin P, Pelletier S et al. Cerebral Erdheim-Chester disease: report of two cases with progressive cerebellar syndrome with dentate abnormalities on magnetic resonance imaging. J Neurol Neurosurg Psychiatry 1998; 65(4): 597–599.
44. Lalitha P, Reddy MC, Reddy KJ. Extensive intracranial juvenile xanthogranulomas. AJNR Am J Neuroradiol 2011; 32(7): E132-E133. Dostupné z DOI: <http://dx.doi.org/10.3174/ajnr.A2209>.
45. Grois N, Fahrner B, Arceci RJ et al. Central nervous system disease in Langerhans cell histiocytosis. J Pediatr 2010; 156(6): 873–881. Dostupné z DOI: <http://dx.doi.org/10.1016/j.jpeds.2010.03.001>.
46. Kitai R, Llena J, Hirano A et al. Meningeal Rosai-Dorfman disease: report of three cases and literature review. Brain Tumor Pathol 2001; 18(1): 49–54.
47. Karcioglu ZA, Sharara N, Boles TL et al. Orbital xanthogranuloma: clinical and morphologic features in eight patients. Ophthal Plast Reconstr Surg 2003; 19(5): 372–381.
48. Courtillot C, Laugier Robiolle S, Cohen Aubart F et al. Endocrine Manifestations in a Monocentric Cohort of 64 Patients With Erdheim-Chester Disease. J Clin Endocrinol Metab 2016; 101(1): 305–313. Dostupné z DOI: <http://dx.doi.org/10.1210/jc.2015–3357>.
49. Namwongprom S, Núñez R, Kim EE et al. Tc-99m MDP bone scintigraphy and positron emission tomography/computed tomography (PET/CT) imaging in Erdheim-Chester disease. Clin Nucl Med 2007; 32(1): 35–38.
50. Lin E. FDG PET/CT for biopsy guidance in Erdheim-Chester disease. Clin Nucl Med 2007; 32(11): 860–861.
51. Steňová E, Steňo B, Povinec P et al. FDG-PET in the Erdheim-Chester disease: its diagnostic and follow-up role. Rheumatol Int 2012; 32(3): 675–678. Dostupné z DOI: <http://dx.doi.org/10.1007/s00296–010–1676-y>.
52. Jendro MC, Zeidler H, Rosenthal H et al. Improvement of Erdheim-Chester disease in two patients by sequential treatment with vinblastine and mycophenolate mofetil. Clin Rheumatol 2004; 23(1): 52–56.
53. Broccoli A, Stefoni V, Faccioli L et al. Bilateral orbital Erdheim-Chester disease treated with 12 weekly administrations of VNCOP-B chemotherapy: a case report and a review of literature. Rheumatol Int 2012; 32(7): 2209–2213. Dostupné z DOI: <http://dx.doi.org/10.1007/s00296–011–1998–4>.
54. Jeon IS, Lee SS, Lee MK. Chemotherapy and interferon-alpha treatment of Erdheim-Chester disease. Pediatr Blood Cancer 2010; 55(4): 745–747. Dostupné z DOI: <http://dx.doi.org/10.1002/pbc.22636>.
55. Bourke SC, Nicholson AG, Gibson GJ. Erdheim-Chester disease: pulmonary infiltration responding to cyclophosphamide and prednisolone. Thorax 2003; 58(11): 1004–1005.
56. Wu Z, Yan J, Hong W et al. A case of Erdheim-Chester disease with bilateral orbital involvement. Yan Ke Xue Bao 2001; 17(3): 163–167.
57. Yano S, Kobayashi K, Kato K et al. A case of Erdheim-Chester disease effectively treated by cyclophosphamide and prednisolone. Nihon Kokyuki Gakkai Zasshi 2007; 45(1): 43–48.
58. Gaspar N, Boudou P, Haroche J et al. High-dose chemotherapy followed by autologous hematopoietic stem cell transplantation for adult histiocytic disorders with central nervous system involvement. Haematologica 2006; 91(8): 1121–1125.
59. Boissel N, Wechsler B, Leblond V. Treatment of refractory Erdheim-Chester disease with double autologous hematopoietic stem-cell transplantation. Ann Intern Med 2001; 135(9): 844–845.
60. Mascalchi M, Nencini P, Nistri M et al. Failure of radiation therapy for brain involvement in Erdheim Chester disease. J Neurooncol 2002; 59(2): 169–172.
61. Miller RC, Villà S, Kamer S et al. Palliative treatment of Erdheim-Chester disease with radiotherapy: a Rare Cancer Network study. Radiother Oncol 2006; 80(3): 323–326.
62. Singh V, Prajeeth CK, Gudi V et al. 2-Chlorodeoxyadenosine (cladribine) induces apoptosis in human monocyte-derived dendritic cells. Clin Exp Immunol 2013; 173(2): 288–297. Dostupné z DOI: <http://dx.doi.org/10.1111/cei.12109>.
63. Blomstrand L, Thor A, Hagberg H. Erdheim-Chester disease presenting as periodontal disease: Experience of treatment with cladribine, interferon-a, local radiotherapy and anakinra. Acta Oncol 2016; 55(2): 248–250. Dostupné z DOI: <http://dx.doi.org/10.3109/0284186X.2015.1023463>.
64. Myra C, Sloper L, Tighe PJ et al. Treatment of Erdheim-Chester disease with cladribine: a rational approach. Br J Ophthalmol 2004; 88(6): 844–847.
65. Perić P, Antić B, Knezević-Usaj S et al. Successful treatment with cladribine of Erdheim-Chester disease with orbital and central nervous system involvement developing after treatment of Langerhans cell histiocytosis. Vojnosanit Pregl 2016; 73(1): 83–87.
66. Rajendra B, Duncan A, Parslew R et al. Successful treatment of central nervous system juvenile xanthogranulomatosis with cladribine. Pediatr Blood Cancer 2009; 52(3): 413–415. Dostupné z DOI: <http://dx.doi.org/10.1002/pbc.21830>.
67. Sheidow TG, Nicolle DA, Heathcote JG. Erdheim-Chester disease: two cases of¨orbital involvement. Eye (Lond) 2000; 14(Pt 4): 606–612.
68. Sutton L, Sutton S, Sutton M. Treatment of necrobiotic xanthogranuloma with 2-chlorodeoxyadenosine. Skinmed 2013; 11(2): 121–123.
69. Adam Z, Řehák Z, Koukalová R et al. PET-CT dokumentovaná kompletní 4letá remise Erdheimovy-Chesterovy nemoci po léčbě kladribinem. Vnitř lék 2014; 60(5–6): 499–511.
70. Braiteh F, Boxrud C, Esmaeli B et al. Successful treatment of Erdheim-Chester disease, a non-Langerhans-cell histiocytosis, with interferon-alpha. Blood 2005; 106(9): 2992–2994.
71. Haroche J, Amoura Z, Trad SG et al. Variability in the efficacy of interferon-alpha in Erdheim-Chester disease by patient and site of involvement: results in eight patients. Arthritis Rheum 2006; 54(10): 3330–3336.
72. Hervier B, Arnaud L, Charlotte F et al. Treatment of Erdheim-Chester Disease with long-term high-dose interferon-α. Semin Arthritis Rheum 2012; 41(6): 1–7. Dostupné z DOI: <http://dx.doi.org/10.1016/j.semarthrit.2011.11.004>.
73. Esmaeli B, Ahmadi A, Tang R et al. Interferon therapy for orbital infiltration secondary to Erdheim-Chester disease. Am J Ophthalmol 2001; 132(6): 945–947.
74. Suzuki HI, Hosoya N, Miyagawa K et al. Erdheim-Chester disease: multisystem involvement and management with interferon-alpha. Leuk Res 2010; 34(1): e21-e24. Dostupné z DOI: <http://dx.doi.org/10.1016/j.leukres.2009.07.026>.
75. Aouba A, Georgin-Lavialle S, Pagnoux C et al. Rationale and efficacy of interleukin-1 targeting in Erdheim-Chester disease. Blood 2010; 116(20): 4070–4076. Dostupné z DOI: <http://dx.doi.org/10.1182/blood-2010–04–279240>.
76. Podestà MA, Graziani G, Reggiani F et al. Improvement of Erdheim-Chester disease-related renal failure after treatment with anakinra. Kidney Res Clin Pract 2014; 33(3): 165–167. Dostupné z DOI: <http://dx.doi.org/10.1016/j.krcp.2014.07.007>.
77. Szturz P, Adam Z, Rehák Z et al. Xanthelasma palpebrarum responding to interleukin-1 blockade. Intern Med J 2014; 44(6): 617–618. Dostupné z DOI: <http://dx.doi.org/10.1111/imj.12441>.
78. Darstein F, Kirschey S, Heckl S et al. Successful treatment of Erdheim-Chester disease with combination of interleukin-1-targeting drugs and high-dose glucocorticoids. Intern Med J 2014; 44(1): 90–92. Dostupné z DOI: <http://dx.doi.org/10.1111/imj.12329>.
79. Courcoul A, Vignot E, Chapurlat R. Successful treatment of Erdheim-Chester disease by interleukin-1 receptor antagonist protein. Joint Bone Spine 2014; 81(2): 175–177. Dostupné z DOI: <http://dx.doi.org/10.1016/j.jbspin.2013.06.013>.
80. Killu AM, Liang JJ, Jaffe AS Erdheim-Chester disease with cardiac involvement successfully treated with anakinra. Int J Cardiol 2013; 167(5): e115-e117. Dostupné z DOI: <http://dx.doi.org/10.1016/j.ijcard.2013.04.057>.
81. Aubert O, Aouba A, Deshayes S et al. Favorable radiological outcome of skeletal Erdheim-Chester disease involvement with anakinra. Joint Bone Spine 2013; 80(2): 206–207. Dostupné z DOI: <http://dx.doi.org/10.1016/j.jbspin.2012.07.005>.
82. Tran TA, Pariente D, Lecron JC et al. Treatment of pediatric Erdheim-Chester disease with interleukin-1-targeting drugs. Arthritis Rheum 2011; 63(12): 4031–4032. Dostupné z DOI: <http://dx.doi.org/10.1002/art.30638>.
83. Cohen-Aubart F, Maksud P, Saadoun D et al. Variability in the efficacy of the IL1 receptor antagonist anakinra for treating Erdheim-Chester disease. Blood 2016; 127(11): 1509–1512. Dostupné z DOI: <http://dx.doi.org/10.1182/blood-2015–09–672667>.
84. Dagna L, Corti A, Langheim S et al. Tumor necrosis factor α as a master regulator of inflammation in Erdheim-Chester disease: rationale for the treatment of patients with infliximab. J Clin Oncol 2012; 30(28): e286-e290. Dostupné z DOI: <http://dx.doi.org/10.1200/JCO.2012.41.9911>.
85. Ferrero E, Belloni D, Corti A et al. TNF-alpha in Erdheim-Chester disease pericardial effusion promotes endothelial leakage in vitro and is neutralized by infliximab. Rheumatology (Oxford) 2014; 53(1): 198–200. Dostupné z DOI: <http://dx.doi.org/10.1093/rheumatology/ket246>.
86. Haroche J, Amoura Z, Charlotte F et al. Imatinib mesylate for platelet-derived growth factor receptor-beta-positive Erdheim-Chester histiocytosis. Blood 2008; 111(11): 5413–5415. Dostupné z DOI: <http://dx.doi.org/10.1182/blood-2008–03–148304>.
87. Janku F, Amin HM, Yang D et al. Response of histiocytoses to imatinib mesylate: fire to ashes. J Clin Oncol 2010; 28: e633-e636. Dostupné z DOI: <http://dx.doi.org/10.1200/JCO.2010.29.9073>.
88. Montella L, Insabato L, Palmieri G. Imatinib mesylate for cerebral Langerhans’-cell histiocytosis. N Engl J Med 2004; 351(10): 1034–1035.
89. Utikal J, Ugurel S, Kurzen H et al. Imatinib as a treatment option for systemic non-Langerhans cell histiocytoses. Arch Dermatol 2007; 143(6): 736–740.
90. Haroche J, Cohen-Aubart F, Emile JF et al. Dramatic efficacy of vemurafenib in both multisystemic and refractory Erdheim-Chester disease and Langerhans cell histiocytosis harboring the BRAF V600E mutation. Blood 2013; 121(9): 1495–1500. Dostupné z DOI: <http://dx.doi.org/10.1182/blood-2012–07–446286>.
91. Franconieri F, Martin-Silva N, de Boyssson H et al. A. Superior efficacy and tolerance of reduced doses of vemurafenib plus anakinra in Erdheim-Chester disease: Towards the paradigm of combined targeting and immune therapies. Acta Oncol 2016; 55(7): 930–932. Dostupné z DOI: <http://dx.doi.org/10.3109/0284186X.2015>.
92. Schirmer JH, Thorns C, Moosig F et al. Treatment failure by canakinumab in a patient with progressive multisystemic Erdheim-Chester disease refractory to anakinra: successful use of vemurafenib. Rheumatology (Oxford) 2015; 54(10): 1932–1934. Dostupné z DOI: <http://dx.doi.org/10.1093/rheumatology/kev237>.
93. Tzoulis C, Schwarzlmüller T, Gjerde IO et al. Excellent response of intramedullary Erdheim-Chester disease to vemurafenib: a case report. BMC Res Notes 2015; 8 : 171. Dostupné z DOI: <http://dx.doi.org/10.1186/s13104–015–1135–7>.
94. Euskirchen P, Haroche J, Emile JF et al. Complete remission of critical neurohistiocytosis by vemurafenib. Neurol Neuroimmunol Neuroinflamm 2015; 2(2): e78. Dostupné z DOI: <http://dx.doi.org/10.1212/NXI.0000000000000078>.
95. Haroche J, Cohen-Aubart F, Emile JF et al. Reproducible and sustained efficacy of targeted therapy with vemurafenib in patients with BRAF(V600E)-mutated Erdheim-Chester disease. J Clin Oncol 2015; 33(5): 411–418. Dostupné z DOI: <http://dx.doi.org/10.1200/JCO.2014.57.1950>.
96. Cohen-Aubart F, Emile JF, Maksud P et al. Marked efficacy of vemurafenib in suprasellar Erdheim-Chester disease. Neurology 2014; 83(14): 1294–1296. Dostupné z DOI: <http://dx.doi.org/10.1212/WNL.0000000000000832>.
97. Benoist N, Mikail N, Deshchamps H et al. Erdheim Chester disease as assessed by modern multimodality imaging. Int J Cardiol 2016; 207 : 235–237. Dostupné z DOI: <http://dx.doi.org/10.1016/j.ijcard.2016.01.165>.
98. Veyssier-Belot C, Cacoub P, Caparros-Lefebvre D et al. Erdheim-Chester disease. Clinical and radiologic characteristics of 59 cases. Medicine (Baltimore) 1996; 75(3): 157–169.
99. Dion E, Graef C, Haroche J et al. Imaging of Thoracoabdominal Involvement in Erdheim-Chester Disease. AJR Am J Roentgenol 2004; 183(5): 1253–1260.
Labels
Diabetology Endocrinology Internal medicineArticle was published in
Internal Medicine

2016 Issue 10
Most read in this issue
- Nová – přímá perorální antikoagulancia: aktuální přehled
- Raloxifen – nevyužitá možnost prevence a léčby postmenopauzální osteoporózy
- Přínos magnetické rezonance pro diagnostiku kardiomyopatií a myokarditidy (1. část)
- Hemosuccus pancreaticus – raritná komplikácia chronickej pankreatitídy