
Atypický priebeh Wilsonovej choroby: kazuistika a prehľad literatúry
Unusual history of Wilson disease: a case report and review of the literature
Wilson disease (WD) belongs to autosomal recessive genetic metabolic disorders with gene mutation ATP7B located on 13th chromosome. The enzyme ATPase plays an important role in WD. It facilitates excretion of copper into bile. This gene is responsible for modification of apoceruloplasmin. In this disease, it leads to insufficient release of copper from organism and accumulation of copper in organs such as liver, brain which can cause dysfunction of a certain organ. According to specific symptoms, we can divide WD into psychiatric, neurologic or hepatic form. The WD usually manifests between 15 and 25 years of age. Hepatic form often occurs sooner, on the contrary, the neurological variant usually occur during the later stages. We present a case report of 45-years-old woman with atypical medical history of WD, in which the diagnostic process was very long and had interdisciplinary character.
Key words:
brain – copper – diagnostic – genetics – liver – panda – Wilson disease
:
František Nehaj 1; Marianna Kubašková 2; Michal Mokáň 1; Juraj Sokol 3; Vladimír Nosáľ 4; Kamil Zeleňák 2; Marián Mokáň 1
:
I. Interná klinika JLF UK a UN Martin, Slovenská republika
1; Rádiologická klinika JLF UK a UN Martin, Slovenská republika
2; Klinika hematológie a transfuziológie, Národné centrum hemostázy a trombózy JLF UK a UN Martin, Slovenská republika
3; Neurologická klinika JLF UK a UN Martin, Slovenská republika
4
:
Vnitř Lék 2017; 63(12): 980-986
:
Case Reports
Wilsonova choroba (WCH) patrí medzi autozomálne recesívne dedičné metabolické ochorenia s mutáciou génu ATP7B na 13. chromozóme. Dôležitú úlohu pri WCH zohráva enzým ATPáza, ktorej hlavnou funkciou je vylučovanie medi do žlče. Tento gén sprostredkováva ATPázu modifikujúcu apoceruloplazmín. Pri tomto ochorení dochádza k nedostatočnému vylučovaniu medi z organizmu, a teda k jej hromadeniu v ľudskom organizme. Meď sa akumuluje predominantne v orgánoch ako je pečeň a mozog, čo môže zapríčiniť poruchu funkcie daného orgánu. Podľa toho, ktoré príznaky prevládajú, rozdeľujeme WCH chorobu na formu neurologickú, často spätú s formou psychiatrickou, hepatálnu a inú. Najčastejšie sa symptómy WCH prejavia v období 15.–25. roku života, v závislosti na klinickej forme. Hepatálna sa často objaví skôr, naopak neurologická forma sa môže prejaviť neskôr. V našom článku prezentujeme kazuistiku 45-ročnej pacientky s atypickým priebehom WCH, ktorej diagnostika bola zdĺhavá a mala interdisciplinárny charakter.
Kľúčové slová:
diagnostika – genetika – meď – mozog – panda – pečeň – Wilsonova choroba
Úvod
Apoceruloplazmín je zodpovedný za tvorbu transportéra medi – ceruloplazmín, ktorý sa podieľa na transporte medi v organizme. Ceruloplazmín sa nepodieľa na vylučovanie medi z pečeňových buniek do žlče, čo predstavuje hlavný mechanizmus patogenézy ochorenia. Defekt génu ATP7B lokalizovaného na 13. chromozóme (13q14.3–q21.1) spôsobuje, že dochádza k poruche inkorporácie medi do apoceruloplazmínu. Táto porucha je dôležitá z diagnostického hľadiska, pretože vedie k nízkej hodnote ceruloplazmínu v sére, ale nepodieľa sa na patogenéze ochorenia. Meď sa hromadí v organizme, vznikajú voľné radikály a klesá redukovaná forma glutationu. Tým dochádza k peroxidácii lipidov membrán, k poškodeniu proteínov obsahujúcich SH skupinu a genetickej informácie. Meď sa pre nedostatočné vylučovanie z organizmu hromadí v pečeni, bazálnych gangliách mozgu, rohovke oka, obličkách i kĺboch [1,2]. Dodnes bolo identifikovaných viac ako 500 mutácií génu ATP7B. Databáza je dostupná na http://www.wilsondisease.med.ualberta.ca/database.asp. Všeobecne uznávaná prevalencia Wilsonovej choroby bola udávaná1 na 30 000 obyvateľov, no podľa posledných genetických štúdií je podstatne vyššia, až 1 na 7 000 obyvateľov. Wilsonovu chorobu (WCH) rozdeľujeme na neurologickú a hepatálnu formu.
Neurologickou formou trpí približne 50 % pacientov. Klinické príznaky začínajú typicky v období 2. a 3. dekády života, no ochorenie sa môže vyskytovať aj detskom ako aj vyššom veku. Z toho vyplýva, že je potrebné zobrať do úvahy možnosť ochorenia aj v detskom veku ako aj v starobe. Existujú 3 základné neurologické prejavy Wilsonovej choroby: parkinsonský syndróm, dystonický syndróm a ataktický syndróm. Väčšina pacientov klinicky prezentuje kombináciu uvedených syndrómov. Hoci je najbežnejšou formou tremoru u pacientov s Wilsonovou chorobou nepravidelný, trhavý, dystonický tremor, je potrebné zdôrazniť, že môžu byť prítomné aj iné typy tremorov (kľudový, statický, intenčný, flapping tremor). Dystónia je prítomná u tretiny pacientov a môže mať charakter generalizovanej, segmentálnej, multifokálnej alebo fokálnej dystónie. Dysartria je často skombinovaná orofaciálnou dyskinéziou s obrazom typického úškrnu (risus sardonicus) a pomalými pohybmi jazyka. Parkinsonský syndróm sa prezentuje hypokinéziou, hypomímiou, šúchavou chôdzou a poruchou jemnej motoriky prstov. Patologické zvýšenie šľachovokosticových reflexov svedčí pre léziu pyramídového systému, no prítomnosť parézy nie je typická pre Wilsonovu chorobu. Epileptické záchvaty sa môžu vyskytovať v akomkoľvek štádiu ochorenia, no vyskytujú sa častejšie počas začiatku liečby [3,4].
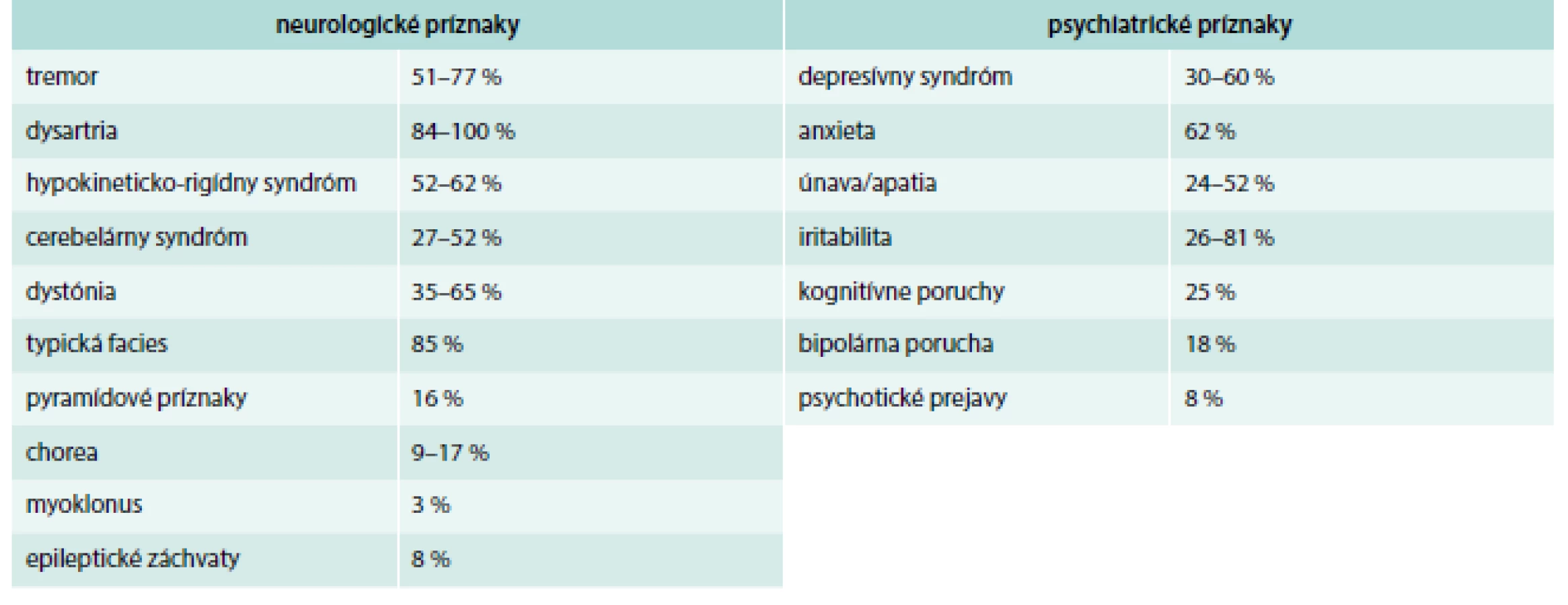
Okrem neurologických prejavov sú u pacientov s Wilsonovou chorobou časté aj psychiatrické príznaky. Približne 20 % pacientov navštívi psychiatra skôr, než sa im stanoví diagnóza Wilsonovej choroby. Spektrum psychiatrických prejavov zahŕňa zvýšenú podráždenosť, úzkosť, odbrzdenosť, depresie, apatiu, chronický únavový syndróm, poruchy spánku, pozornosti, pamäti, nálady a osobnostné zmeny [3–6]. V rámci neurologického vyšetrenia treba vylúčiť v rámci diferenciálnej diagnostiky esenciálny tremor, Parkinsonovu chorobu, cervikálnu dystóniu, idiopatickú generalizovanú dystóniu, blefarospazmus, Huntingtonovu chorobu, spinocereberálnu ataxiu a iné psychiatrické ochorenia [2–5]. Prehľad neurologických a psychiatrických príznakov pri WCH je uvedený v tab. 1 [7].
Hepatálnou formou trpí 20–30 % pacientov. Môže sa prejaviť ako akútna hepatitída (66 %), fulminantné zlyhanie pečene, chronická aktívna hepatitída, cirhóza pečene, steatóza pečene. Prirodzený priebeh ochorenia bez liečby nie je doposiaľ detailne objasnený, pričom pri zahájení liečby hepatálnej formy sa neurologické prejavy nikdy nerozvinú. Naopak, časť pacientov s neurologickou formou a manifestáciou medzi 25.–35. rokom života má v anamnéze eleváciu hepatálnych enzýmov v detskom veku (pred 10. rokom života). Medzi ďalšie sprievodné prejavy patrí ikterus, hemolytická anémia, koagulopatia, ascites, hepatomegália, edémy, portálna hypertenzia. Progresia ochorenia vedie až k transplantácii pečene, ktorá je indikovaná len pri akútnom hepatálnom zlyhaní [4,8]. Chronická hepatálna lézia s vývojom do cirhózy sa väčšinou pri liečbe stabilizuje a transplantácia je indikovaná len vo výnimočných prípadoch.
Podľa Svetovej zdravotníckej organizácie (World health organisation – WHO) môžeme pacientov rozdeliť do 4 skupín:
- forma pečeňová s prejavom akútnej hepatitídy s ikterom
- forma pečeňová ako chronické pečeňové ochorenie
- forma neurologická so súčasným výskytom pečeňovej cirhózy
- forma neurologická s prítomnou asymptomatickou pečeňovou steatózou alebo pečeňovou fibrózou [8]
Z endokrinologického hľadiska môže dôjsť k poruchám menštruačného cyklu, k predčasnému klimaktériu, príp. spontánnym potratom. Liečbou WCH sú však tieto zmeny zvratné. Prejaviť sa môže však aj ako renálna insuficiencia z dôvodu ukladania medi do proximálnych tubulov. Je sprevádzaná aminoacidúriou, hyperkalciúriou, hyperfosfátúriou, hyperurikosúriou, fosfátúriou a tiež poruchou vylučovania cukrov. Častým javom je tvorba nefrolitov. Poruchy kostného metabolizmu sa manifestujú ako osteoporóza, osteomalácia, osteoartróza, alebo degeneratívne zmeny na kĺboch, najmä kolenných [9,10].
Diagnostika
Základom každého vyšetrenia je cielená anamnéza. V biochemickom náleze nachádzame elevované hepatocelulárne enzýmy (alanínaminotransferáza – ALT, aspartátaminotransferáza – AST), zvýšený bilirubín. Dôležitá je nízka hladina ceruloplazmínu – v sére pod 0,1 g/l; 80–90 % pacientov (norma 0,20–0,60 g/l). Zvýšené hodnoty cerulopalzmínu môžu byť počas gravidity, po podaní estrogénov, pri zápalových procesoch. Meď v sére je z veľkej časti viazaná na ceruloplazmín (90–95 %), a preto s jeho poklesom klesá aj hladina medi a zvyšuje sa hladina voľnej medi (> 2 µmol/l). Odpad medi močom za 24 hod býva tiež zvýšený (> 1–1,6 µmol/l/24 hod – norma 0,242–0,63).
Pri penicilamínovom teste sa pacientovi pred zberom moču počas 24 hod podá 500 mg penicilamínu každých 12 hod. Tým sa podporí vylučovanie medi močom a jej odpad je až niekoľkonásobne vyšší. Dôležitá je počas tohto testu strava s malým množstvom medi. Tento test je však preukazný len v detskej populácii. Senzitivita tohto testu je až 92 % a špecificita 93 % u pacientov so symptómami.
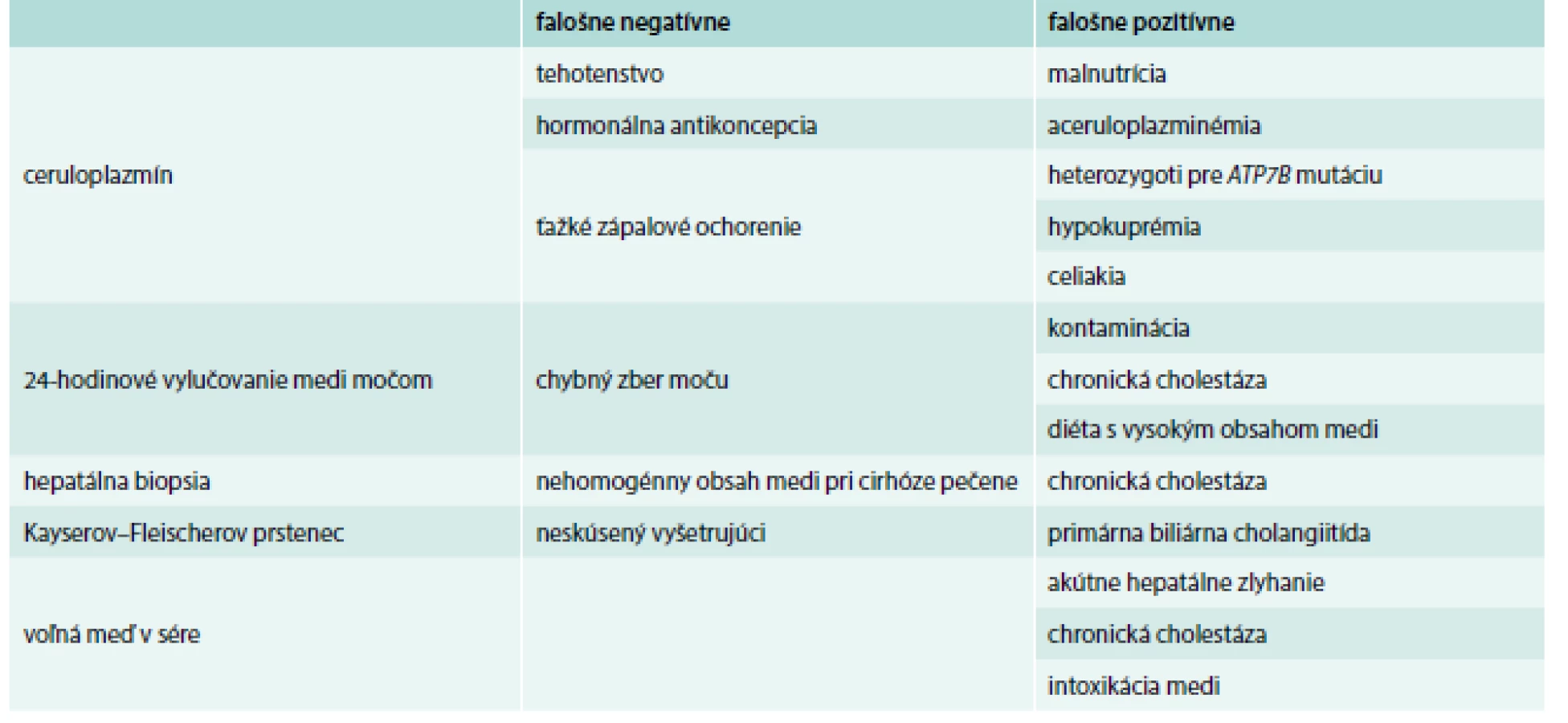
Diagnostickým testom je biopsia pečene a obsah medi v sušine. U zdravých jedincov je obsah medi v sušine < 50 µg/l (0,8 µmol/g), u pacientov bez symptómov je obsah medi > 250 µg/g a u symptomatických medzi 450–1 200 µg/g. Za nešpecifické hodnoty sa považuje rozmedzie 50–250 µg/g. Takéto vyšetrené koncentrácie majú senzitivitu 83,3 % a špecificitu 98,6 %. Tie bývajú u heterozygotov a aj u niektorých pacientov. Falošne negatívny výsledok (tab. 2) býva pri nerovnomernom rozložení medi v pečeni, kedy sa biopsiou nezachytí reprezentatívna vzorka [1,7,8].
U pacientov s WCH môžeme nájsť pomocou zobrazenia mozgu magnetickou rezonanciou (MRI) hyperintenzívne zmeny (glióza, nekróza a cystické zmeny s lokálnym edémom) v T2 vážených obrazoch. Zmeny nachádzame v oblasti nucleus caudatus, putamen, talamu, ponsu, mezencefala, mozočka, globus pallidus, kde môžu byť prítomné aj hypointenzívne zmeny (možné paramagnetický efekt depozít feritínu).
Môžu sa objaviť aj demyelinizačné zmeny (supratentoriálne v bielej hmote), ktoré môžu imitovať zmeny pri sclerosis multiplex. Prítomná je cerebrálna, kortikálna aj kmeňová atrofia. Ako patognomický príznak sa udáva nález obrazu medvedíka pandy a príznak jasného claustra, no tieto sa vyskytujú iba u 12 % a 4 % pacientov. Oveľa častejšie sa vyskytujú T2 hyperintenzívne zmeny charakteru lézií bielej hmoty v tectum mesencephali a ponse charakteru centrálnej pontínnej demyelinizácie. Prítomná je cerebrálna, kortikálna aj kmeňová atrofia [11].
Oftalmologické vyšetrenie štrbinovou lampou so zameraním na rohovku je dôležité pre nález typického zlatohnedého Kayserovho–Fleischerovho prstenca, ktorý je patognomickým príznakom ochorenia. Kayserov–Fleischerovov prstenec vzniká v dôsledku ukladania medi v descemetovej membráne rohovky, najmä v hornej časti. Vzácnejšie sa vyskytne slnečnicová katarakta z dôvodu akumulácie medi v puzdre šošovky. V neposlednom rade netreba zabudnúť na genetické vyšetrenie, kde v prípade pozitívneho nálezu je potrebné doplnenie genetického vyšetrenia aj u prvostupňových príbuzných – súrodenci a deti pacienta. Genetické vyšetrenie je jednou z hlavných diagnostických metód, ktoré sa sústreďuje nielen na najčastejšie sa vyskytujúce mutácie, ale aj iné odchýlky v géne pre WCH [1,8,12–14].
Liečba
Cieľom liečby je navodenie negatívnej bilancie medi. To znamená vyplaviť naakumulovanú meď z organizmu, znížiť jej príjem v potravinách, zabrániť opätovnému hromadeniu v ľudskom organizme. Negatívna bilancia sa dosahuje predovšetkým liečbou, ktorá je celoživotná. Cieľom liečby je prevencia poškodenia pečene a centrálneho nervového systému (CNS) u asymptomatických pacientov, u klinicky rozvinutej choroby to však môže byť zlepšenie, eventuálne stabilizácia klinického stavu.
Homozygoti, aj keď sú bezpríznakoví, by mali byť liečení. Heterozygoti (nosiči) nevyžadujú liečbu.
Medikamentózne sa používa penicilamín (dimetylcystein). Tento liek viaže meď a vylučuje sa močom. V úvodnej dávke sa podáva 150 mg/deň. Postupne sa dávka zvyšuje na 600–1 200 mg/deň po dobu 4–6 mesiacov. Súčasne sa s ním podáva pyridoxín (vitamín B6 – pre jeho vyššiu spotrebu pri terapii penicilamínom) v dávke 40–60 mg/deň. Zlepšenie príznakov nastane o 3–6 mesiacov. Dĺžka liečby a dávky penicilamínu sú individuálne, záleží na konkrétnej situácii. Niektorí pacienti sú liečení vysokou dávkou natrvalo, ktorá sa riadi biochemickým a klinickým nálezom. Ďalšou liečebnou alternatívou je liečba zinkom v dávke 3-krát 50 mg, ktorý sprostredkováva syntézu metalothioneínu v bunkách čreva. Zinok znižuje resorpciu medi črevom a zvýši sa vylučovanie medi stolicou. Liečba zinkom sa uprednostňuje u presymptomatických pacientov a pri neuropsychiatrickej forme, zatiaľ čo pri hepatálnej sa uprednostňuje liečba penicilamínom. Nežiadúce účinky liečby penicilamínu sa vyskytujú u 20 % pacientov v priebehu 1. týždňov liečby, ktoré sú vo väčšine prípadov mierne alebo dôjde k zhoršeniu už existujúcich príznakov. Patrí k nim horúčka, exantém, leukopénia, trombocytopénia. Lymfadenopatia – syndróm podobný systémovému lupus erythematosus, či imunokomplexová glomerulonefritída s rozvojom nefrotického syndrómu sú extrémne vzácne. V prípade, ak sa objavia závažné nežiadúce účinky, k liečbe penicilamínom s kortikosteroidmi, ale sa zvolí iná forma liečby. Nežiaduce účinky zinku sú zriedkavé, môže sa objaviť gastritída, sideropénia.
U pacientov, ktorí netolerujú penicilamin, sa používa trientin (900–1 200 mg/deň). Mechanizmus účinku má podobný s penicilamínom, avšak na vylučovaní medi sa podieľa menej. Výhodou je však nižší výskyt nežiaducich účinkov a o tomto lieku sa uvažuje, že je na neurologické postihnutie vhodnejší v porovnaní s penicilamínom.
Podobne ako zinok pôsobí tetrathiomolybdenát, ktorý tvorí komplexy spolu s meďou a albumínom v čreve a krvi. Takýmto princípom bráni jej vstrebávaniu a viaže voľnú meď v krvi.
Počas medikamentóznej liečby sa sleduje vylučovanie medi močom. Tá po začatí liečby výrazne stúpne. V chronickej fáze by hodnota medi vylúčenej za 24 hod mala byť v rozmedzí 3–8 µmol. Hodnoty pod 3 µmol nasvedčujú buď pre hypokuprémiu spôsobenú liečbou, príp. pre nonkomplianciu pacienta. Následne vyšetrená vyššia hodnota voľnej medi v sére svedčí práve pre nonkomplianciu pacienta. V prípade terapie zinkom by vylučovanie medi močom nemalo presiahnuť hodnotu 1,2 µmol v priebehu 24 hod a koncentrácia zinku v moči by sa mala pohybovať okolo 2 mg/24 hod, pričom koncentrácia voľnej sérovej medi by nemala byť ani zvýšená ani znížená. U žien by sa mala obmedziť hormonálna antikoncepcia (s obsahom estrogénov, aj vnútromaternicové teliesko, ktoré môže obsahovať meď). Transplantácia pečene je indikovaná pri akútnom a chronickom pečeňovom zlyhaní, ktoré neodpovedá na medikamentóznu terapiu [1,8,12,13].
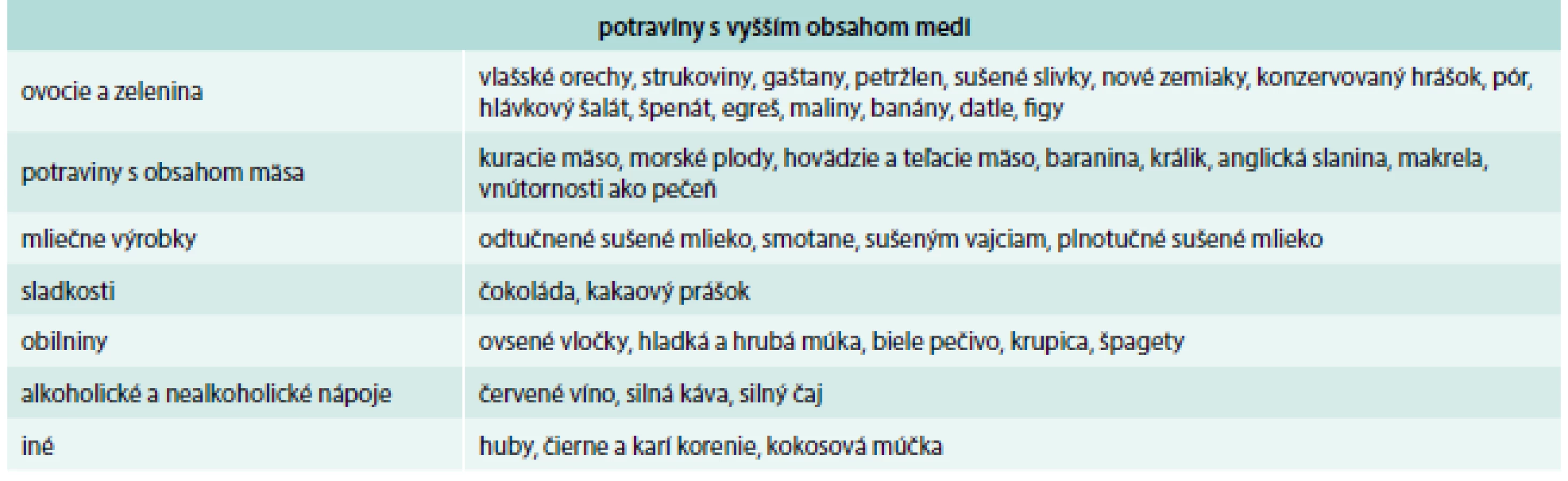
Potraviny, ktoré obsahujú vyšší obsah medi, sú uvedené v tab. 3, nižší obsah medi v tab. 4 a nízky obsah medi v tab. 5.


Kazuistika
45-ročná pacientka bez závažného predchorobia, bez pozitívnej rodinnej anamnézy na genetické metabolické ochorenia, bola prvýkrát hospitalizovaná pre dlhodobé abdominoalgie a dyspeptické ťažkosti. Bola realizovaná výpočtová tomografia (CT) abdomenu s nálezom zhrubnutej steny jejunálnych kľučiek v dĺžke asi 30 cm so zúženým lumenom. Enteroskopicky bola odobratá vzorka, histologicky sa nepotvrdilo autoimunitné ochorenie typu morbus Crohn, lymfóm ani iný neoplastický proces.
S odstupom času sa potvrdilo, že vyššie popísané okolnosti prípadu nesúviseli so základným ochorením pacientky. Do diferenciálnej diagnostiky boli zapojené ďalšie chirurgické a internistické disciplíny s nejednoznačným diagnostickým záverom. Pacientkine ťažkosti boli v priebehu vyšetrení spochybnené, absolvovala psychiatrické vyšetrenie s podávaním psychiatrickej liečby, po ktorej ťažkosti neustúpili.
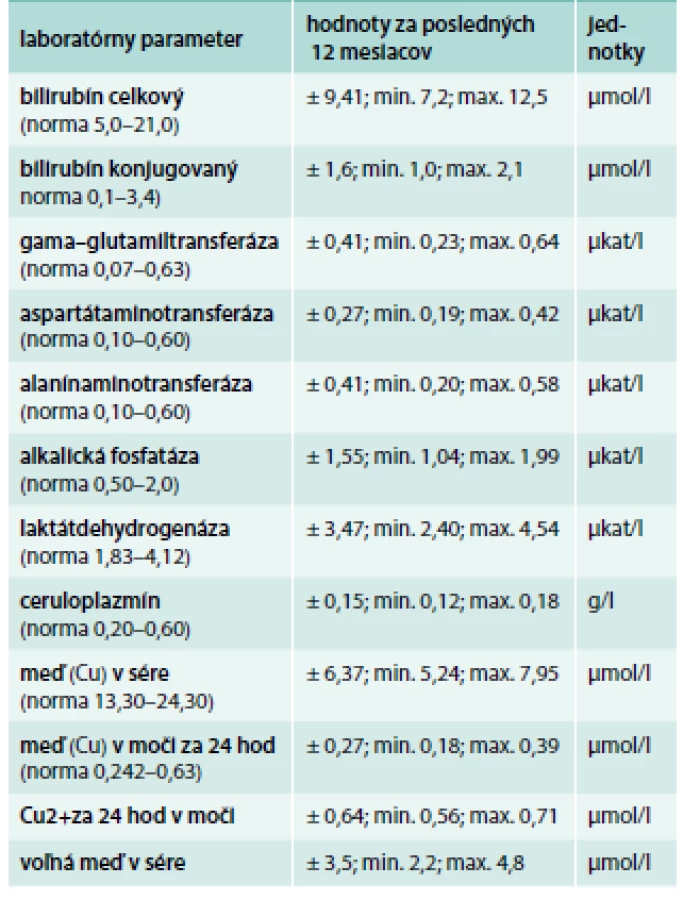
V objektívnom náleze bol prítomný statický a intenčný tremor horných končatín trvajúci približne od 35. roku života, ktorý sa s pribúdajúcim vekom postupne zhoršoval. Pacientka udávala rozmazané videnie a vertigo. V laboratórnom obraze bola znížená hladina ceruloplazmínu a voľnej medi v sére, odpady medi močom boli znížené (tab. 6) [7].
Na základe uvedených nálezov a príznakov, bolo vyjadrené podozrenie na WCH. Ultrasonografia potvrdila steatózu pečene, ktorá sa intermitentne prejavovala ľahkou biochemickou aktivitou.
Doplnilo sa očné vyšetrenie, ktoré bolo bez nálezu zlatohnedého prstenca – Kayserov-Fleischerov prstenec. Bolo doplnené zobrazenie mozgu magnetickou rezonanciou (MRI) a výsledkom bol patognomický nález obrazu medvedíka pandy (obr).

Pre pozitivitu výsledkov laboratórnych parametrov a klinického obrazu bolo indikované a realizované genetické vyšetrenie so záverom potvrdenia morbus Wilson a bolo zistené, že probandka je zložený heterozygot (c.3207C>A;p.His1069Gln)/(c.1995G>Ap.Met665Ile) génu ATP7B. Genetické vyšetrenie bolo realizované aj u prvostupňových príbuzných (sestra, brat, dcéra s syn). U syna pacientky bolo dokázané nosičstvo mutácie c.1995G>Ap.Met665Ile.
Po skompletizovaní výsledkov bola pacientka konzultovaná so špecializovaným ústavom genetických metabolických porúch v Prahe s navrhnutím liečby, ktorú z pracovných a rodinných dôvodov odmietla. Penicilamínový test vzhľadom na negativistický postoj pacientky nebol realizovaný. Pacientka je v dispenzarizácii spádového gastroenterológa bez progresie základného ochorenia.
Diskusia
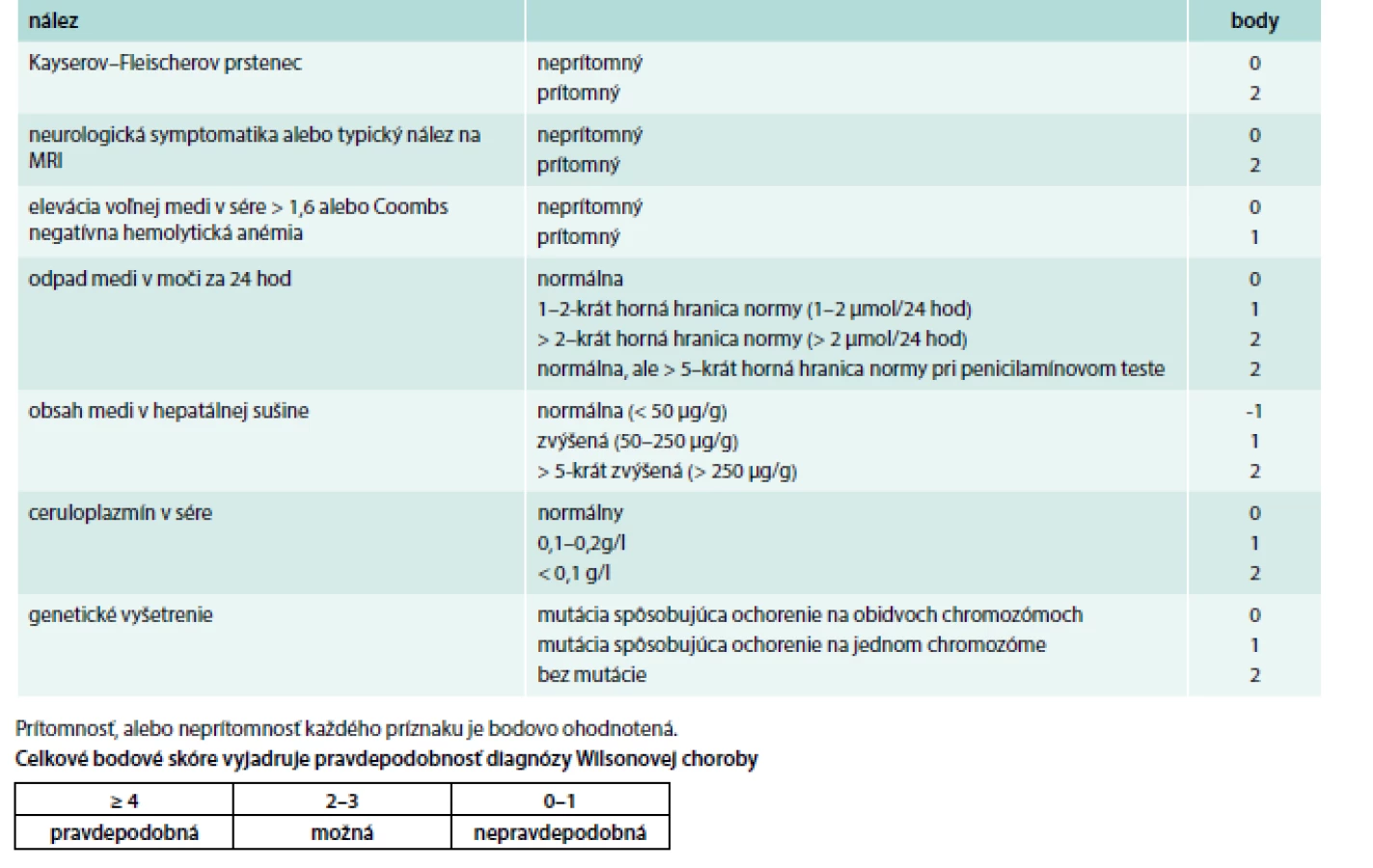
Prvé príznaky WCH sa najčastejšie prejavia medzi 15.–25. rokom života. Pri diferenciálnej diagnostike treba myslieť na hepatitídu, malnutríciu a ďalšie ochorenia. Aceruloplazminémia a hypokuprémiu sú extrémne vzácne. Všeobecne u pacientov od 5–50 rokov s akýmkoľvek extrapyramídovým, cereberálnym, prípadne psychiatrickým ochorením, a najmä pri výskyte hepatopatie treba v rámci diferenciálnej diagnózy myslieť aj na WCH. Pre pravdepodobnosť určenia diagnózy možno použiť Ferenciho, prípadne Leipzig skóre, ktorého súčasťou je aj zhodnotenie neurologického postihnutia. Prítomnosť alebo neprítomnosť každého príznaku je bodovo ohodnotené a celkové skóre vyjadruje pravdepodobnosť diagnózy WCH (tab. 7).
Pri ťažkostiach v diagnostickom rozhodovaní existujú doporučené postupy, ktoré nám uľahčujú stanoviť a potvrdiť finálnu diagnózu (schéma) [7]. Vo všeobecnosti diferenciálna diagnóza závisí od veku pacienta a forme manifestácie.

Záver
Genetické metabolické ochorenie ako WCH postihuje pacientov v mladom, produktívnom veku a zásadným významom ovplyvňuje kvalitu života. Diagnóza býva nielen zložitá a zdĺhavá, ale často aj náhodná. Popísaný prípad pacientky je zaujímavý a atypický tým, že prvé symptómy a príznaky sa u pacientky prejavili až v 35. roku života. V neposlednom rade treba zdôrazniť, aby sa zľahčovanie príznakov a symptómov pacienta, ktorý môže trpieť závažnou avšak ojedinelou chorobou, nezamieňala za psychiatrickú diagnózu.
Táto práca bola podporená grantami VEGA 1/0187/17.
MUDr. Juraj Sokol, PhD.
juraj.sokol@me.com
Klinika hematológie a transfuziológie,
Národné centrum hemostázy a trombózy JLF UK a
UN Martin,
Slovenská republika
www.jfmed.uniba.sk
Doručeno do redakce 16. 4. 2017
Přijato po recenzi 28. 8. 2017
Sources
1. Roberts EA, Schilsky ML. Diagnosis and treatment of Wilson disease: an update. Hepatology 2008; 47(6): 2089–2111. Dostupné z DOI: <http://dx.doi.org/10.1002/hep.22261>.
2. Mareček Z, Brůha R. Wilsonova choroba. Vnitř Lék 2013; 59(7): 578–583.
3. Lorincz MT. Neurologic Wilson‘s disease. Ann N Y Acad Sci 2010; 1184 : 173–187. Dostupné z DOI: <http://dx.doi.org/10.1111/j.1749–6632.2009.05109.x>.
4. Merle U, Schaefer M, Ferenci P et al. Clinical presentation, diagnosis and long-term outcome of Wilson‘s disease: a cohort study. Gut 2007; 56(1): 115–120.
5. Akil M, Brewer GJ. Psychiatric and behavioral abnormalities in Wilson’s disease. Adv Neurol 1995; 65 : 171–178.
6. Svetel M, Potrebic A, Pekmezovic T et al. Neuropsychiatric aspects of treated Wilson‘s disease. Parkinsonism Relat Disord 2009; 15(10): 772–775. Dostupné z DOI<http://dx.doi.org/10.1016/j.parkreldis.2009.01.010>.
7. Dušek P, Růžička E. Neurologická forma Wilsonovy nemoci. Postgraduální medicína 2011; 13(5): 472–478.
8. Bruha R, Marecek Z, Pospisilova L et al. Long-term follow-up of Wilson disease: natural history, treatment, mutations analysis and phenotypic correlation. Liver Int 2011; 31(1): 83–91. Dostupné z DOI: <http://dx.doi.org/10.1111/j.1478–3231.2010.02354.x>.
9. Azizi E, Eshel G, Aladjem M. Hypercalciuria and nephrolithiasis as a presenting sign in Wilson disease. Eur J Pediatr 1989; 148(6): 548–549.
10. Menerey KA, Eider W, Brewer GJ et al. The arthropathy of Wilson’s disease: clinical and pathologic features. J Rheumatol 1988; 15(2): 331–337.
11. Saatci I, Topcu M, Baltaoglu FF et al. Cranial MR findings in Wilson‘s disease. Acta Radiol 1997; 38(2): 250–258.
12. Holscher S, Leinweber B, Hefter H et al. Evaluation of the symptomatic treatment of residual neurological symptoms in Wilson disease. Eur Neurol 2010; 64(2): 83–87. Dostupné z DOI: <http://dx.doi.org/10.1159/000316066>.
13. Weiss KH, Gotthardt DN, Klemm D et al. Zinc Monotherapy Is Not as Effective as Chelating Agents in Treatment of Wilson‘s Disease. Gastroenterology 2011; 140(4): 1189–1198. Dostupné z DOI: <http://dx.doi.org/10.1053/j.gastro.2010.12.034>.
14. Gromadzka G, Schmidt HH, Genschel J et al. p. H1069Q mutation in ATP7B and biochemical parameters of copper metabolism and clinical manifestation of Wilson‘s disease. Mov Disord 2006; 21(2): 245–248.
Labels
Diabetology Endocrinology Internal medicineArticle was published in
Internal Medicine

2017 Issue 12
Most read in this issue
- The current role of warfarin
- Citalopram and QT prolongation
- Drug induced tendon injury
- Unusual history of Wilson disease: a case report and review of the literature