
Dvacet let cesty nefrologa do hlubin toxicity fosforu
20 years of nephrologist‘s journey into the depths of phosphorus toxicity
This article focuses on phosphate toxicity in kidney disease and namely in kidney failure. Twenty years ago, the first article documenting the link between hyperphosphatemia and mortality, was published. Shortly after, the association between phosphate and cardiovascular complications in kidney failure was demonstrated. Phosphate itself plays an active role in vascular calcifications. It induces phenotypic changes in vascular smooth muscle cells giving rise a matrix with consequent phosphate and calcium accumulation. The body is fighting again phosphate load. Initially, these mechanisms are adaptive (PTH and FGF-23 elevation). In kidney failure, they become toxic. Dietary measures, gastrointestinal phosphate binders and dialysis elimination are used for correction of hyperphosphatemia. Despite all observations, experimental and clinical studies with many discoveries, the relation between mineral and bone disorder and cardiovascular damage, remains still not fully solved.
Keywords:
cardiovascular complications – FGF-23 – hyperphosphatemia – chronic kidney disease – mediocalcinosis – mineral and bone disorder (CKD-MBD) – phosphate – PTH
Authors:
Sylvie Dusilová Sulková 1,2
Authors‘ workplace:
Hemodialyzační středisko FN Hradec Králové
1; Katedra interních oborů LF UK Praha, pracoviště Hradec Králové
2
Published in:
Vnitř Lék 2020; 66(1): 44-50
Category:
Overview
Práce je zaměřena na toxicitu fosforu při nemocech a zejména selhání ledvin. Před 20 lety byla poprvé publikována souvislost mezi hyperfosfatemií a mortalitou. Krátce poté byla zjištěna souvislost mezi fosforem a kardiovaskulárními komplikacemi při selhání ledvin. Fosfor má aktivní roli v kalcifikaci cév, mění fenotyp buněk hladké cévní svaloviny a do vzniklé matrix se ukládá spolu s vápníkem. Akumulaci fosforu se organismus dlouho brání, avšak zprvu adaptivní mechanismy (elevace FGF-23 a PTH) se při selhání ledvin stávají rovněž toxické. Pro snížení koncentrace fosforu v organismu využíváme dietní opatření, vazače fosforu v zažívacím traktu, a dialyzační eliminaci. Přes všechna pozorování, experimentální i klinické studie s mnoha objevy v předchozích 20 letech, vztah mezi poruchou minerálové a kostní poruchy a kardiovaskulárním poškozením stále není dořešen.
Klíčová slova:
fosfor – FGF-23 – hyperfosfatemie – kalcifikace – kardiovaskulární komplikace – mediokalcinóza – minerálová a kostní porucha při nemocech ledvin (CKD-MBD) – PTH
Úvod
Známým a obligatorním důsledkem chybějící funkce ledvin je hromadění katabolitů, neboli uremických toxinů (se známou uremickou toxicitou) či retinovaných látek (dosud bez prokázané přímé toxicity). Cílem náhrady funkce ledvin je tyto látky odstranit alespoň do té míry, která je slučitelná s dalším životem.
Nejlepší náhradou trvalého selhání ledvin je samozřejmě transplantace. Nejčastější metodou však je a bude hemodialýza (HD) (1). Avšak ta nahrazuje jen funkci vylučovací, navíc pouze částečně a intermitentně. Peritoneální dialýza (PD) je sice kontinuální, ale eliminace katabolitů je rovněž jen částečná. Nefyziologičnost dialýzy, resp. života s dialýzou, přispívá významně k tomu, že dialyzovaní pacienti často trpí komplikacemi, se kterými se jiní pacienti vůbec nesetkají!
Obligatorní komplikací při selhání ledvin je sekundární hyperparatyreóza. Vzniká v důsledku chybění vylučovací a současně regulační a hormonální funkce ledvin. Podle současných poznatků dominuje chybění eliminace toxinů, konkrétně zde jde zejména o fosfor (2, 3).
Velmi často též vidíme komplexní poškození cév a myokardu. Kardiovaskulární komplikace (4–7) jsou dokonce hlavní příčinou úmrtí pacientů s nemocemi i selháním ledvin. K četným známým tzv. „tradičním“ rizikovým faktorům přistupují další, spojované přímo se selháním ledvin („uremia-related“) či s dialyzační léčbou („dialysis related“). Takovýchto tzv. „netradičních“ rizik je při funkčních renálních poruchách velmi velký počet, navíc jejich působení není izolované, ale v komplikovaných propojeních včetně terminálních dysregulací. Jedním z hlavních uremických toxinů, přímo i nepřímo spojených s kardiovaskulárním závažným poškozením, je opět fosfor (3).
Vysoké kardiovaskulární riziko při chronickém onemocnění ledvin (chronic kidney disease – CKD) a zejména při terminálním selhání (end- -stage renal disease – ESRD) je známé mnoho let (8). Avšak možnost, že by toto riziko bylo spojené s toxicitou fosforu, byla dlouho opomíjena. Ještě v roce 1998, kdy Block et al poprvé popsal zvýšené riziko mortality u pacientů s vyšší fosfatemií, možnost kauzality vůbec nezvažoval (9).
První zmínka o možném příčinném vztahu mezi hyperfosfatemií a poškozením cév a myokardu se objevila v písemnictví v roce 1999 (10). Autoři již v názvu svého článku označili fosfor za „tichý zabiják“ (silent killer) dialyzovaných pacientů, analyzovali důvody, ze kterých vyvodili, že „pro dialyzovaného pacienta je kostní nemoc mnohem menší zlo než kardiovaskulární důsledky, které z jeho kostní nemoci plynou“ (přeloženo volně). Takto začala cesta nefrologů do hlubin uvedených v názvu této práce.
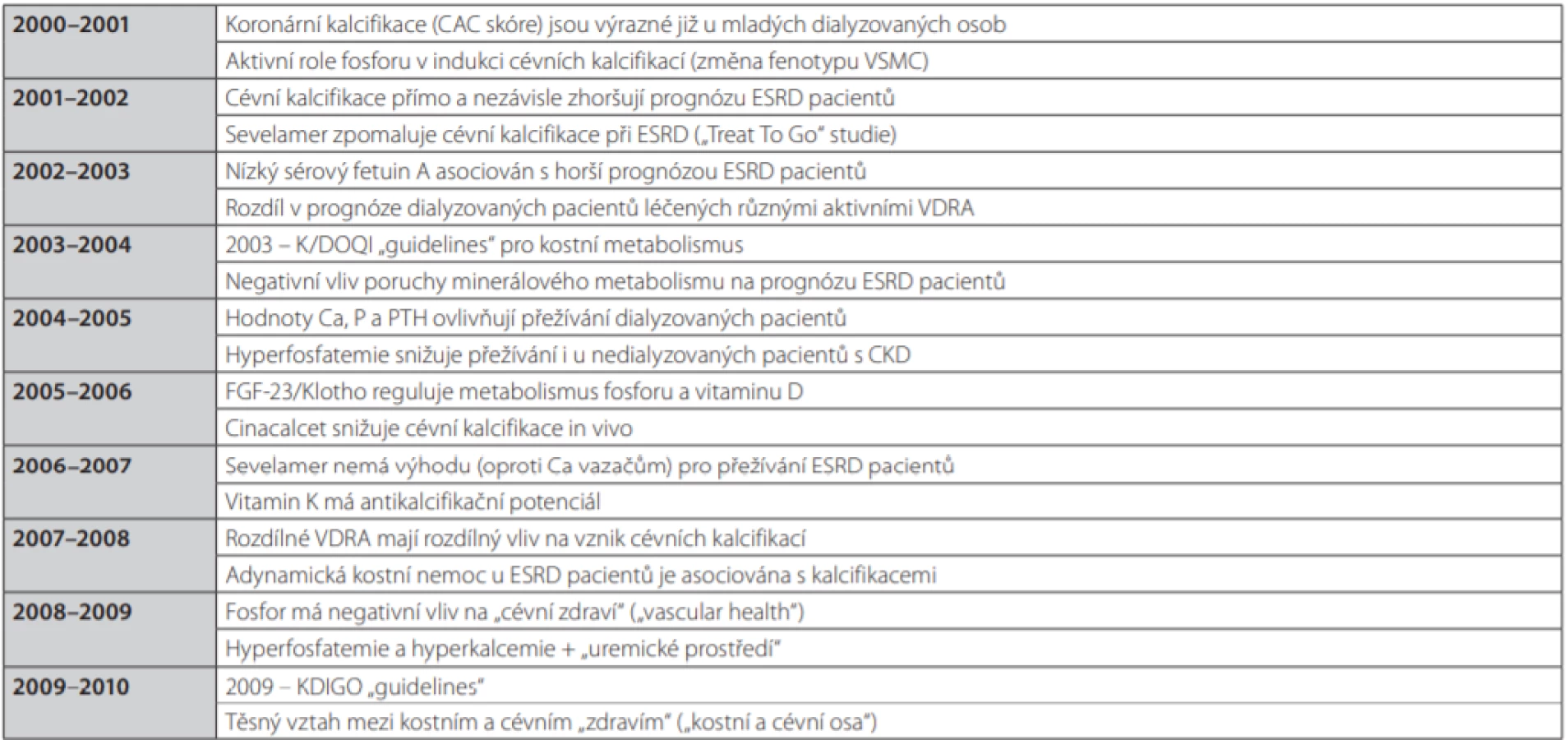
Cílem předkládaného textu je přiblížit poznatky, které postupně, avšak definitivně prokázaly přímou kauzální souvislost mezi kostní a minerálovou poruchou (reprezentovanou zejména hyperfosfatemií a hyperparatyreózou) a kardiovaskulárními komplikacemi, které představují hlavní příčinu morbidity i mortality dialyzovaných pacientů, a též uvést současný přehled možných a dostupných řešení. Šíři nových dílčích témat a jednotlivých poznatků ilustruje částečně Tab. 1 (11).
K vybraným okruhům, které budou zmíněny, patří role fosforu při vzniku a progresi sekundární hyperparatyreózy a minerálové a kostní nemoci; další a jiné projevy fosfátové toxicity; základní poznatky o fosfatoninech (PTH a FGF-23) a jejich vzájemném vztahu i vztahu k aktivnímu vitaminu D; dále současné terapeutické možnosti pro zabránění akumulace fosforu. Zmíněna bude i podstata nejen funkčních, ale i morfologických změn kardiovaskulárního systému, zejména aktivní role fosforu pro vznik mediokalcinózy. Pro úplnost upozorníme na krajní závažnou komplikaci, opět s klíčovou rolí fosforu v patogenezi, na kalcifylaxi.
Fosfor v patogenezi a progresi sekundární hyperparatyreózy
Sekundární hyperparatyreóza (SHPT) je očekávaným důsledkem chronického onemocnění a zejména chronického selhání ledvin (2, 12–14). Patogenetické mechanismy, bez ohledu na komplexnost a složitost, lze zjednodušeně rozdělit do 2 linií:
- chybějící vylučovací funkce ledvin,
- chybějící endokrinní funkce ledvin.
Chybění endokrinní funkce vede k deficitu kalcitriolu. Přitom kalcitriol reguluje přepis genetické informace pro tvorbu PTH v buňkách příštítných tělísek. Pokud chybí vazba kalcitriolu na jaderný vitamin D receptor (VDR), přepisuje se gen pro PTH více, než je potřeba. Tvorba PTH se zvyšuje. Naopak, kalcitriol i parikalcitol po aktivaci VDR a následující složité intracelulární kaskádě intenzitu genové transkripce snižují. Pokud chybí endokrinní funkce, není aktivní vitamin D v ledvinách tvořen a vzniká podklad pro hyperparatyreózu, a sice velké množství přepsané genové informace. Genový transkript, vzniklý přepisem genetické informace, má určitou životnost, během které se z transkriptu přepisuje vlastní PTH. Pokud je transkriptu málo (funguje kalcitriol ve vazbě na VDR), avšak jeho životnost je prodloužena, stále není hyperparatyreóza zažehnána. Vitalitu transkriptu pro PTH prodlužuje zejména hypokalcemie (!) a hyperfosfatemie (!), a to změnou balance mezi ochrannými faktory a endonukleázami (12, 13, 15).
Nyní k druhé linii, chybějící vylučovací funkci. Nejdůležitější roli zde hraje fosfor. Pro organismus je zřejmě tak toxický, že již při malém snížení glomerulární filtrace se aktivují kontraregulačně působící mechanismy (12, 15, 16). Jejich úkolem je zvrátit riziko retence fosforu. Mechanismus spočívá v aktivní inhibici zpětného vstřebávání fosforu v renálních tubulech. Látky, které zvyšují exkreční frakci (frakční exkreci) fosforu, se nazývají fosfatoniny. Dva „fyziologické“ fosfatoniny (PTH a FGF-23) budou popsány v dalším textu. Jejich aktivita je zprvu funkční a přirovnání k „trade-off“ termínu – „něco za něco“ – je vlastně správné. Organismus platí za to, že bude zbaven fosforu. Cena je splatná v budoucnosti a není malá. Při terminální ztrátě vylučovací funkce ledvin „obchod“ končí, ale aktivita fosfatoninů nikoliv. Obě výše zmíněné dráhy vzniku a fixace hyperparatyreózy jsou podle současných poznatků pevně propojeny „můstkem“, který představuje FGF-23.
Z výše uvedeného vyplývá význam fosforu v patogenezi SHPT: s elevací PTH je spojen přímo (mechanismus „trade-off“) i nepřímo (deficit kalcitriolu v důsledku FGF-23). Navíc, fosfor sám o sobě prodlužuje životnost genového transkriptu pro PTH a patří ke stimulátorům patologické hyperplazie PT (2, 13, 15).
Uremická toxicita, uremické toxiny. Fosfor je uremický toxin
Konkrétních uremických toxinů a retinovaných molekul je mnoho a jsou stále dál zkoumány (17). Zjednodušeně se dělí na látky s malou molekulovou hmotností (urea, kreatinin), které se při dialýze dobře eliminují, látky se středně velkou molekulou (β2 -mikroglobulin, relativní molekulová hmotnost 11 800), které se běžnou dialýzou neodstraňují, ale přestupují difuzí a hlavně konvekcí přes vysokopropustné (high-flux) dialyzační membrány (metoda on-line hemodiafiltrace) a látky, resp. toxiny, vázané na bílkoviny (např. indoxylsulfát), které nejsou eliminací odstraněny, resp. jejich odstranění je jen velmi malé (18). K velmi významným uremickým toxinům se řadí i fosfor (3, 16, 19). Jeho eliminační charakteristika při dialýze je specifická: malá molekula sice umožňuje přestup přes membránu, limitující je však vícekompartmentová distribuce v organizmu. Rychlost přestupu fosforu z ostatních kompartmentů do krve je nižší, než eliminace dialýzou. Proto základním požadavkem je zde délka procedury.
Tehdy blíže neurčená „toxicita“ fosforu byla dokumentována již před více než 20 lety (8). Analýza dat dvou velkých kohort dialyzovaných pacientů (rok 1990 a rok 1993) ukázala, že hyperfosfatemie je nezávislým rizikovým faktorem mortality. Zvýšení koncentrace fosforu nad 6,5mg/dl (nad 2,01mmol/l, převodní koeficient z mg/dl na mmol/l je 0,3228) bylo po zohlednění („adjustaci“) demografických a dalších dat spojeno s rizikem o 27 % vyšším než při normofosfatemii (2,4–6,5mg/dl). Se zvyšující se koncentrací fosforu dále stoupalo. Prognosticky nepříznivé byly sice i vysoké koncentrace PTH, ale mortalita asociovaná s hyperfosfatemií byla významnější a navíc na hodnotě PTH nezávislá. Sérová koncentrace kalcia mortalitu neovlivňovala. I když autoři v závěru zdůraznili nutnost korekce hyperfosfatemie, vlastní příčinu vyšší mortality ve spojení s hyperfosfatemií považovali za nejasnou. Jen pro zajímavost (pro srovnání s dnes zjišťovanými koncentracemi v dialyzačních programech) uvádíme tehdejší konkrétní hodnoty: průměrná fosfatemie 6,2mg/dl (2,0mmol/l); ve 30% byla fosfatemie nad 7mg/dl (nad 2,26mmol/l) a u 10% pacientů nad 9mg/dl (nad 2,9mmol/l) (8). Vidíme, že současné koncentrace se liší jen snížením extrémního hodnot.
Koncentrace vápníku v této práci neukázaly žádný vztah k mortalitě. Závěry pozdějších studií byly různorodé. Jako rizikové se ukázaly buď až velmi vysoké (méně často i velmi nízké) koncentrace kalcia v krvi. Sami zastáváme stanovisko, že kalcemie sama o sobě není prognosticky jednoznačná. Koncentrace kalcia v krvi není spolehlivá, neodráží zásoby kalcia v organismu (20). Korekce hypokalcemie u dialyzovaných pacientů není zcela bezpečná a u některých pacientů může být i kontraproduktivní (ukládání do mimokostních tkání, zejména do cév, spolu s fosforem).
Přímá i nepřímá toxicita fosforu pro myokard, a zejména pro cévní stěnu, je zmíněna v samostatné části textu.
Možnosti a limity prevence a léčby hyperfosfatemie
Po objevu toxicity fosforu se rozběhly početné studie zaměřené na možnosti dosažení normofosfatemie, ať již eliminací při dialýze, snížením obsahu fosforu v potravě či zabráněním vstřebání fosforu z potravy pomocí vazačů fosfátů v zažívacím traktu. Pochopitelně, nejblíže k úspěchu je aplikace všech tří postupů souběžně (3).
Dieta s omezením fosforu znamená vynechání potravin s konzervanty a aditivy (potravinová „éčka“ s fosforem), velkým omezením mléčných výrobků včetně sýrů a dalšími mnohými doporučeními. Obecně se preferuje příjem bílkovin rostlinného (neživočišného) původu. Limitem pro praxi je riziko malnutrice – každá potrava s obsahem bílkovin obsahuje fosfor, a přísná restrikce fosforu může skončit až deficitem živin (21).
Vazače fosfátů v zažívacím traktu jsou známy téměř od počátku dialyzačního léčení. Tehdejší pacienti měli nejen vysoké sérové koncentrace fosforu (jejich kardiotoxický význam tehdy znám nebyl), ale spolu s nimi i mimokostní a mimocévní depozita sloučenin fosforu a vápníku (dnes tento nález prakticky již nevidíme). Prvním vazačem fosforu byl kalcium karbonát. Sám o sobě sice snížil fosfatemii, ale nevyřešil problém kalcifikací. Proto byl široce a na dlouhou dobu nahrazen velmi účinným hydroxidem hliníku (aluminium hydroxid, tehdejší přípravek Aludrox). Po rozpoznání často fatální aluminiové toxicity (pozorované na konci 80. let minulého století i u nás) byl hydroxid hliníku opuštěn, u nás naštěstí rychle a úplně.
Rozvoj přinesl několik dalších možností (22, 23). Všechny stávající vazače fosfátů jsou prokazatelně účinné. Případné odlišnosti jsou dány aditivními charakteristikami. Konkrétní volba vazače fosfátů je dána rozhodnutím lékaře, ale i preferencí pacienta. Vazače se užívají zásadně s jídlem (během jídla), kdy je největší dostupnost fosforu pro navázání. Užívání před či po jídle, či dokonce nalačno, je účinné méně, resp. vůbec ne.
Současné přípravky lze rozdělit podle toho, zda obsahují kalcium (kalciové vs nekalciové) (22, 23). Nejde jen o kalcium karbonát (dosud užívaný), ale i kombinovaný vazač obsahující vápník spolu s hořčíkem. Nekalciové vazače lze dále dělit podle toho, zda obsahují či neobsahují kovovou složku. Bez kovů je vazač založený na bázi pryskyřice. K ostatním patří sloučeniny obsahující kov, který se vstřebává extrémně málo (lanthan) či s obsahem železa. Všechny vazače fosfátů působí shodně – principem je navázání fosforu na jednu složku původního vazače, a následnou eliminaci střevem. Aluminium hydroxid obsahoval hliník, jehož vstřebatelnost byla nepatrná, avšak bohužel hliník je eliminován ledvinami, a proto za určitých okolností k akumulaci hliníku docházelo. Analogický současný vazač, využívající lantan, je bezpečnější nejen z ohledu řádově menší absorpce lantanu v porovnání s hliníkem, ale i z hlediska eliminační dráhy (hepatální eliminace).
Vazače fosfátů ani dietní opatření nijak neovlivní endogenní zdroje hyperfosfatemie, vzniklé redistribucí, např. při osteoresorpci při hyperparatyreóze. Navíc, při účinném snížení fosforu v GIT jsou více exprimovány kanály, které aktivně transportují fosfor ze střevního lumen do oběhu (24). Z těchto i z dalších důvodů jsme sami velmi opatrní v posuzování pacienta s hyperfosfatemií z hlediska „adherence“ k léčbě.
Dialyzační eliminace je další účinnou možností. Připomínáme však, že hemodialyzační eliminace je intermitentní, a jen částečná, a proto je klíčovým faktorem čas – délka procedury. Rychlost transferu z intersticia je totiž vždy menší než rychlost eliminace z krve do dialyzačního roztoku.
Stávající preventivní i léčebné možnosti jsou sice široké a účinné, přesto je téma akumulace fosforu v organismu se svými důsledky stále vysoce aktuální (3, 6, 25, 26). I po 20 letech, věnovaných této tematice v celé hloubce i šířce, jsme stále na cestě, nikoliv v cíli.
Cílové koncentrace fosforu u hemodialyzovaných pacientů – je normofosfatemie dosažitelná?
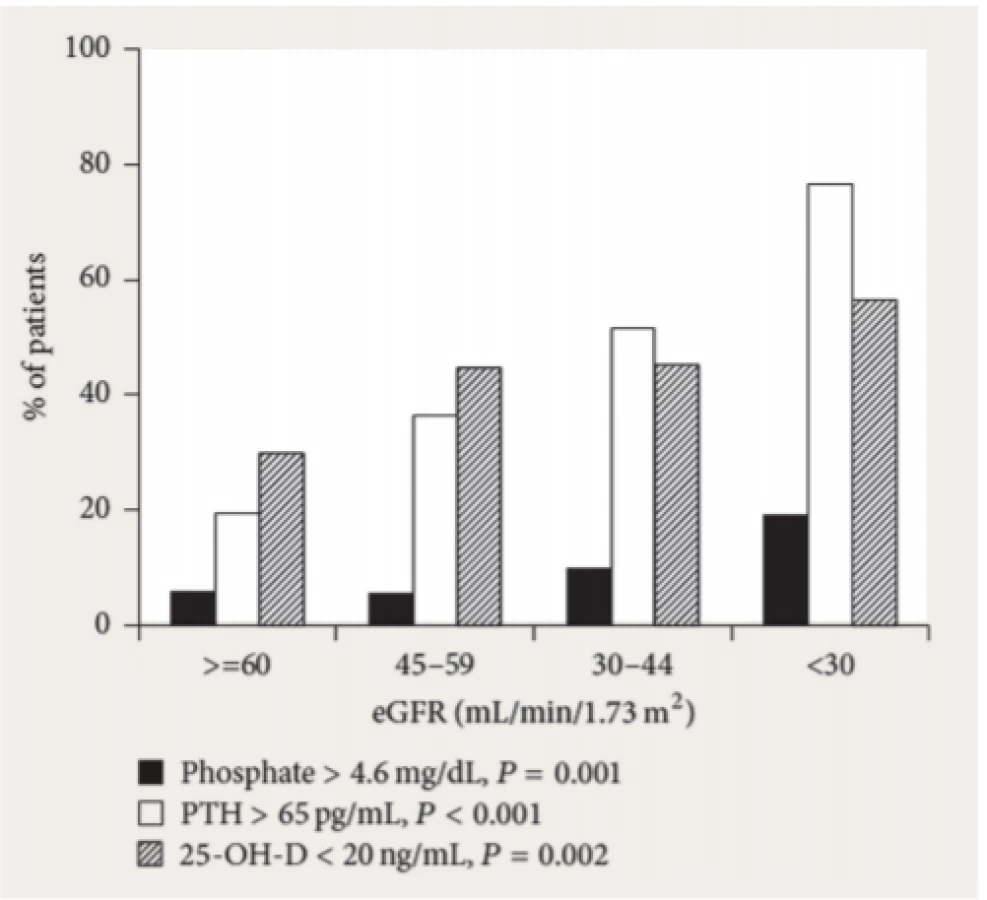
Jaké vlastně mají být cílové koncentrace fosforu? Stanovení se provádí z odběru krve před dialýzou, naměřená hodnota by tedy měla odpovídat maximální možné. Nadnárodní doporučení skutečně považují za „cílovou“ hodnotu tu koncentraci, která nepřesahuje horní referenční mez laboratoře. Stávající poznatky totiž neumožňují kritéria zvolnit, akumulace fosforu skutečně není žádoucí. V současné době je podle nadnárodních doporučení akceptovaná predialyzační koncentrace 1,78 mmol/l (26, 27). A to přesto, že víme, že je to obtížné, jak dokládá analýza dat z více zemí (28).
V režimech častějších a/nebo delších dialýz je úprava fosfatemie mnohem snazší než v konvenčním rozvrhu 3 dialýz týdně. Ti pacienti, kteří se dialyzují vícekrát týdně, mají fosfatemii obvykle v normě a nevyžaduje ani vazače, dokonce se snižují dietní limity. I tato pozitivní zkušenost dokumentuje výhodnost domácí hemodialýzy s alternativními rozvrhy (29).
Kostní a minerálová porucha při chronickém onemocnění ledvin (CKD-MBD)
Termín CKD-MBC (chronic kidney disease – mineral and bone disorder; minerálová a kostní porucha při chronickém onemocnění) byl použit cíleně, poprvé v roce 2006 (30). Definuje a zastřešuje komplexní povahu laboratorních a klinických změn minerálového a kostního metabolismu při CKD, se současným vztahem k cévním, potažmo též myokardiálním změnám, jejichž hlavní podstatou jsou cévní kalcifikace.
Vlastní členění zahrnuje:
- definici poruchy rozpoznatelné z odchylek určených laboratorních ukazatelů, tj. sérové koncentrace Ca, P, PTH, ALP (event. kostního izoenzymu ALP) a též 25D;
- definici vlastní kostní nemoci, založenou výhradně na kostní histomorfometrii (proto bohužel těžko a málo využitelnou v praxi);
- cévní kalcifikace.
V pozdějších schématech je přiřazována sekundární hyperparatyreóza jako uniformní nadstavba, resp. vzájemné propojení všech tří komponent CKD-MBD (30).
V letech 2009 a 2017 byly publikovány a aktualizovány doporučené postupy (clinical practice guidelines), týkající se právě CKD-MBD (28, 31). Mimo jiné obsahují cílové hodnoty jednotlivých ukazatelů; jejich určení se však opírá o „evidence-based“ tvrdá data, což je z hlediska praktického využití někdy problematické. Z nových poznatků je vřazeno mezi vyšetřovací možnosti i vyšetření kostní denzity (DXA). Dříve bylo toto vyšetřování „nedoporučováno“, a to proto, že hodnoty postrádaly interpretaci cíleně platnou pro CKD a dialyzované pacienty.
Dnes je náhled jiný (28). Nízká kostní denzita neodliší typ renální osteopatie, neboť i těžká hyperparatyreóza může mít denzitu normální. Riziko fraktur při nízké kostní denzitně však platí i pro dialyzované osoby, i když příčiny pravděpodobně jsou nejen „renální“. Byl navržen termín „osteoporóza asociovaná s CKD“ (resp. se selháním ledvin). Podle našeho názoru a našich zkušeností patří tyto stavy do společné péče klinického osteologa a nefrologa; po odlišení vlastní „klasické“ renální osteopatie přichází rozvaha o jiných možných příčinách osteoporózy, včetně věku, inaktivity a hormonálních změn, a též i podílu předchozí či stávající terapie kortikoidy, což je u nefrologických pacientů časté. V těchto situacích léčbu vede na našem pracovišti osteolog (32).
Třetí komponenta CKD-MBD, tj. kalcifikace, bude zmíněna samostatně (viz dále).
Fosfor a cévní kalcifikace
Definice a texty věnované problematice CKD-MBD se cíleně zaměřují i na klinický význam a detekci cévních a dalších kalcifikací (19, 28, 31). Pokud jsou zjištěny, je třeba pacienta považovat za kardiologicky vysoce rizikového.
V cévních kalcifikacích kupodivu není rozhodující samotný vápník, ale – fosfor! Působení fosforu v genezi kalcifikací, umístěných naprosto nefyziologicky v cévní stěně (vrstva svalové hladké svaloviny), není pasivní (prosté ukládání, spolu s vápníkem), ale aktivní (3, 33, 34). Fosfor dokáže aktivně změnit fenotyp buněk hladké cévní svaloviny v buňky podobné buňkám kostním. Ty produkují „matrix“, a do té se fosfor (spolu s vápníkem) ukládá. Výsledkem je tzv. mediokalcinóza tepen. Nejde o nějaký ojedinělý či málo zřetelný nález. Naopak – u pacientů v dialyzačním programu (někdy již v predialýze) zaznamenáváme na RTG snímcích viditelné (kalcifikované) cévy, resp. arterie až do periferie. Mediokalcinóza koronárních tepen je příčinou tzv. „microvessel disease“, „onemocnění malých cév“. Při myokardiální lokalizaci je klinický obraz totožný jako při ischemii na podkladě aterosklerózy. Mediokalcinóza, na rozdíl od aterosklerózy (která je u dialyzovaných pacientů rovněž silně vyjádřena), zhoršuje dodávku kyslíku tím, že cévní stěna ztrácí pružnost a adaptaci na aktuální koronární průtok (5–7, 33, 34).
Právě aktivní role fosforu v přímém poškození cévní stěny je podstatou toho, že fosfor, resp. jeho akumulace, či dokonce hyperfosfatemie, jsou skutečně považovány za toxické. Dominanta fosforu v procesu kalcifikace cévní stěny je velká a potřebný vápník je „nalezen“ vždy, bez ohledu na koncentraci vápníku v krvi.
Adaptivní a maladaptivní role fosfatoninů
Základní charakteristika zprvu pozitivních obou fosfatoninů již byla popsána (12). Oba působí aktivně na transportní mechanismus fosforu v renálních tubulech, který inhibují (odtud název „fosfatoniny“). V dalších účincích však je jejich role rozdílná: zatímco PTH zvyšuje renální hydroxylaci 25-hydroxycholekalciferolu, FGF-23 ji snižuje. FGF-23 a PTH mají tedy shodný účinek ve vztahu k bilanci fosforu. Naopak, FGF-23 a PTH mají rozdílný účinek ve vztahu k bilanci kalcia. Zatímco kalcium snižuje parathormon, na FGF-23 má vliv opačný (při kalciové zátěži FGF-23 stoupá) (12).
Jak známo, tvorba kalcitriolu je řízena renální 1α-hydroxylázou. Oba fosfatoniny tento enzym regulují, avšak nejsou samy. Výsledná koncentrace kalcitriolu v krvi je odrazem nejméně 4 souběžně působících vlivů:
- zachovalý renální parenchym,
- dostupnost prekurzoru, tj. dostatek vitaminu D v organismu,
- aktivita příštítných tělísek,
- koncentrace a aktivita FGF-23.
Zatímco první 3 vlivy tvorbu kalcitriolu podporují a podněcují, resp. k ní přímo přispívají, poslední zmíněný (FGF-23) působí jednoznačně a silně negativně – tvorbu kalcitriolu tlumí (degraduje enzym zodpovědný za tvorbu).
Při finální maladaptaci patří FGF-23 i PTH mezi významné uremické toxiny. Představují tzv. „netradiční“ („uremia related“) rizikové faktory poškozující kardiovaskulární systém, s velmi vážnými důsledky (35–7).
Parathormon je mimo jiné permisivním faktorem fibrotizace myokardu (38, 39). Fibrotizace sama jako důsledek nepoměru mezi hypertrofovaným myokardem a původním cévním zásobení. Při takto vzniklé „mikroischemii“ reaguje srdce vznikem mikrojizev, dochází k fibróze tkáně myokardu. Na této změně se aktivně účastní i PTH.
K doloženým a/nebo stále studovaným účinkům a důsledkům vysokých koncentrací FGF-23 u pacientů s onemocněním, resp. selháním ledvin patří predikce budoucí refrakterní hyperparatyreózy, rychlejší progrese CKD a obecně nepříznivá prognóza. Je doložen přímý kardiovaskulární toxický vliv, jeho molekulární podstata se zkoumá (12, 15, 39–41).
K problematice kalcia
V počátcích dialyzační léčby nebyla z kalcia žádná obava, naopak (bilance kalcia při selhání ledvin byla považována za negativní). Vazače fosfátů byly na kalciové bazi (prvním dostupným vazačem fosfátů byl uhličitan vápenatý, kalciumkarbonát). Koncentrace (difuzibilního) kalcia v dialyzačním roztoku 1,75mmol/l byla běžná. Ani sekundární hyperparatyreóza neupozornila na žádné riziko, neboť byla (a je) provázena normokalcemií.
Po dlouhou dobu ani léčba aktivními metabolity vitaminu D neupozornila na to, že kalciová bilance při selhání ledvin již vlastně dávno není negativní. Naopak! Pokud používáme léky s obsahem kalcia, navyšujeme koncentraci kalcia v dialyzačním roztoku, a zvyšujeme přívod kalcia metabolity vitaminu D, je bilance vždy pozitivní. Velikost takto „dodaného“ kalcia však nelze určit, a nelze ji ani odhadnout (vstřebávání kalcia z kalciových přípravků je silně variabilní). Doporučené koncentrace kalcia v krvi u dialyzovaných pacientů jsou identické s laboratorním referenčním rozmezím (28). Jejich dosažení však u části pacientů je problematické, při souběžné malnutrici či zánětu až nemožné (20, 27).
Kalciová bilance po podání vitaminu D je pozitivní. Nativní vitamin D ji zvýší nejméně. Kalcitriol má vyšší hyperkalcemizující potenciál než parikalcitol. Nejvyšší hrozba byla časově shodná s obdobím, kdy byl (přechodně) dostupný intravenózní kalcitriol; záhy byl nahrazen právě parikalcitolem, který používáme stále, ale opatrně.
Pozitivní kalciová bilance by mohla být pozitivní pro metabolismus kosti. Při selhání ledvin však bohužel často nevíme, zda dodané kalcium bude skutečně aponováno do kosti (viz doprovodná sekundární hyperparatyreóza a na druhé straně viz posun rovnováhy prokalcifikačních a protikalcifikačních faktorů z hlediska kalcifikace cév) (3, 33, 34).
Pozitivní kalciová bilance se účastní patogeneze mimokostních kalcifikací měkkých tkání a kalcifikací stěny arterií všech kalibrů. Podílí se na poškození kardiálních myocytů. Spolu s fosforem zvyšuje tuhost cévní stěny a mechanismem srdečního přetížení (afterload) přetěžuje srdce (20).
I při selhání ledvin je někdy nutné kalcium do organismu doplnit. Typicky při „fenotypu osteomalacie“, kdy je kalcium spíše nižší a je vyšší kostní ALP. V pozadí je deficit vitaminu D. Další situací je syndrom „hladové kosti“ („hungry bone syndrome“) (42). Naopak, při hypokalcemii asociované s hypoparatyreózou, jsme zdrženliví. Veškeré dodané kalcium zůstává v organismu, dialyzační procedura s nižším kalciem není řešením, neboť odstraní jen aktuálně volné kalcium. Cévní kalcifikace jsou již ireverzibilní, a ve své podstatně progresivní.
Souvislost mezi hyperfosfatemií a vitaminem D
Velká část této problematiky byla již zmíněna či alespoň naznačena. Ačkoliv se to může zdát nepravděpodobné a pro mnohé velmi překvapivé, nízké hladiny nativního vitaminu D jsou spojeny s vyšším kalcifikačním rizikem (43). Pouze takové „zacházení“ s vitaminem D (myšleno se všemi jeho metabolity, analogy, a selektivně působícími aktivátory receptoru pro vitamin D, tj. VDR aktivátory), které nerespektuje požadavky bezpečnosti a pouze směřuje k „účinnosti“, je prokalcifikační. V těchto případech jde přímo o riziko potencované (souběžně pozitivní bilance kalcia a fosforu). Aditivním rizikovým faktorem je malnutrice, zánět, předchozí resp. již přítomné kalcifikace, špatně rozvržené dávkování resp. celá léčebná strategie, a pochopitelně i špatně kontrolovaná fosfatemie a pozitivní kalciová bilance plynoucí z dalších zdrojů.
I když vitamin D zvyšuje bilanci vápníku a fosforu, nemají vitamin D a jeho metabolity a analoga samy o sobě přímý prokalcifikační vliv. Prokalcifikačně však může dopadnout neoptimální zacházení s preparáty vitaminu D (viz výše). Pro hladiny vitaminu D platí tzv. „U“ křivka rizika (nežádoucí jsou hladiny nejen vysoké, ale i nízké) (27, 43). Pokud však probíhá léčba vitaminem D a fosfatemii není věnována pozornost, vytvářejí se cévní kalcifikace vzhledem k vyšší dostupnosti fosforu výrazně více.
Krátce ke kalcifylaxi
Vzhledem k mimořádné klinické závažnosti (mortalita i při léčbě stále více než 60%, někdy uváděno i 80%) zmíníme i toto téma, neboť i sem sahá přesah toxicity fosforu (přehledně viz (44)). I když patogeneze a rizikové faktory jsou nejen komplexní, ale i široké a navzájem nespojité, podstatou je vždy porucha metabolismu vápníku a fosforu, bez ohledu na to, zda je viditelná, či nikoliv.
Vždy zjišťujeme mikrocirkulační poruchu, vzniklou obvykle v terénu mediokalcinózy (!), lokalizované periferně, s predispozicí k oblastem s tukovým podkožím (stehna, bérce, ale i břicho, a jiná místa, vzácně i vnitřní orgány). Podstatou je souběh mikroischemie při chybějící mikroperfuzi v kombinaci s trombotickým uzávěrem návratové mikrovaskulatury. Nekrotická tkáň je dříve nebo později infikována, průnik antibiotik do tkáně do nekróz je silně limitován. Kromě okamžitého přerušení expozice warfarinu (44, 45) kontrolujeme hladiny i bilanci fosforu i kalcia a pátráme po hyperparatyreóze, kterou neprodleně a radikálně řešíme. Můžeme však vidět i kalcifylaxi sdruženou naopak s hypoparatyreózou. Z důvodu bezpečnosti pro pacienta je optimální cestou předání pacienta na pracoviště, které má erudici, zkušenosti a potřebné zázemí pro léčbu.
Tato sice velmi vzácná, avšak přesto se vyskytující komplikace zejména (ale nikoliv pouze) u dialyzovaných pacientů je zde zmíněna jako dokreslení toho, jak daleko může patofyziologická abnormalita asociovaná se selháním ledvin, doprovázená i dalšími okolnostmi, ve své extrémní podobě vést!
Závěr
Hlavní lékařsky orientovanou pracovní náplní (nejen) dialyzačních nefrologů není „jen“ stanovení renální diagnózy a její léčba, dlouhodobá nefrologická dispenzarizace či rozvaha o metodě náhrady funkce ledvin při jejich selhání. Většinu své pracovní invence zaměřuje nefrolog, stejně jako lékař jiných odborností a specializací, na komplexní péči o celkový zdravotní stav svěřených pacientů.
Zejména v dialyzační nefrologii se setkáváme s unikátními patofyziologickými cestami, odrážejícími skutečnost (dříve prakticky opomíjenou), že spolu s klesající funkcí ledvin se aktivují regulační mechanismy, zprvu sice účinné, avšak posléze jednoznačně destruktivní. V této chvíli jsou však již fixované, obvykle podmíněny i morfologickými změnami daného systému. Touto cestou vznikají unikátní a bohužel nepříznivé komplikace, jejichž klinická manifestace i význam nakonec dominují nad (dialýzou či transplantací léčitelnými a léčenými) projevy selhání ledvin.
Na toxicitu fosforu může být zprvu nahlíženo jen jako na sklíčko v mozaice barevného okna chrámu, které právě se zájmem studujeme. Rozsáhlá a aktivní role fosforu v popsaném přesahu minerálové a kostní patofyziologie při selhání ledvin do patologie dalších orgánů a systémů (zde spojení s kardiovaskulárním systémem) ukazuje, jak i při sebelepší dialyzační technice nedostihneme následky, které selhání ledvin vyvolává. Vlastní selhání ledvin je totiž mnohem a mnohem komplikovanější, než by plynulo z pouhé retence. O prognóze pacienta se selháním ledvin nakonec nerozhoduje chybění životně důležitého orgánu, ale přesah patofyziologických vazeb do závažných poškození jiných orgánů. Další uremické toxiny jsou zkoumány, avšak příběh toxicity fosforu ještě nekončí.
Podpořeno projekty LFHK Progres Q40-14 a MZ ČR–RVO FNHK, 00179906; kód 8100.
KORESPONDENČNÍ ADRESA AUTORA:
prof. MUDr. Sylvie Dusilová Sulková, DrSc., MBA,
Hemodialyzační středisko FN
Sokolská tř. 581,
500 05 Hradec Králové
Cit. zkr: Vnitř Lék 2020; 66(E-1): e19–e25
Článek přijat redakcí: 16. 4. 2019
Článek přijat k publikaci: 15. 11. 2019
Sources
1. U.S. Renal Data System. USRDS 2018 Annual Data Report: Atlas of End-Stage Renal Disease in the United States 2018. www.usrds.org
2. Cunningham J, Francesco Locatelli F et al. Secondary Hyperparathyroidism: Pathogenesis, Disease Progression, and Therapeutic Options. Clin J Am Soc Nephrol 2011; 6: 913–921.
3. Vervloet M, Sezer S, Massy ZA, et al. on behalf of the ERA-EDTA Working Group on Chronic Kidney Disease-Mineral and Bone Disorders and the European Renal Nutrition Working Group: The role of phosphate in kidney disease. Nat Rev Nephrol 2016; 13: 27–38.
4. Shefold JC, Filipatos G, Hasenfuss G, et al. Heart failure and kidney dysfunction: epidemiology, mechanism and management. Nat Rev Nephrol 2016; 12: 610–623.
5. Herzog CA, Asinger RW, Berger AK. Cardiovascular disease in chronic kidney disease. A clinical update from Kidney Disease: Improving Global Outcomes (KDIGO). Kidney Int 2011; 80: 572–586.
6. Gansevoort RT, Correa-Rotter R, Hemmelgarn BR, et al. Chronic kidney disease and cardiovascular risk: epidemiology, mechanisms, and prevention. Lancet 2013; 382: 339–352.
7. Cozzolino M, Mangano M, Stucchi A, et al. Cardiovascular disease in dialysis patients. Nephrol Dial Transplant 2018; 33(suppl. 3): iii28–iii34.
8. Lazarus JM, Lowrie EG, Hampers CL, et al. Cardiovascular disease in uremic patients on hemodialysis. Kidney Int 1995; Suppl. 2: 167–175.
9. Block GA, Hulbert-Shearon TE, Levin NW, et al. Association of serum phosphorus and calcium x phosphate product with mortality risk in chronic hemodialysis patients: a national study. Am J Kidney Dis 1998; 31: 607–617.
10. Amann K, Gross ML, London GM, et al. Hyperphosphatemia – a silent killer of patients with renal failure? Nephrol Dial Transplant 1999; 14: 2085–2087.
11. Brandenburg
12. Silver J, et al. FGF-23 and secondary hyperparathyroidism in chronic kidney disease. Nat Rev Nephrol 2013; doi:10.1038/nrneph.2013.147
13. Goodman WG, Quarles LD. Development and progression of secondary hyperparathyroidism in chronic kidney disease: lessons from molecular genetics. Kidney Int 2008; 74: 276–288.
14. Týcová I, Sulková SD, Štěpánková J, et al. Molecular patterns of diffuse and nodular parathyroid hyperplasia in long-term hemodialysis. Am J Physiol Endocrinol Metab 2016; 311: E720–E729.
15. Silver J, Kilav R, Naveh-Many T Mechanisms of secondary hyperparathyroidism. Am J Physiol Renal Physiol 2002; 283: F367–F376.
16. Komaba H, Fukagawa M Phosphate-a poison for humans? Kidney Int 2016; 90: 753–763.
17. Vanholder R, Argiles A, Baurmeister U, et al. Uremic toxicity: present state of the art. Int J Artif Organs 2001; 24: 695–725.
18. Hyšpler R, Tichá A, Šafránek R, et al. Indoxyl Sulfate Elimination in Renal Replacement Therapy: Influence of Citrate- versus Acetate-Buffering Component during Bicarbonate Dialysis. Dis Markers 2018; 15: 3985–3986.
19. Ritter CS, Slatopolsky E Phosphate Toxicity in CKD: The Killer among Us. Clin J Am Soc Nephrol 2016; 11: 1088–1100.
20. Moe SM Calcium as a cardiovascular toxin in CKD-MBD. Bone 2017; 100: 94–99.
21. Lynch K, Lynch R, Curhan GC, et al. Prescribed dietary phosphate restriction and survival among hemodialysis patients. Clin J Am Soc Nephrol 2011; 6: 620–629.
22. Sekercioglu N, Thabane L, Diaz-Martinez JP, et al. Comparative Effectiveness of Phosphate Binders in Patients with Chronic Kidney Disease: A Systematic Review and Network Meta-Analysis. PLoS One 2016; 11(6): e0156891.
23. Floege J. Phosphate binders in chronic kidney disease: a systematic review of recent data. J Nephrol 2016; 29: 329–340.
24. Isakova T, Ix JH, Sprague SM, et al. Rationale and Approaches to Phosphate and Fibroblast Growth Factor 23 Reduction in CKD. J Am Soc Nephrol 2015; 26: 2328–2339.
25. Mehta R, Isakova T Continued search for therapies to favourably modify phosphate and FGF-23 levels in CKD. Clin J Am Soc Nephrol 2017; 12: 1911–1913.
26. Ketteler M, Block GA, Evenepoel P, et al. Executive summary of the 2017 KDIGO Chronic Kidney Disease-Mineral and Bone Disorder (CKD-MBD) Guideline Update: what’s changed and why it matters. Kidney Int 2017; 92: 26–36.
27. Cozzolino M. CKD-MBD KDIGO guidelines: how difficult is reaching the “target”? Clin Kidney J 2018; 11: 70–72.
28. KDIGO 2017 clinical practice guideline update for the diagnosis, evaluation, prevention, and treatment of Chronic Kidney Disease-Mineral and Bone Disorder (CKD-MBD) Kidney Int Suppl 2017; 7: 1–59.
29. Lopot F, Švára F, Polakovič . „Revival domácí dialýzy”. Aktuality v nefrologii 2018; 24: 46–55.
30. Moe S, Drueke T, Cunningham J, et al. Definition, evaluation, and classification of renal osteodystrophy: a position statement from Kidney Disease: Improving Global Outcomes (KDIGO). Kidney Int 2006; 69: 1945–1953.
31. KDIGO clinical practice guideline for the diagnosis, evaluation, prevention, and treatment of chronic kidney disease-mineral and bone disorder (CKD-MBD). Kidney Int 2009; 76: (Suppl. 113): S3–S130.
32. Brunerová L, Palicka V, Sulkova SD. Commentary on management of osteoporosis in advanced CKD: common view of a nephrologist and a bone specialist. Endocr Pract 2019; 25: 193–196.
33. Lau WL, Pai A, Moe SM, et al. Direct effects of phosphate on vascular cell function. Adv Chronic Kidney Dis 2011; 18: 105–112.
34. Cozzolino M, Ciceri P, Galassi A. The Key Role of Phosphate on Vascular Calcification. Toxins 2019; 11: 213.
35. Disthabanchong S. Phosphate and cardiovascular disease beyond chronic kidney disease and vascular calcification. Int J Nephrology 2018; doi.org/10.1155/2018/3152806
36. Razzaque MS The FGF23-Klotho axis: endocrine regulation of phosphate homeostasis. Nat Rev Endocrinol 2009; 5: 611–619.
37. Grabner A, Mazzaferro S, Cianciolo G. Fibroblast Growth Factor 23: Mineral Metabolism and Beyond. Contrib Nephrol 2017; 190: 83–95.
38. Rodriguez M, Lorenzo V. Parathyroid hormone, a uremic toxin. Semin Dial 2009; 22: 363–368.
39. Navarro-García JA, Fernández-Velasco M, Delgado C, et al. PTH, vitamin D, and the FGF-23-klotho axis and heart: Going beyond the confines of nephrology. Eur J Clin Invest 2018; 48: doi: 10.1111/eci.12902.
40. Gutiérrez OM, Mannstadt M, Isakova T. Fibroblast growth factor 23 and mortality among patients undergoing hemodialysis. N Engl J Med 2008; 359: 584–592.
41. Agarwal I, Ide N, Ix JH. Fibroblast growth factor-23 and cardiac structure and function. J Am Heart Assoc 2014; 3: e000584.
42. Ho LY, Wong PN, Sin HK, et al. Risk factors and clinical course of hungry bone syndrome after total parathyroidectomy in dialysis patients with secondary hyperparathyroidism. BMC Nephrol 2017; 18: 12.
43. Wang J, Zhou JJ, Robertson GR, et al. Vitamin D in Vascular Calcification: A Double-Edged Sword? Nutrients 2018; 10. pii: E652.
44. Nigwekar SU, Thadhani R, Brandenburg VM. Calciphylaxis. N Engl J Med 2018; 378: 1704–1714.
45. Nigwekar SU, Kroshinsky D, Nazarian RM. Calciphylaxis: risk factors, diagnosis, and treatment. Am J Kidney Dis 2015; 66: 133–146.
Labels
Diabetology Endocrinology Internal medicineArticle was published in
Internal Medicine

2020 Issue 1
Most read in this issue
- Sarkopenická obezita – aktuální přehled problematiky
- Chronický stres, psychická nepohoda a deprese zvyšují četnost infekčních, autoimunitních, ale i maligních nemocí
- Odešel velký člověk a lékař prof. MUDr. Vítězslav Kolek, DrSc., FCCP
- Bolesti a deformace dolní čelisti – projev fibrózní dysplazie čelisti