
Pendredův syndrom u pacientů s hypotyreózou: genetická diagnostika, fenotypová variabilita a výskyt fenokopií
Pendred Syndrome Among Patients with Hypothyroidism: Genetic Diagnosis, Phenotypic Variability and Occurrence of Phenocopies
Background.
Pendred syndrome (OMIM274600) is one of the causes of congenital hypothyroidism due to thyroid dyshormonogenesis. It is an autosomal recessive disease classically characterized by dyshormonogenetic goitre and sensorineural deafness. It is caused by mutations in PDS/SLC26A4 gene encoding for pendrin – an anion transporter, mostly expressed in the thyroid gland and the inner ear. The thyroid impairment in Pendred syndrome develops only in 80% of affected individuals in form of a euthyroid or hypothyroid goitre, which is rarely present at birth, when it can be diagnosed by the neonatal screening for congenital hypothyroidism.
The study was aimed to identify patients with Pendred syndrome among children with congenital or postnatal non-autoimmune hypothyroidism and subsequently confirm the diagnosis by finding mutations in the PDS/SLC26A4 gene.
Methods and Results.
We examined two-hundred thirty-six Caucasians with hypothyroidism diagnosed by screening or developing later in childhood. The clinical diagnosis of Pendred syndrome was based on the laboratory and ultrasonographic signs of thyroid dyshormonogenesis (elevated TSH, low T4/fT4, goitre or normal thyroid volume) in association with sensorineural hearing loss. In subjects clinically diagnosed as Pendred syndrome, we sequenced all 21 exons of the PDS/SLC26A4 gene and their flanking intron-exon junctions.
Among 236 children, nine fulfilled the diagnostic criteria of Pendred syndrome. In four, the diagnosis was confirmed by identification of mutations in the PDS/SLC26A4 gene, the remaining five patients were concluded phenocopies.
Conclusions.
Our study confirms the high phenotypic variability of thyroid impairment in Pendred syndrome and underlines the necessity of a molecular-genetic investigation for establishing the diagnosis in regard of the great number of phenocopies.
However, from the endocrinologist’s point of view, the genetic testing is only reasonable in patients with congenital hypothyroidism due to dyshormonogenesis in association with sever to profound sensorineural hearing loss.
Key words:
congenital hypothyroidism, dyshormonogenesis, pendrin, SLC26A4, Pendred syndrome
Authors:
K. Banghová 1; E. Al Taji 2; D. Novotná 3; J. Zapletalová 4; O. Hníková 2; J. Čáp 5; J. Klabochová 6; M. Kúseková 7; J. Lebl 1
Authors‘ workplace:
Pediatrická klinika UK 2. LF a FN Motol, Praha
1; Klinika dětí a dorostu UK 3. LF a FN KV, Praha
2; II. dětská klinika LF MU a FN, Brno
3; Dětská klinika LF UP a FN, Olomouc
4; 2. Interní klinika FN, Hradec Králové
5; Nemocnice U sv. Jiří, Plzeň
6; Detská Fakultná Nemocnica Košice
7
Published in:
Čas. Lék. čes. 2008; 147: 616-622
Category:
Original Article
Overview
Východisko.
Pendredův syndrom (OMIM274600) je jednou z příčin kongenitální hypotyreózy na podkladě dyshormonogeneze. Jedná se o autozomálně recesivní onemocnění definované jako asociace senzorineurální hluchoty a dyshormonogenetické strumy. Onemocnění je způsobeno mutacemi v PDS/SLC26A4 genu kódujícím pendrin – aniontový transportér, který je exprimován hlavně ve štítné žláze a vnitřním uchu. Dyshormonogeneze se u Pendredova syndromu projeví jen u 80 % pacientů jako eutyreoidní nebo hypotyreoidní struma a jen zřídka je klinicky významná již při narození, kdy může být diagnostikována novorozeneckým skríninkem.
Cílem studie bylo identifikovat pacienty s klinickou diagnózou Pendredova syndromu mezi dětmi s vrozenou nebo postnatálně vzniklou non-autoimunitní hypotyreózou a následně diagnózu potvrdit nalezením mutací v genu pro pendrin.
Metody a výsledky.
Vyšetřili jsme 236 dětí s hypotyreózou zjištěnou novorozeneckým skríninkem nebo později v dětství. Klinická diagnóza Pendredova syndromu byla založena na laboratorních a sonografických známkách tyreoidální dyshormonogeneze (vysoké TSH, nízké T4/fT4, struma nebo normální velikost štítné žlázy) ve spojení se senzorineurální poruchou sluchu. U pacientů s klinickou diagnózou Pendredova syndromu bylo přímou sekvenací vyšetřeno všech 21 exonů a přilehlých intronových úseků PDS/SLC26A4 genu. Z 236 dětí splňovalo diagnostická kritéria Pendredova syndromu devět. Molekulárně-genetické vyšetření PDS/SLC26A4 genu prokázalo složeně heterozygotní nosičství mutací u čtyř pacientů, ostatních pět bylo uzavřeno jako fenokopie Pendredova syndromu.
Závěry.
Naše studie potvrzuje vysokou fenotypovou variabilitu postižení štítné žlázy u pacientů s Pendredovým syndromem a poukazuje na potřebnost genetického vyšetření k potvrzení klinické diagnózy vzhledem k častému výskytu fenokopií. Z endokrinologického hlediska však má molekulárně-genetické vyšetření genu pro pendrin smysl zřejmě jen u pacientů s vrozenou hypotyreózou na podkladě dyshormonogeneze, která je spojena se středně těžkou až těžkou senzorineurální poruchou sluchu.
Klíčová slova:
kongenitální hypotyreóza, dyshormonogeneze, pendrin, SLC26A4, Pendredův syndrom.
Kongenitální hypotyreóza je nejčastější vrozenou endokrinní poruchou s frekvencí 1 : 3000 až 1 : 4000 novorozenců (1). Ve 20–25 % je její příčinou dyshormonogeneze, tj. porucha biosyntézy tyreoidálních hormonů způsobená poruchou transportu jódu, jeho organifikace, poruchou syntézy tyreoglobulinu nebo poruchou dehalogenace (1). V posledních deseti letech byla objasněna molekulární podstata některých těchto poruch a narůstá počet známých genů, jejichž porucha může dyshormonogenezi způsobovat. Jedním z nich je gen pro pendrin, transmembránový protein s funkcí aniontového transportéru ve štítné žláze a ve vnitřním uchu (2). Je známý pod názvy PDS (Pendred syndrome gene) nebo SLC26A4 (member 4 of solute carrier family 26) (3). Jeho mutace vedou ke klinickému obrazu Pendredova syndromu. Pendredův syndrom patří mezi méně časté příčiny tyreoidální dyshormonogeneze s poruchou organifikace jódu. Dědí se autozomálně recesivně a jeho incidence se odhaduje na 7,5–10 : 100 000 (4–7).
Klinický obraz Pendredova syndromu je variabilní. Obvykle dominuje vrozená porucha sluchu, která se projeví v časném dětství a má senzorineurální (tj. percepční) charakter (4). Tato porucha je většinou oboustranná, někdy asymetrická a zpravidla těžká (> 60 dB). V průběhu života progreduje, ale tíže postižení může kolísat. Je provázena strukturálními malformacemi vnitřního ucha, které lze radiologicky prokázat. Nejčastější je rozšíření vestibulárního akvaduktu (EVA) spojené s dilatací membranózního labyrintu (ductus a saccus endolymphaticus), méně často na CT spánkové kosti nacházíme tzv. Mondiniho kochleu (8).
Porucha funkce štítné žlázy je méně konstantním projevem Pendredova syndromu, a to jak inter-, tak intrafamiliárně. Důvodem této fenotypové variability mohou být zřejmě jak exogenní faktory (např. rozdílný příjem jódu ve stravě), tak rozdílný vliv jednotlivých mutací PDS/SLC26A4 genu na funkci pendrinu nebo přítomnost jiných dosud neznámých kompenzačních mechanizmů (např. jiných apikálních transportérů).
Dysfunkce štítné žlázy se proto u Pendredova syndromu projeví jenom u přibližně 80 % pacientů, a to ve formě strumy – většinou eufunkční (9). Stejně jako u ostatních dyshormonogenetických strum je i v tomto případě struma zprvu zpravidla difuzní, ale postupně podléhá nodulární přestavbě s rizikem maligní transformace. Některé studie uvádějí, že dyshormonogenetické strumy mají signifikantně vyšší výskyt folikulárních karcinomů než strumy jiné etiologie, což se vysvětluje chronickou stimulací folikulárních buněk zvýšenými hladinami tyreotropinu (10).
Jen asi 30 % pacientů s Pendredovým syndromem má subklinickou nebo manifestní hypotyreózu. Různorodý je i věk při prvním průkazu hypotyreózy – kolísá od narození až po dospělost, převažuje ale druhá dekáda života, zejména období kolem puberty (9). Jen v jednotlivých případech lze hypotyreózu diagnostikovat již novorozeneckým skríninkem (9, 11).
Vzhledem k vyšší penetranci postižení sluchu než štítné žlázy vycházela většina dosavadních studií Pendredova syndromu ze souborů pacientů se sluchovou poruchou. Cílem naší studie bylo identifikovat pacienty s Pendredovým syndromem a mutací v genu pro pendrin mezi pacienty s hypotyreózou, a tím vytvořit nový pohled na toto onemocnění – z hlediska endokrinologa.
SOUBOR NEMOCNÝCH A POUŽITÉ METODY
Pacienti
Do studie jsme zahrnuli pacienty ze tří různých kohort. První a největší skupinu tvořilo 197 dětí a adolescentů s kongenitální hypotyreózou diagnostikovanou novorozeneckým skríninkem v České republice v letech 1985–2005. Výsledky této části studie jsme již samostatně publikovali (12). Druhou skupinu 35 jedinců tvořilo pět pacientů s kongenitální hypotyreózou, narozených před zavedením skríninku, a 30 pacientů s hypotyreózou diagnostikovanou klinicky v pozdějším dětském věku. Třetí skupina představovala čtyři pacienty s klinickou diagnózou Pendredova syndromu, jejichž lékaři nás přímo požádali o molekulárně-genetické vyšetření. Jeden z nich byl diagnostikován novorozeneckým skríninkem na Slovensku a u tří se hypotyreóza manifestovala klinicky v pozdějším věku.
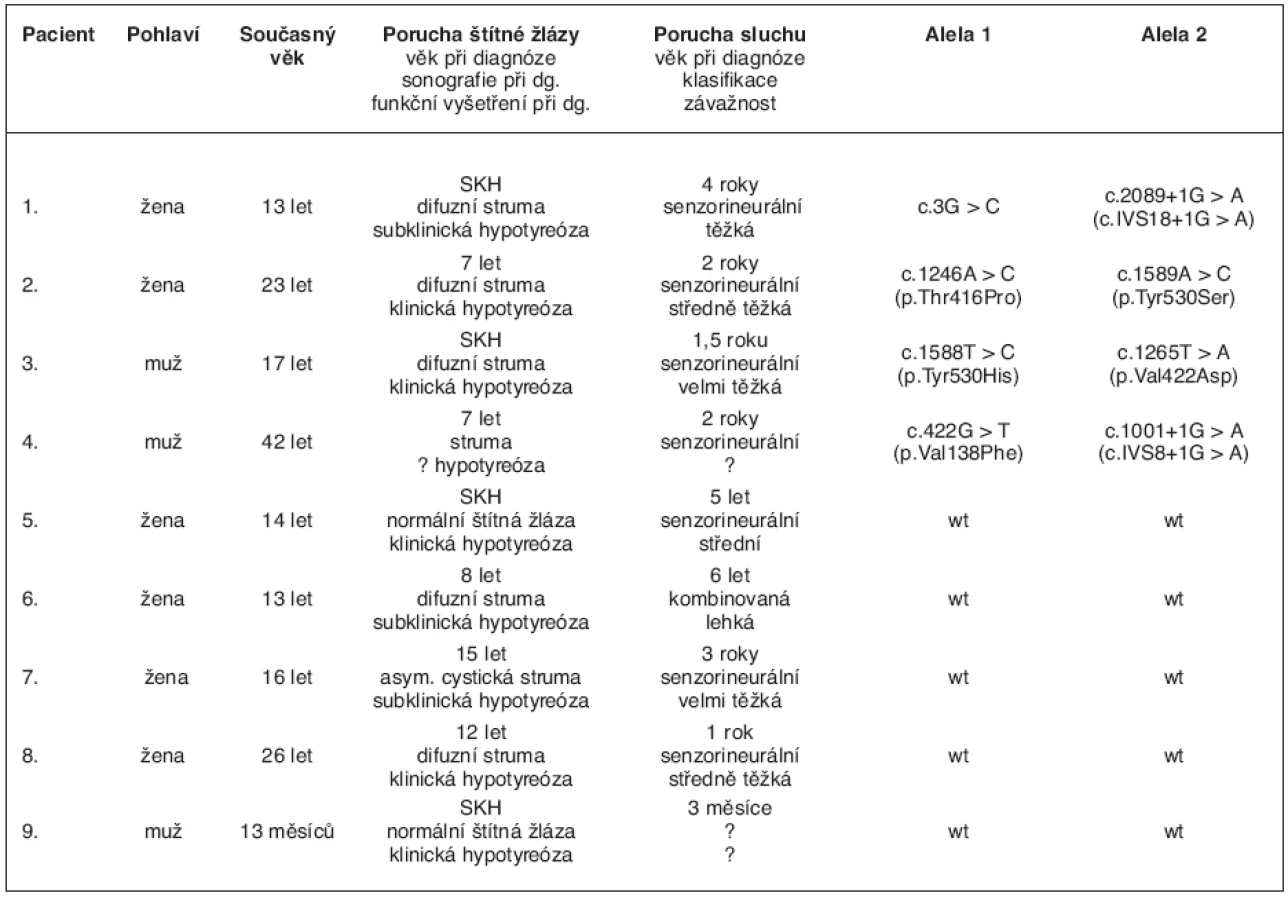
U všech 236 probandů poskytl ošetřující lékař údaje o anamnéze, o ultrazvukovém vyšetření štítné žlázy před léčbou a během substituční léčby, o hladinách hormonů štítné žlázy, o poruše sluchu a věku při její diagnóze a o případném audiometrickém vyšetření. Pět pacientů z prvních dvou skupin splňovalo klinická kritéria pro diagnózu Pendredova syndromu – měli poruchu štítné žlázy na podkladě dyshormonogeneze a současně senzorineurální poruchu sluchu. Spolu se čtyřmi pacienty ze třetí skupiny jsme tak pro molekulárně-genetické vyšetření PDS/SLC26A4 genu měli k dispozici devět pacientů s klinicky definovaným Pendredovým syndromem. Z nich čtyři byli zachyceni novorozeneckým skríninkem a u pěti se hypotyreóza projevila v pozdějším věku. Jejich fenotypová a genotypová data shrnuje tabulka 1.
Metody
DNA byla získána za pomocí standardizovaných metod z leukocytů periferní krve nebo ze slin sesbíraných do DNA Self-Collection Kits (DNA Genotec Inc, Ontario, Canada). Dvacet jedna exonů a přilehlých intronových úseků PDS/SLC26A4 genu bylo vyšetřeno metodou přímé sekvence. Použit byl kapilární sekvenátor ABI PRISM 310. Podmínky PCR a použité primery jsou publikovány ve studii Borck et.al. (13). Všechny nalezené mutace byly potvrzeny obousměrnou sekvenací.
U probandů s pozitivním nálezem mutací jsme dodatečně vyšetřili jejich dostupné rodinné příslušníky. Dále u nich bylo provedeno HR-CT pyramid se zaměřením na vestibulární akvadukt s cílem posoudit případné strukturální anomálie vnitřního ucha. Vestibulární akvadukt byl považován za rozšířený, pokud jeho průměr přesáhl 1,5 mm.
Etické aspekty
Studie byla schválena etickou komisí 3. lékařské fakulty Univerzity Karlovy v Praze. Pacienti nebo jejich zákonní zástupci potvrdili svůj souhlas s genetickým vyšetřením podpisem informovaného souhlasu.
Výsledky
Sekvenční analýzou PDS/SLC26A4 genu jsme prokázali mutace u čtyř z devíti pacientů. U dvou z nich byla hypotyreóza diagnostikována novorozeneckým skríninkem, u dvou později na základě klinických příznaků. Celkově jsme identifikovali osm mutací, všechny ve složeně heterozygotním stavu.
1. pacient
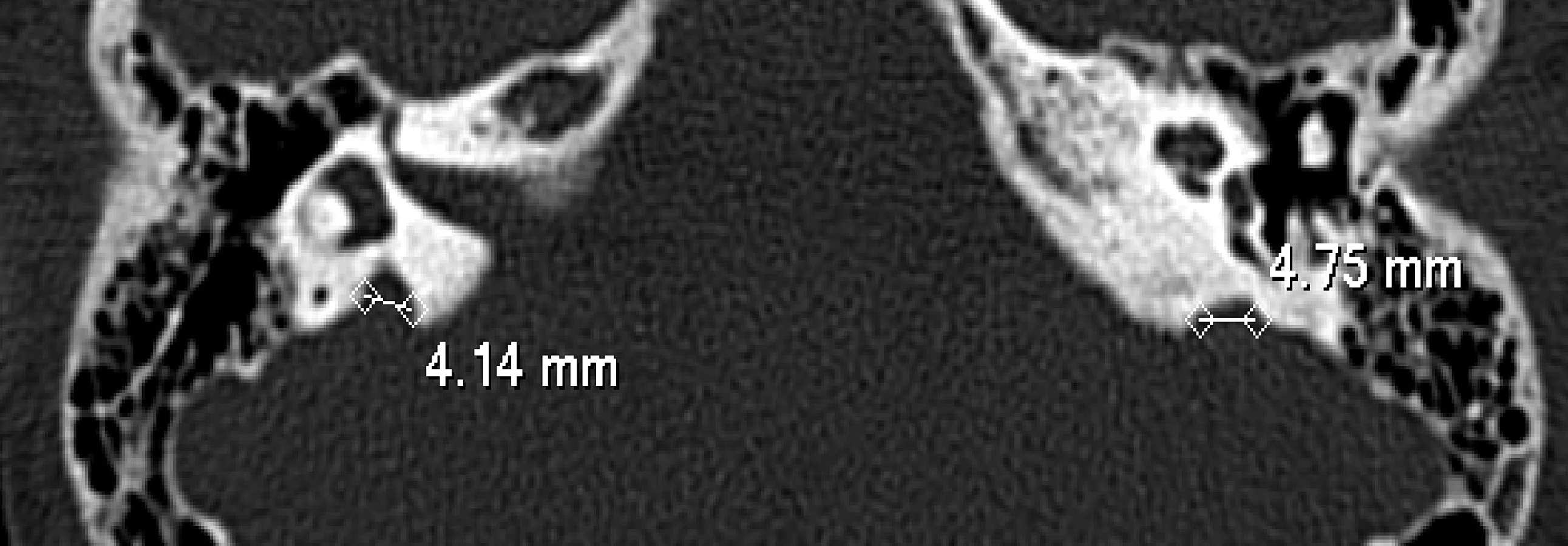
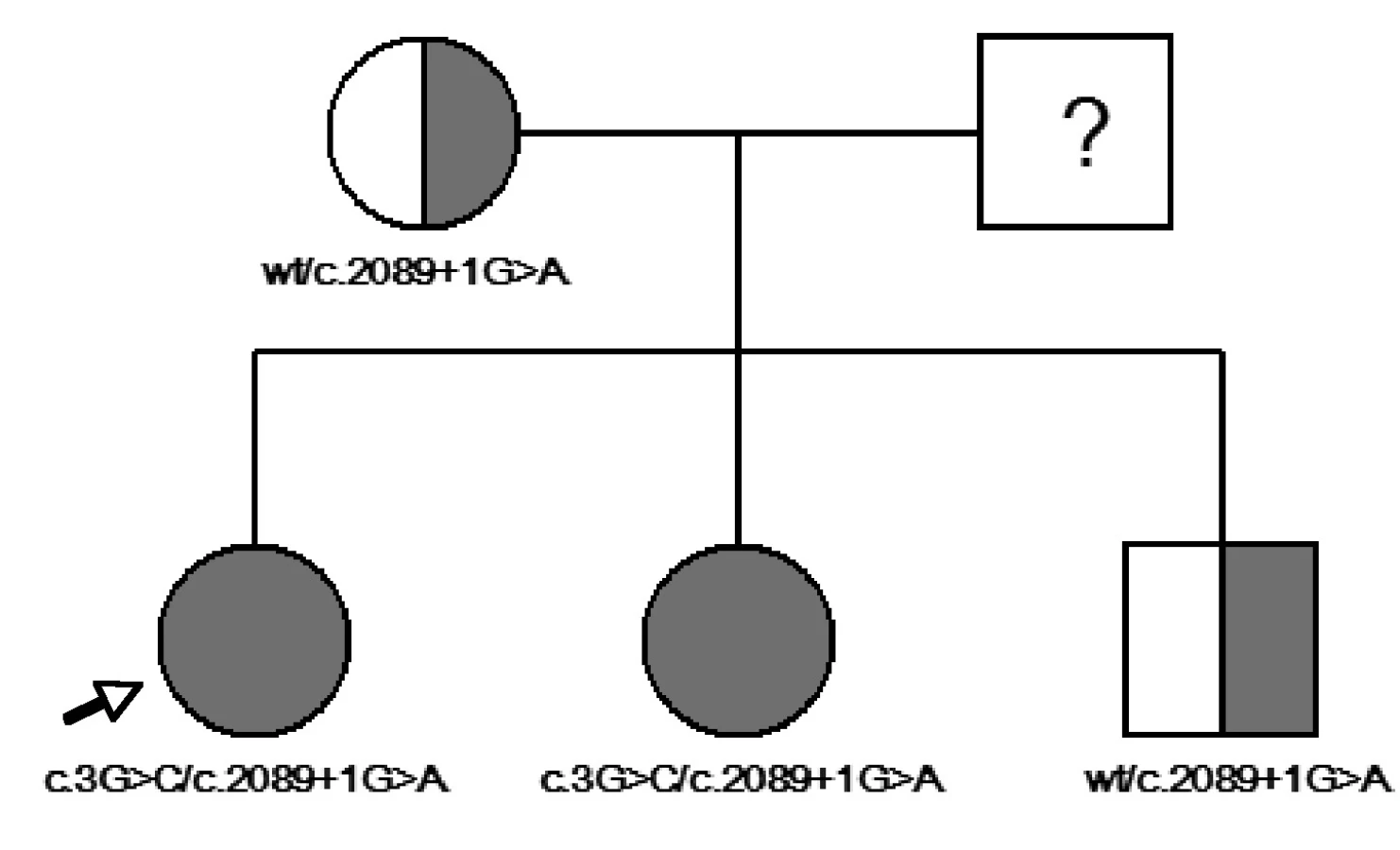
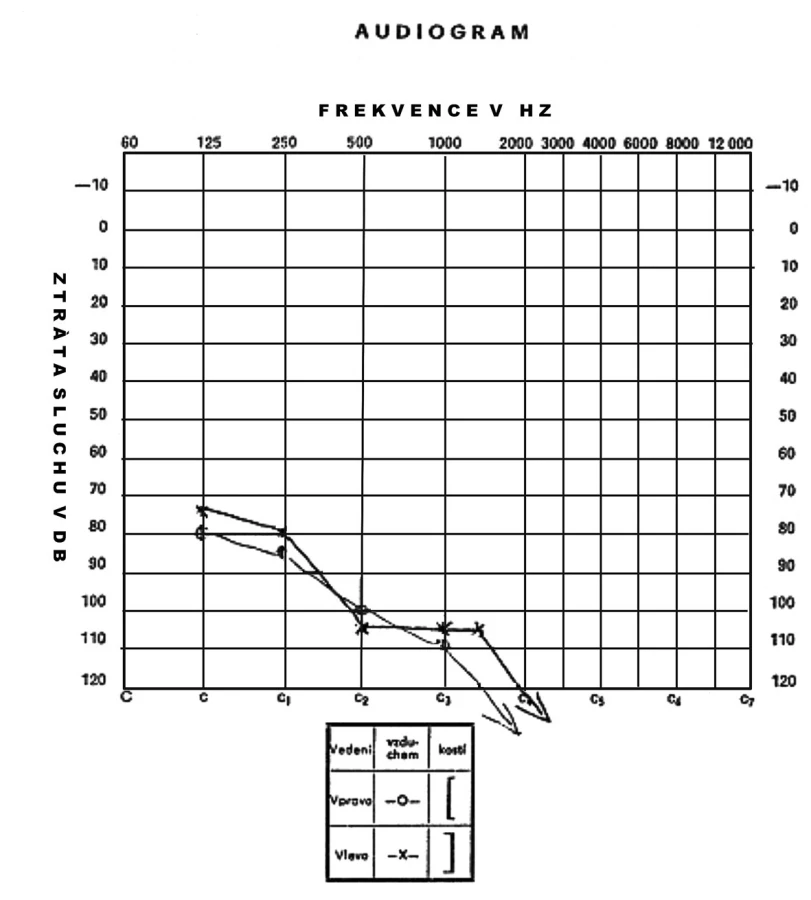
Nyní 13letá dívka byla zachycena novorozeneckým skríninkem, který prokázal subklinickou hypotyreózu (TSH 71 mIU/l, tT4 114 nmol/l). Ultrazvukové vyšetření štítné žlázy provedené po narození ukázalo malou difuzní strumu. Substituční léčba L-tyroxinem zahájená ve věku 14 dní vedla rychle k dosažení trvalého eutyreoidního stavu, a to bez dalšího růstu strumy. Porucha sluchu byla zjištěna ve věku čtyř let a byla klasifikována jako senzorineurální a těžká (70 dB oboustranně). Klinickou diagnózu Pendredova syndromu potvrdil nález dvou mutací ve složeně heterozygotním stavu – mutace ve startovacím kodonu genu v exonu 2 (c.3G>C) (12) a mutace ve splice site intronu 18 (c.2089 + 1G>A = c.IVS18 + 1G>A) (14). HR-CT vyšetření vnitřního ucha prokázalo bilaterální rozšíření vestibulárního akveduktu – vpravo 4,75 mm, vlevo 4,14 mm (obr. 1).
U 10leté sestry této probandky jsme sice zjistili stejný genotyp, ale fenotyp obou dívek se lišil. U sestry se velmi těžká senzorineurální hluchota (120 dB vpravo, 110 dB vlevo) manifestovala v předškolním věku, ale skrínink vrozené hypotyreózy byl negativní. Funkce štítné žlázy vyšetřená v 10 letech byla normální, jen ultrazvukové vyšetření ukázalo malou difuzní strumu. Vestibulární akvedukt byl oboustranně rozšířen (vpravo 4,75 mm, vlevo 4,66 mm). Matka a mladší bratr obou sester jsou nositeli heterozygotní mutace v intronu 18. Nemají žádné fenotypové projevy. Otec vyšetřen nebyl, ale velmi pravděpodobně je nositelem druhé mutace (obr. 2).
2. pacient
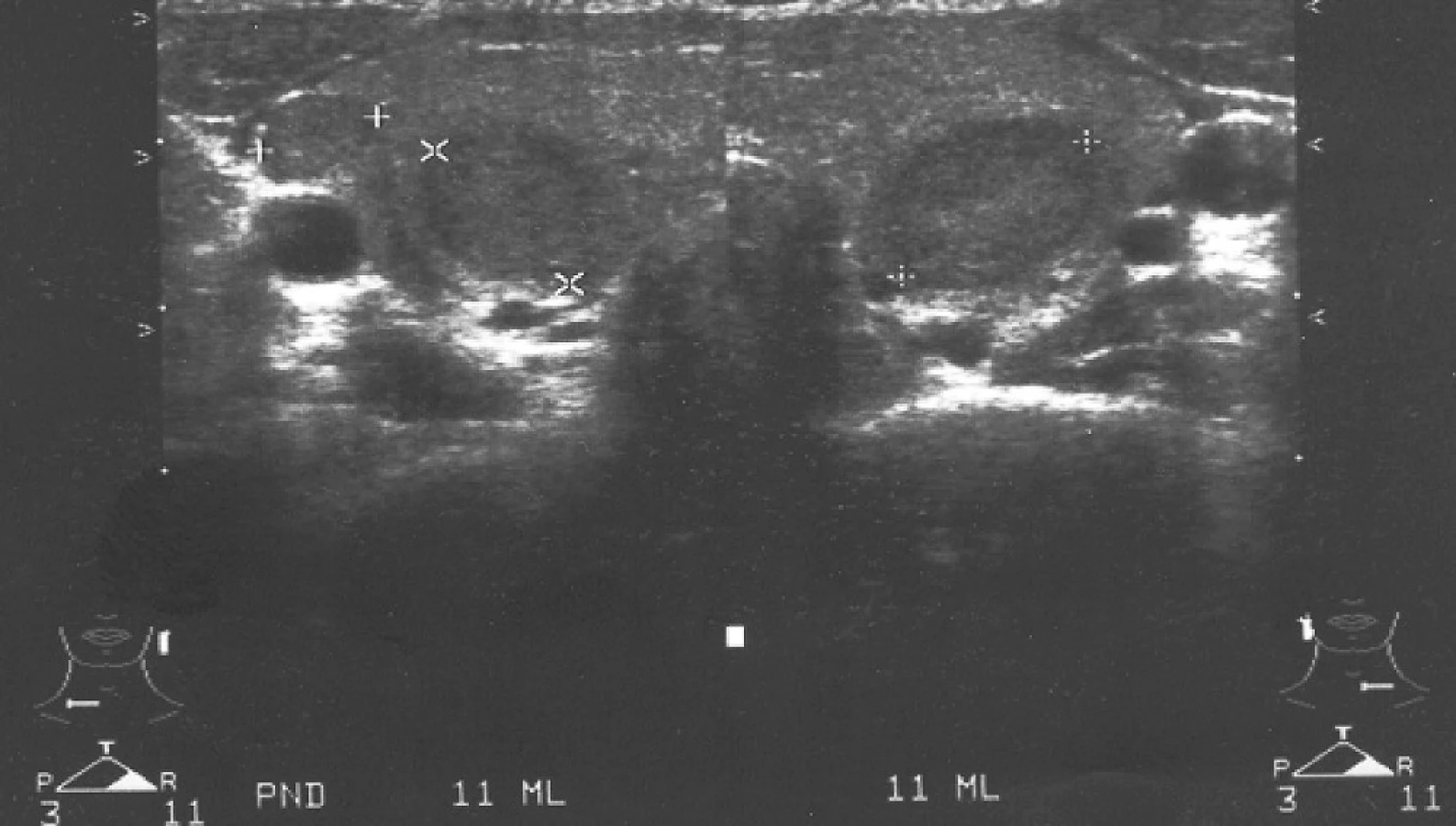
Případ této 23leté ženy jsme již samostatně publikovali (15). Pacientka je jediným dítětem zdravých nepříbuzných rodičů, skrínink vrozené hypotyreózy byl negativní. Ve věku dvou let si rodiče všimli, že dívka špatně slyší. Audiometrické vyšetření ukázalo senzorineurální středně těžkou nedoslýchavost (65 dB oboustranně). Postižení štítné žlázy se projevilo v sedmi letech věku malou strumou. Laboratorně byla prokázána hypotyreóza (TSH 14,8 mIU/l, fT4 6,53 pmol/l) a byla zahájena substituční léčba L-tyroxinem. Molekulárně-genetické vyšetření genu pro pendrin odhalilo složeně heterozygotní nosičství dvou substitucí c.1246A>C (= p.Thr416Pro) v exonu 10 (16) a c.1589A>C (= p.Tyr530Ser) v exonu 14 (17). Oba rodiče nesou jednu z mutací v heterozygotním stavu. I přes dobrou spolupráci a dlouhodobě normální hladiny tyreoidálních hormonů došlo u této ženy v průběhu života k progresi strumy. Pro nodulární přestavbu (obr. 3) u ní byla provedena aspirační biopsie tenkou jehlou s nálezem adenomu a byla indikována totální tyreoidektomie, kterou podstoupila ve věku 22 let. Následné histologické vyšetření neprokázalo známky malignity. Porucha sluchu se zatím neprohloubila. V období studie bylo u ní provedeno HR-CT vyšetření vnitřního ucha s nálezem bilaterální dilatace vestibulárního akveduktu.
3. pacient
Hypotyreóza byla u tohoto nyní 17letého chlapce zjištěna novorozeneckým skríninkem (neonatální TSH 38 mIU/l, tT4 9 nmol/l). V té době byla ultrazvukem zjištěna malá difuzní struma. Léčba L-thyroxinem byla zahájena 15. den života a vedla k postupnému relativnímu zmenšování strumy až do věku 10 let, kdy byla sonograficky štítná žláza normální velikosti. Porucha sluchu se manifestovala ve věku 1,5 roku a byla klasifikována jako senzorineurální a velmi těžká (110 dB vlevo a 100 dB vpravo) (obr. 4). Molekulárně-genetické vyšetření prokázalo genotyp c.1588T>C (= p.Tyr530His) / c.1265T>A (= p.Val422Asp) (12). Rodinná anamnéza je neznámá, neboť chlapec byl brzy po narození předán k adopci.
4. pacient
Muž, 42 let, s vrozenou senzorineurální poruchou sluchu zjištěnou ve věku dvou let byl v péči endokrinologa od 10 let pro non-autoimunitní hypofunkční strumu. Hormonální hladiny před zahájením substituční léčby nejsou známé. V průběhu životu došlo k progresi a nodulární přestavbě strumy, pro kterou byl dvakrát operován. Při obou parciálních strumektomiích histologické vyšetření neprokázalo známky malignity. Molekulárně-genetické vyšetření potvrdilo diagnózu Pendredova syndromu nálezem dvou známých mutací v genu pro pendrin – c.422G>T (= p.Val138Phe) v exonu 4 (14) a c.1001+1G>A (= c.IVS8 + 1G>A) ve splice site intronu 8 (14, 17). Bratr pacienta se léčí pro hypofunkční strumu a senzorineurální poruchu sluchu, genetické vyšetření však u něj nebylo možné provést. Rodiče obou bratrů jsou zdrávi.
5. pacient
U nyní 14leté dívky prokázal novorozenecký skrínink klinickou hypotyreózu s hladinou TSH 150 mIU/l a tT4 15 nmol/l. Velikost štítné žlázy byla při prvním sonografickém vyšetření po narození na horní hranici normy. V průběhu hormonální substituce zahájené ve věku 17 dní života se velikost žlázy postupně normalizovala. Senzorineurální neprogredující ztráta sluchu v rozsahu 50 dB oboustranně byla zjištěna ve věku pěti let. V rodinné anamnéze nebyla nalezena zmínka o poruše sluchu, sestra otce se léčila pro hypofunkční strumu. Molekulárně-genetické vyšetření genu pro pendrin v tomto případě neprokázalo žádnou mutaci.
6. pacient
Dnes 13letá dívka byla v péči ORL lékaře od šesti let pro lehkou oboustrannou a symetrickou (35 dB) kombinovanou poruchu sluchu. Skrínink vrozené hypotyreózy byl negativní. V osmi letech byla zjištěna difuzní struma spojená se subklinickou non-autoimunitní hypotyreózou. Zpočátku byla léčena jodidem, později byla převedena na L-tyroxin. Na této terapii byla eufunkční s neměnným morfologickým obrazem strumy. Postupně se ale rozvinula porucha růstu na podkladě deficitu růstového hormonu a bylo zjištěno pravostranné zdvojení kalichopánvičkového systému a atypické kulovité rozšíření horního pólu sleziny. Mentální vývoj odpovídal věku. U dívky jsme nenalezli žádnou mutaci v genu pro pendrin. Rodinná anamnéza byla negativní z hlediska poruchy sluchu i onemocnění štítné žlázy.
7. pacient
Senzorineurální porucha sluchu se u nyní 16leté dívky manifestovala v raném dětském věku. Audiometrické vyšetření prokázalo velmi těžkou nedoslýchavost (praktickou hluchotu) pravého ucha a těžkou nedoslýchavost (85 dB) vlevo. Na preventivní prohlídce ve věku 15 let byla praktickým lékařem zjištěna tužší hmatná struma spojená se subklinickou hypotyreózou (TSH 6,64 mIU/l, fT4 7,52 pmol/l). Ultrazvukové vyšetření zobrazilo asymetrickou strumu s cystickými strukturami v pravém laloku. Vzhledem k tomuto nálezu byla provedena aspirační biopsie tenkou jehlou. Tekutina z pseudocysty obsahovala řídký koloid, rozpadlé erytrocyty a benigní tyreocyty. Sonografický obraz zůstal po následující roky nezměněn. Mutace v genu pro pendrin nalezena nebyla.
8. pacient
Prvním projevem u této dnes 26leté ženy byla porucha sluchu zjištěná ve věku jednoho roku vyšetřením metodou BAEP (brainstem auditory evoked potentials), které bylo indikováno pro opožďující se psychomotorický vývoje dítěte. Později byla audiometrickým vyšetřením porucha sluchu klasifikována jako senzorineurální a středně těžká (60 dB oboustranně). Ve věku 12 let byla dívka hospitalizována k vyšetření poruchy růstu a únavového syndromu. V klinickém obrazu byla patrná malá struma a myxedém, laboratorně byla prokázána těžká klinická hypotyreóza a ultrazvukové vyšetření potvrdilo difuzní strumu. Substituční terapie L-tyroxinem vedla k rychlému dosažení eutyreoidního stavu bez další progrese strumy. U dívky byla v dětství také zjištěna vrozená srdeční vada (defekt síňového septa) a hemodynamicky nevýznamná trikuspidální insuficience. Ani u ní molekulárně-genetické vyšetření neprokázalo mutaci v genu pro pendrin.
9. pacient
Třináctiměsíční chlapec byl diagnostikován novorozeneckým skríninkem kongenitální hypotyreózy ve Slovenské republice (hodnoty skríninku TSH > 195 mIU/l, fT4 < 2,8 pmol/l). Ultrazvukový nález odpovídal kritériím dyshormonogeneze – byla nalezena symetrická štítná žláza normální velikosti a lokalizace. Porucha sluchu byla zjištěna ve věku tří měsíců metodou otoakustických emisí. U dítěte nebyly patrné jiné vrozené vývojové vady či onemocnění a rodinná anamnéza byla negativní. Molekulárně-genetické vyšetření neprokázalo mutaci v žádném z 21 exonů genu pro pendrin.
Diskuze
Pendredův syndrom, klinicky definovaný jako asociace senzorineurální nedoslýchavosti a dyshormonogenetické strumy, vede k postižení dvou různých systémů – endokrinního a sluchového – na podkladě stejného molekulárního mechanizmu. Postižení pacienti proto potřebují komplexní mezioborovou péči, na které se podílejí endokrinologové a otorinolaryngologové (18). Porucha sluchu většinou bývá prvním klinickým symptomem a manifestuje se u všech případů. Pendredův syndrom je nejčastější příčinou syndromické hluchoty a je zodpovědný až za 10 % případů vrozené nedoslýchavosti (4, 5). Proto se doporučuje provést genetický skrínink genu pro pendrin u každého pacienta s vrozenou percepční poruchou sluchu asociovanou s radiologicky prokázanou strukturální abnormitou vnitřního ucha typickou pro toto onemocnění.
Z hlediska endokrinologa je stanovení diagnózy Pendredova syndromu ztíženo vysokou fenotypovou variabilitou a skutečností, že porucha štítné žlázy se u přibližně 20 % pacientů nemusí projevit vůbec (9). Na druhé straně je známo, že nedoslýchavost je u dětí s vrozenou hypotyreózou nejčastější sdruženou funkční vadou, která může být způsobena mutací v genu pro pendrin, ale častěji přímým vlivem nedostatku hormonů štítné žlázy na pre - i postnatální vývoj sluchového aparátu (19). Tato skutečnost vede k výskytu fenokopií Pendredova syndromu.
Vysokou fenotypovou variabilitu a častý výskyt fenokopií potvrzuje i naše studie, která při diagnostice Pendredova syndromu vycházela primárně z pacientů endokrinologických ambulancí s poruchou funkce štítné žlázy zjištěnou novorozeneckým skríninkem kongenitální hypotyreózy nebo později v dětství. Z devíti vyšetřených pacientů jsme diagnózu genetickým vyšetřením potvrdili u čtyř, u zbylých pěti se tedy pravděpodobně jednalo o fenokopie, opomineme-li jiné, vzácnější možné příčiny Pendredova syndromu, jako jsou například mutace v oblasti promotoru PDS/SLC26A4 genu či defekty jiných, doposud neznámých genů. Při zpětném detailním pohledu na fenotyp pacientů lze klinickou diagnózu Pendredova syndromu zpochybnit pouze u 6. pacientky, u které se porucha sluchu manifestovala až ve věku šesti let a byla pouze lehká (35 dB), což je pro tuto diagnózu atypické. Dále lze spekulovat, že její další sdružené vývojové vady (zdvojení kalichopánvičkového systému, atypická slezina) jsou stejně jako u 8. pacientky (vrozená srdeční vada) způsobeny jiným společným patogenetickým mechanizmem. U ostatních pacientů bez nalezené mutace (5. a 7. pacientka a 9. pacient) fenotyp věrně kopíruje Pendredův syndrom a patogeneze společného postižení štítné žlázy a sluchu zůstává nejasná.
Studie potvrzuje vysokou fenotypovou variabilitu z hlediska věku manifestace i závažnosti postižení štítné žlázy. Pozoruhodný je záchyt dvou dětí s pozitivním genetickým vyšetřením a hypotyreózou diagnostikovanou již časně po narození pomocí novorozeneckého skríninku (1. a 3. pacient). Podobné případy jsou popisovány ojediněle, a to i ve studiích s většími skupinami probandů. Tyto případy zdůrazňují důležitost časného vyšetření sluchu u dětí s kongenitální hypotyreózou, které je dnes zajištěno povinným skríninkovým vyšetřením sluchu metodou otoakustických emisí do tří měsíců věku dítěte (20).
U 1. pacientky je zajímavá rodinná anamnéza s intrafamiliární variabilitou syndromu, kdy identický genotyp byl u dvou sester spojen s odlišnou klinickou manifestací postižení štítné žlázy. Stejná mutace u obou nepochybně stejně narušila funkci pendrinu a život v jedné rodině pravděpodobně zajistil srovnatelný přísun jódu ve stravě. Proto zbývá pouze spekulace o jiných mechanizmech, které individuálně ovlivňují rozvoj poruchy štítné žlázy – např. funkce dalších jodidových transportérů, mimo jiné nedávno objeveného hAIT (human apical iodide transporter). K objasnění této problematiky budou nutné další studie.
Z hlediska fenotypové variability nelze opomenout ani odlišný dlouhodobý vývoj strumy, patrný u našich pacientů s Pendredovým syndromem. Zatímco u 1. pacientky zůstal morfologický obraz strumy neměnný, u 3. pacienta došlo k relativní regresi strumy do normální velikosti. Méně příznivý byl vývoj strumy u 2. a 4. pacienta, přičemž u obou byla stejně jako u prvních dvou pacientů substituční léčba L-tyroxinem dle informace ošetřujícího lékaře dostatečná a účinná. U obou však došlo k progresi strumy s nodulární přestavbou, která si vyžádala chirurgickou intervenci. Zatímco u 2. pacientky byla při první operaci provedena totální tyreoidektomie, 4. pacient podstoupil nodulektomii, kterou však pro další hyperplazii reziduální tkáně bylo nutné opakovat. Podobný vývoj je u dyshormonogenetických strum známý a je pravděpodobně důsledkem chronické stimulace tyreotropinem. Progrese a vývoj nodulární hyperplazie nebo adenomů přináší riziko maligní transformace folikulárních buněk, které je u strum na podkladě dyshormonogeneze dle některých údajů vyšší než u strum jiné etiologie (10). Proto při léčení těchto pacientů má být kladen zvláštní důraz na striktní udržení eufunkčního stavu a pravidelná zobrazovací vyšetření štítné žlázy.
Závěr
Studie potvrzuje vysokou fenotypovou variabilitu postižení štítné žlázy u pacientů s Pendredovým syndromem a poukazuje na potřebnost genetického vyšetření k potvrzení klinické diagnózy tohoto onemocnění vzhledem k častému výskytu fenokopií. Z endokrinologického hlediska však doporučujeme provést molekulárně-genetické vyšetření genu pro pendrin pouze u pacientů s vrozenou hypotyreózou na podkladě dyshormonogeneze asociovanou se středně těžkou až těžkou senzorineurální poruchou sluchu.
Studie Pendredova syndromu je podpořena výzkumným záměrem MSM 0021620814.
Děkujeme MUDr. Ondřejovi Cinkovi, Ph.D. za odborné laboratorní vedení projektu.
MUDr. Karolína Banghová
Pediatrická klinika 2. LF UK a FNM
V Úvalu 84, 150 06 Praha 5
fax: +420 224 432 020, e-mail: k_banghova@yahoo.com
Sources
1. Park, S. M., Chatterjee, V. K.: Genetics of congenital hypothyroidism. J. Med. Genet., 2005, 42, s. 379–389.
2. Scott, D. A., Wang, R., Kreman, T. M. et al.: The Pendred syndrome gene encodes a chloride-iodide transport protein. Nat. Genet., 1999, 21, s. 440–443.
3. Everett, L. A., Glaser, B., Beck, J. C. et al.: Pendred syndrome is caused by mutations in a putative sulphate transporter gene (PDS). Nat. Gene, 1997, 17, s. 411–422.
4. Fraser, G. R.: Association of Congenital Deafness with Goitre (Pendred‘s Syndrome) a Study of 207 Families. Ann. Hum. Genet., 1965, 28, s. 201–249.
5. Reardon, W., Trembath, R. C.: Pendred syndrome. J. Med. Genet., 1996, 33, s. 1037–1040.
6. Banghová, K., Al Taji, E., Lebl, J.: Pendrin a jeho úloha v patogenezi kongenitální hypotyreózy a dalších onemocnění. DMEV, 2006, 9, s. 80–84.
7. Astl, J., Veselý, D., Jablonický, P.: Pendredův syndrom – poznámky k problematice vrozené autosomálně recesivní percepční nedoslýchavosti spojené se strumou. Otorinolaryng. a Foniat., 2004, 53, s. 55–59.
8. Phelps, P. D., Coffey, R. A., Trembath, R. C. et al.: Radiological malformations of the ear in Pendred syndrome. Clin. Radiol., 1998, 53, s. 268–273.
9. Reardon, W., Coffey, R., Chowdhury, T. et al.: Prevalence, age of onset, and natural history of thyroid disease in Pendred syndrome. J. Med. Genet., 1999, 36, s. 595–598.
10. Medeiros-Neto, G., Stanbury, J. B.: In: Medeiros-Neto G, Stanbury JB (eds) Inherited Disorders of the Thyroid system. Boca Raton: CRC press, 1994, s. 909–916.
11. Gaudino, R., Garel, C., Czernichow, P., Leger, J.: Proportion of various types of thyroid disorders among newborns with congenital hypothyroidism and normally located gland: a regional cohort study. Clin. Endocrinol. (Oxf.), 2005, 62, s. 444–448.
12. Banghova, K., Al Taji, E., Cinek, O. et al.: Pendred syndrome among patients with congenital hypothyroidism detected by neonatal screening: identification of two novel PDS/SLC26A4 mutations. Eur. J. Pediatr., 2008, 167, s. 777–783.
13. Borck, G., Roth, C., Martine, U. et al.: Mutations in the PDS gene in German families with Pendred‘s syndrome: V138F is a founder mutation. J. Clin. Endocrinol. Metab., 2003, 88, s. 2916–2921.
14. Blons, H., Feldmann, D., Duval, V. et al.: Screening of SLC26A4 (PDS) gene in Pendred‘s syndrome: a large spectrum of mutations in France and phenotypic heterogeneity. Clin. Genet., 2004, 66, s. 333–340.
15. Banghova, K., Cinek, O., Al Taji, E. et al.: Thyroidectomy in a patient with dyshormonogenetic goitre - a case of Pendred syndrome confirmed by finding mutations in the PDS/SLC26A4 gene. 2008. J. Pediatr. Endocrinol. Metab. (in press).
16. Napiontek, U., Borck, G., Muller-Forell, W. et al.: Intrafamilial variability of the deafness and goiter phenotype in Pendred syndrome caused by a T416P mutation in the SLC26A4 gene. J. Clin. Endocrinol. Metab., 2004, 89, s. 5347–5351.
17. Pryor, S. P., Madeo, A. C., Reynolds, J. C. et al.: SLC26A4/PDS genotype-phenotype correlation in hearing loss with enlargement of the vestibular aqueduct (EVA): evidence that Pendred syndrome and non-syndromic EVA are distinct clinical and genetic entities. J. Med. Genet., 2005, 42, s. 159–165.
18. Laštůvka, P., Taudy, M., Vrabec, P.: Pendredův syndrom u dospívající dívky. Otorinolaryngol, 2001, 50, s. 92–94.
19. Debruyne, F., Vanderschueren-Lodeweyckx, M., Bastijns, P.: Hearing in congenital hypothyroidism. Audiology, 1983, 22, s. 404–409.
20. Věstník MZ ČR. Metodický návod zajištění celoplošného novorozeneckého laboratorního screeningu a následné péče, oddíl 2 - kongenitální hypotyreóza. 2002, s. 4–11.
Labels
Addictology Allergology and clinical immunology Angiology Audiology Clinical biochemistry Dermatology & STDs Paediatric gastroenterology Paediatric surgery Paediatric cardiology Paediatric neurology Paediatric ENT Paediatric psychiatry Paediatric rheumatology Diabetology Pharmacy Vascular surgery Pain management Dental HygienistArticle was published in
Journal of Czech Physicians

- Advances in the Treatment of Myasthenia Gravis on the Horizon
- Possibilities of Using Metamizole in the Treatment of Acute Primary Headaches
- Metamizole at a Glance and in Practice – Effective Non-Opioid Analgesic for All Ages
- Metamizole vs. Tramadol in Postoperative Analgesia
- Spasmolytic Effect of Metamizole
Most read in this issue
- Diferenciální diagnostika refrakterní refluxní choroby jícnu
- Hostilita jako rizikový faktor řady onemocnění a možnosti jejího ovlivnění
- Lenalidomid (Revlimid) v léčbě mnohočetného myelomu – první zkušenosti v České republice
- Pendredův syndrom u pacientů s hypotyreózou: genetická diagnostika, fenotypová variabilita a výskyt fenokopií