
Oxid dusnatý u obezity a metabolického syndromu
Nitric oxide in patients with obesity and metabolic syndrome
Nitric oxide in its pleiotropic role interacts with many diverse systems and beside others acts in pathophysiology of obesity and metabolic syndrome. Our review tends to summarize available basic publications aimed at the impact of NO on mitochondrial respiration, insulin resistance mainly in hepatocyte and the impact of NO on other factors of glucose metabolism. In this review, the authors try to shed light to pathophysiology of impaired NO bioavailability during diabetes and obesity too.
Key words:
nitric oxide, mitochondrial respiration, insulin resistance.
:
J. Hodis; N. Gaier; H. Farghali
:
Univerzita Karlova v Praze, 1. lékařská fakulta, Farmakologický ústav
:
Čas. Lék. čes. 2009; 148: 34-38
:
Review Article
Oxid dusnatý (NO) ve své pleiotropní roli ovlivňuje mnohé systémy. Mimo jiné se účastní v patofyziologii metabolického syndromu a obezity. Článek podává přehled základních publikací, které poukazují na efekt NO na mitochondriální respiraci, inzulínovou rezistenci zvláště v jaterní buňce a další faktory glukózového metabolismu, stejně jako hledá příčinu zhoršení biologické dostupnosti NO u diabetu a obezity.
Klíčová slova:
oxid dusnatý, mitochodriální respirace, inzulínová rezistence.
Oxid dusnatý (NO)
NO jako volný radikálový plyn vzniká během enzymatické reakce mezi L-argininem, O2 a NADPH za tvorby citrulinu a vzniku NADP+ pomocí syntázy oxidu dusnatého (NOS; EC 1.14.13.39). Známý mechanismus účinku hlavně zahrnuje relaxaci cév. Nezávisle na relaxačním efektu na cévy bylo zjištěno, že NO (hlavně vzniklé z nNOS a eNOS) je endogenní aktivátor rozpustné guanylátcyklázy, tvořící cGMP. NOS je rodina enzymů, která se skládá ze dvou kalcium-calmodulin-dependentních, konstitutivních izoforem, neuronální NOS (nNOS, NOS-I) a endoteliální NOS (eNOS, NOS-III), a kalcium-independentní, inducibilní NOS (iNOS, NOS-II) (1, 2). Část izoformy eNOS se nachází hlavně v membránové frakci, kdežto izoforma iNOS jak v rozpustné, tak v membránové frakci (3).
Rozklad a přenos
Oxid dusnatý reaguje s kyslíkem za tvorby N2O4, který se dále spojuje s vodou za tvorby směsi dusitanových a dusičnanových anionů. Dusitanové aniony jsou oxidovány na dusičnanové oxyhemoglobinem. Oxid dusnatý je inaktivován hemem a volným radikálem – superoxidem. Zhášeči – scavengery superoxidu – jako například superoxid dismutáza, chrání NO, zvyšují intenzitu jeho účinku a prodlužují jeho trvání. Naopak interakce oxidu dusnatého se superoxidem vede k tvorbě peroxynitritu a následnému poškození tkání. Účinek peroxynitritu je regulován obsahem glutathionu v buňce. Za fyziologických podmínek reaguje glutathion s NO za vzniku S-nitrosoglutathionu (GSNO), mnohem stabilnější formy NO. Nitrosoglutathion může sloužit jako přenašeč NO s dlouhodobou životností (4).
Obezita
V současnosti je obezita definována jako BMI (body mass index = váha v kg/výška v m2) přes 30, nadváha pak BMI mezi 25 a 30 (definici dle obvodu pasu viz níže). Optimální životní prognóza je v pásmu BMI 20 mezi 22. V USA stoupla prevalence obezity a nadváhy u dospělých nad 20 let na 54,9 % za posledních 10 let. Přitom životní prognóza se u lidí obézních v jejich 20 letech věku snižuje až o 20 let. Vysoká prevalence (15%) obezity a nadváhy amerických dětí ve věku 6–19 let ukazuje na špatný trend do budoucna (5). Obezita s sebou nese hlavně několikanásobné zvýšení morbidity na další onemocnění, a to již od BMI 27 (hypertenze 2,9×, infarkt myokardu 1,9×, cévní mozkové příhody 3,1×, diabetes mellitus 2 2,9×, dna 2,5, hyperlipidémie 1,5×, kolorektální karcinom 1,3×, rakovinu krčku nebo těla dělohy 1,6×, rakovinu prsu 1,2×)(6).
Role NO u obezity
Mnoho výzkumných týmů předpokládá, že NO hraje významnou roli v regulaci energetické rovnováhy, neboť například podání nespecifického inhibitoru NO syntázy – N nitro-L-arginin-metyl esteru (L-NAME) vede k poklesu váhy a sníží příjem potravy u myši (7).Tento efekt je vyšší u myší na tučné dietě. Tam dochází i ke snížení velikosti adipocytů po L-NAME stejně jako snížení akumulace mastných kyselin v adipocytu. V souvislosti s tím L-NAME blokuje efekt vysokotučné diety na obsah triglyceridů (TAG) v játrech, zvyšuje glukózovou toleranci a in vivo i inzulínovou senzitivitu. Autoři předpokládají, že tento efekt je díky zvýšené ztrátě energie, neboť L-NAME zvyšuje hladinu mRNA uncoupling proteinů 1 a 3 (UCP1,3), a to jak ve svalu, tak v hnědé tukové tkáni. Zajímavé je, že L-NAME zároveň zvyšuje expresi peroxisome proliferator-activated receptoru γ (PPAR-γ) ve svalu (8). Navíc tvorba NO v mitochondrii pomocí mitochondriální syntázy redukuje vnitřní dýchání, a tím i spotřebu kyslíku (9). To se nejspíše děje inhibicí mitochondriální respirace skrze přímou vazbu NO na cytochrom C-oxidázu (cGMP-independentní dráhou) (10). Vazba NO na cytochrom C-oxidázu je reverzibilní a kompetitivní s kyslíkovou molekulou. Stechiometrický poměr inhibice mezi O2 a NO je 1 : 2000, tedy na inhibici 30 pM O2 je třeba 60 nM NO, jak prokazuje práce v in vitro pokusech. Autoři však použili fyziologické hladiny NO a O2 a považují tuto stechiometrii za možnou i v podmínkách in vivo (11). Pozdější práce však ukazují na jinou stechiometrii O2/NO = 1 : 30, stejně jako se změnila původní představa vazby NO na Fea3 2+ a3 podjednotky cytochrom C-oxidázy (CcO) na novější představu o vazbě na CuB2+ část CcO (12). Zůstává platné, že NO je asi 200× silnějším inhibitorem CcO než CO (13). V těchto pokusech problematický je i fakt, že NO je de - facto scavengerem volného O., dále že NO samotné je u nNOS a eNOS zpětnovazebným inhibitorem další produkce NO a naposled, že NOS enzym i v nedimerizované formě, a tedy ve formě neschopné tvořit NO je schopen přesto oxidovat NADPH na NADP+, což dohromady značně komplikuje měření stechiometrie oxidoredukčních dějů a spotřebu O2.
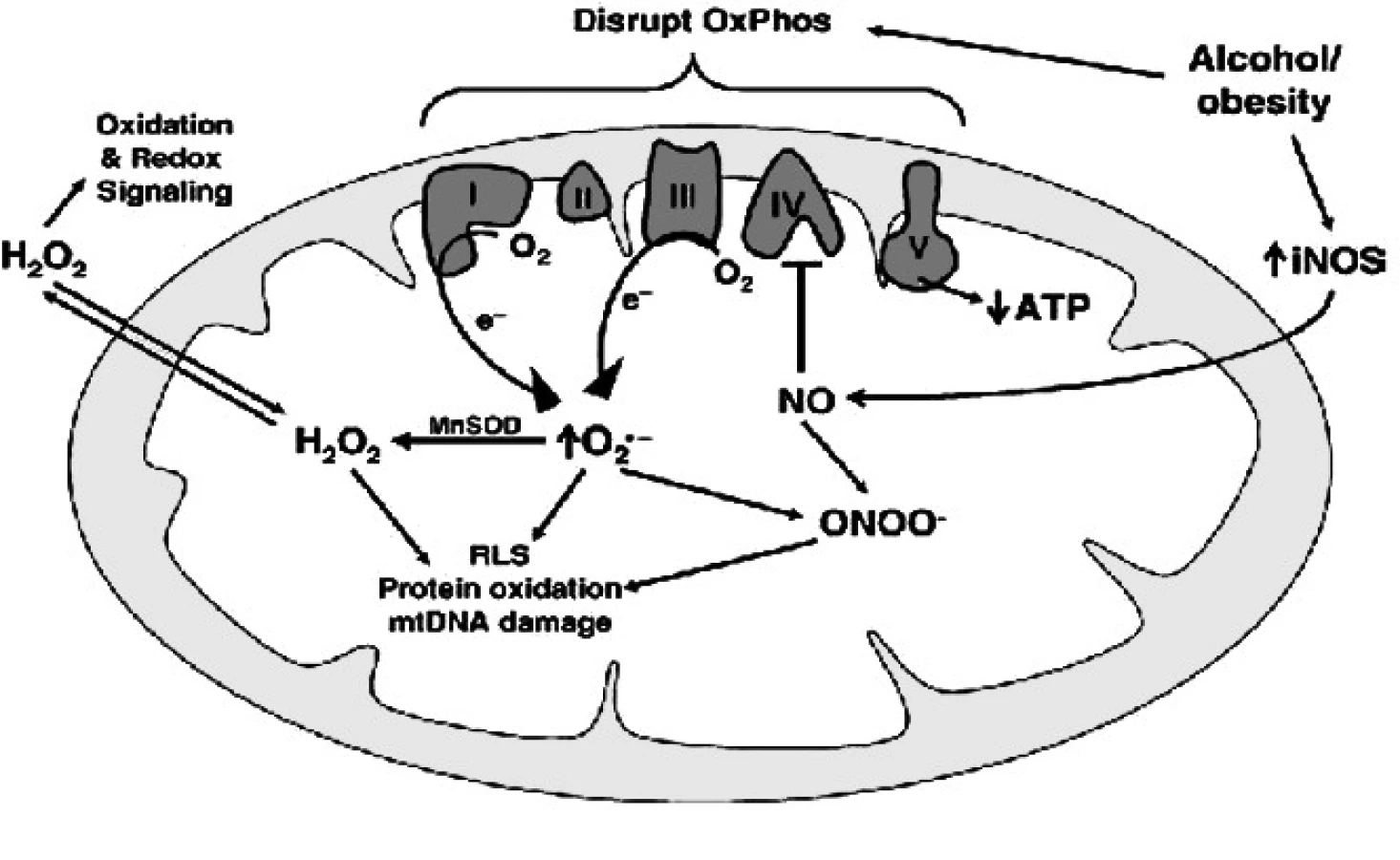
Zdá se, že roli hraje i substrát, který používá mitochondrie na tvorbu ATP. (Například Schweizer a Richter popsali blok CcO navozený NO u pyruvátu a malátu jako substrátu tvorby ATP (14), zatímco jinde můžeme najít záznam, že u substrátu s vazbou na FADH2 (succinát nebo palmitoyl-l-carnitin) vede přidání NO k zefektivnění oxidativní fosforylace zvýšení ATP produkce za snížení úniku protonů a zachování nebo snížení spotřeby O2, a tím přibližuje stechiometrii reakce spalování substrátu vázaného na NADH, jako je malát a glutamát (15). Mnozí autoři pak poukazují na fakt, že při NO navozené blokádě CcO dochází ke zvýšenému vstupu Ca2+ do mitochondrií, což může být i podkladem buněčné smrti navozené velkým excesem NO vytvořeného iNOS například při sepsi.
Na druhé straně někteří autoři zjistili fakt, že NO vede k zvýšení mitochondriální biogeneze v hnědých tukových buňkách, a to zprostředkovaně přes aktivaci cGMP. Lze tedy předpokládat, že množství mitochondrií se zvyšuje, i když jejich oxidační schopnost je nízká, a toto všechno vede ke zvýšení spotřeby potravy in vivo (16). V jiných publikacích lze nalézt i pozitivní roli NO na snižování zásob tuku – u eNOS knock-outovaných myší se nachází nižší počet mitochondrií v hnědé tukové tkáni (BAT), snížení exprese UCP 1 a PPAR-γ. Nalézáme zde i defektní výdej energie, zvýšení hmotnosti, inzulínovou rezistenci a hypertenzi (17). Ovlivnění jednotlivých složek není vázáno na sebe v řetězci, ale je nezávislé, neboť jiní autoři popsali, že hnědé tukové buňky s poruchou PPAR-γ normálně exprimují UCP1 (18, 19).
Existují i spekulace o centrálním efektu NO na chuť k jídlu. NO samotný uvolněný z eNOS a iNOS údajně nepřímo stimuluje transport inzulínu skrze hematoencefalickou bariéru. A bylo prokázáno, že podávání inzulínu do CNS indukuje hyperglykémii, hypoinsulinémii a anorexii, snižuje v hypothalamu expresi neuropeptidu Y a snižuje tělesnou váhu. Tedy NO by tak nepřímo zvýšeným transportem inzulínu do mozku mohl vést ke snížení váhy. Naopak NO uvolněný z nNOS v buňkách endotelu mozkových cév inhiboval transport inzulínu do mozku a měl ve svém důsledku efekt opačný (20).
Metabolický syndrom
Definice metabolického syndromu podle Mezinárodní federace pro diabetes obsahuje: pozitivní anamnézu obezity centrálního typu (definované jako obvod pasu > 94 cm u evropských mužů a > 80 cm u evropských žen s určitou odlišností u jiných národů) a navíc některé další 2 znaky z následujících 4:
- A. zvýšená hladina TAG (triglyceridů) > 1,7 mmol/l (150 mg/dl) nebo již zavedená specifická terapie této hyperlipidémie a/nebo
- B. snížený HDL - cholesterol < 1,03 mmol/l (40 mg/dl) u mužů a < 1,29 mmol/l (50 mg/dl) u žen nebo specifická terapie této dyslipidémie a/nebo
- C. zvýšení krevního tlaku: systolického > 130 nebo diastolického > 85 a nebo léčba hypertenze v anamnéze a/nebo
- D. zvýšená glykemie nalačno (FPG) > 5,6 mmol/l (100mg/dl) nebo předchozí anamnéza diabetes mellitus 2. typu.
Nebezpečí metabolického syndromu netkví pouze ve zvýšení kardiovaskulární morbidity (nadváha více než 20 kg vede k více jak šestinásobnému zvýšení prevalence ischemické choroby srdeční), ale zároveň v riziku vzniku diabetes mellitus 2. typu. Míru tohoto rizika popisuje pro prognózu 10 let dotazník FINDRISC bez obtíží dostupný na http://www.diabetes.fi/tiedoston_katsominen.php?dok_id=567 (21).
Jestliže je metabolický syndrom založen, mimo jiné, hlavně na inzulínové rezistenci, můžeme se ptát, kde v těle je rozhodující tkáň pro její vznik. Dominantní roli u inzulínorezistence dle některých autorů hrají játra; sval a tuk nejsou pro vznik inzulínorezistence rozhodující (22).
Oxid dusnatý u metabolického syndromu – vliv NO na inzulínorezistenci
Bylo publikováno několik prací, které spojovaly vznik inzulínorezistence s indukcí iNOS stejně jako s hladinou volných mastných kyselin, prozánětlivých cytokinů, oxidativním stresem. NO je schopen ovlivňovat inzulínovou signalizaci v několika krocích: snižovat Akt aktivitu přes s-nitrosylaci specifických cysteinových zbytků, vést k degradaci Irs1 v buňkách příčně pruhovaného svalu. NOS 2 knock-outované myši jsou chráněny proti inzulínové rezistenci příčně pruhovaného svalstva, což je spojeno se zvýšením PI3K-Akt aktivity. Během zánětu pak právě NOS2 zprostředkovává inzulínovou rezistenci kosterního svalstva například při sepsi s předpokládaným mechanismem založeným na s-nitrosylaci inzulínového receptoru, Irs1 a Akt (23). Během zánětu s indukovaným iNOS a až 1000× vyšší produkcí NO spolu s výskytem oxidativních radikálů dochází, dle některých autorů, k posttranslačním změnám proteinů (S-nitrosylaci cysteinových zbytků a tyrosinovou nitrací) i působením peroxynitritu. Tyto změny jsou samozřejmě nezávislé jak na cGMP, tak na hladině kalcia. Oxidativní stres výrazně zvyšuje S-nitrosylaci a inaktivaci nebo rozklad signálních proteinů inzulínové dráhy. V játrech obézních, leptin-deficitních myší je 2,5× zvýšena exprese iNOS ve srovnání s kontrolou a zvyšuje se i hladina nitrotyrosinu – markeru nitračního stresu. Při podání iNOS inhibitoru L N6 (1-Iminoetyl)lysinu(L-NIL) dochází jak ke zvýšení transkripce a translace Irs-1, tak bylo zaznamenáno i zvýšení transkripce Irs2. Toto následně vede ke zlepšení inzulínové signální dráhy přesIRS – PI3K a současně ke zlepšení kontroly glykémie a inzulínosenzitivity (nižší hladině glykémie nalačno a hladině lačného inzulínu). Tyto změny však nejsou spojeny se změnami cGMP, což vypadá, jakoby endoteliální a neuronální NOS nehrály v inzulínorezistenci roli. Podobný efekt na Irs-1 v příčně pruhovaném svalu u obézní, diabetické (leptin-deficientní) myši, jako je po blokátoru L-NIL, lze však dosáhnout i genetickým poškozením genu pro iNOS. I u pacientů s diabetem typu 2 se setkáváme se zvýšenou expresí iNOS stejně jako S-nitrosylovanými proteiny a tyrosinovou nitrací (24). Jak naznačují další práce, ovlivnění glukozového metabolismu pomocí NO nemusí být jen na základě ovlivnění inzulínové dráhy samotné. V nedávno publikované vlastní studii jsme zjistili, že blokátory NOS jako aminoguanidine (silněji) a L-NAME (méně) jsou schopné snížit výrazně glykogenolýzu indukovanou adrenalinem spolu se snížením produkce NO (25). V jiné práci Albuszies et al. zjišťuje inaktivaci phosphoenolpyruvát karboxykinázy – hlavního enzymu glukoneogeneze u iNOS+/+ myší za septického šoku, zatímco iNOS knock-outované myši mají tento enzym plně aktivní (26).
Na rozdíl od předchozích prací jiní autoři poukazují na opačný trend, kdy zvýšení NO se zdá ve vztahu k metabolickému syndromu výhodné a to zvláště jeho produkt peroxonitrit a označují naopak L-NAME (blokátor NOS) jako induktor inzulínorezistence: Například ve studii Guarina et al. různé donory NO byly schopny snížit inzulínorezistenci vyvolanou L-NAME. Zajímavý je závěr, že zatímco NO donor SIN-1(3-morpholinosydnonimine) snižuje jaterní inzulínorezistenci po podání do portální žíly, působí neenzymatický vzestup NO a superoxidu (O2-) (je donorem NO/O2- napodobujíc tak lépe aktivitu iNOS), jiný NO donor – SNP, uvolňuje sice rychle a spontánně NO, ale bez O2- . V prvém případě tak dochází k tvorbě GSNO v játrech. Ve druhém případě není inzulínorezistence ovlivněna. Zajímavý je i fakt, že inzulínorezistence pomocí SIN-1 je ovlivněna pouze podáním tohoto donoru do portální žíly, zatímco intravenózní systémové podání obou nebo jednoho každého donoru nevede k žádnému efektu na inzulínorezistenci (27). Podobný efekt intraportálně podaného L NAME a SIN-1 popsal ve své práci Moore a kol., kde je popsán i vazodilatační efekt na portální žílu u dávky SIN-1 3 mg/ml, zatímco dávka 10× nižší vazodilataci portální žíly nezpůsobuje (28).
Vliv metabolického syndromu a obezity na NO
Možné mechanismy ovlivnění NO produkce a biodostupnosti
Je známo, že mnohé cytokiny jsou spojovány s metabolickým syndromem a obezitou. Například TNFα je spojován v pozitivní korelaci s centrální obezitou i metabolickým syndromem (29) a je dokázáno, že je schopen indukovat iNOS (30). Naopak negativní korelace je prokázána u adiponectinu (31) a jeho vliv na NOS byl taktéž prokázán (32).
Změna v transkripci
Zvýšení trankripce iNOS jak v humánních, tak potkanních hepatocytech na základě cytokinové signalizace se děje prostřednictvím transkripčních faktorů zvláště nuclear factor-κB (NF-κB). Aby byl nuclear factor-κB (NF-κB) aktivován, vyžaduje však translokaci z cytoplazmy do jádra a tato translokace je silně závislá na redoxním stavu buňky. To je možná důvodem, proč je odlišná odpověď na LPS v Kupfferových buňkách jater, kde je nezávislá na hladině intracelulárního glutathionu a naopak v hepatocytech a buněčných liniích makrogágů, kde je na hladině glutathionu silně závislá. Jednotlivé druhy buněk se liší i odpovědí na dexametazon, kde v hepatocytech dexametazon tlumí LPS indukovanou produkci NO, kdežto v Kupfferových buňkách nikoliv. Podle některých posledních výsledků se zdá, že v Kupfferových buňkách je produkce nuclear factor-κB (NF-κB) konstitutivní, kdežto v hepatocytech je až následně indukovaná LPS (33).

Potranslační změny
Zdá se, že efekt cytokinů je založen nejen na ovlivnění transkripce NOS, ale i na fosforylaci enzymu. Autoři Nishimura et al. popsali efekt deficitu adiponektinu u adiponektin knock-out myší na fosforylaci eNOS se zvýšením produkce NO (34). Autoři Ellger et al. zkoumali efekt modelu diabetu u dlouhodobě kriticky nemocných zvířat na dostupnost volného NO. Zjistili, že dostupnost NO byla přímo závislá na glykémii, nesouvisela s inzulinémií. Inzulín nehrál ani roli v indukci exprese iNOS nebo eNOS, což si lze vysvětlit inhibicí PI3K dráhy inzulínové signalizace jak zánětem, tak hyperglykémií. Zdá se, že snížení produkce NO při hyperglykémii není ovlivněno ve fázi transkripce NOS, protože NOS exprese byla naopak zvýšená a jedná se spíše o posttranslační změny. Autoři předpokládají, že se jedná spíše o zpětnou negativní vazbu, kdy NO tlumí další aktivitu eNOS (vyšší oxidací hemového železa uvnitř NOS molekuly) než změnu kofaktorů (BH4, NADPH nebo glutathionu), kterou nezaznamenali. Jiným mechanismem snížení biodostupnosti NO u diabetu je i možné snížení substrátu-argininu, kde byla nalezena slabá, ale pozitivní korelace mezi NOS-aktivitou a hladinou argininu (35). O ovlivnění dostupnosti NO v endotelu při diabetu referuje přehledně článek Creagera a Lushera. Hyperglykémie snižuje biodostupnost oxidu dusnatého (NO) a prostacyklinu (PGI2) a zvyšuje se syntéza vazokonstrikčních prostanoidů a endothelinu (ET-1), a to rozličnými mechanismy:
Možný mechanismus snížení (na endotelu závislé) vazodilatace při diabetes mellitus (36):
Snížená syntéza NO/senzitivita vůči NO
- snížená dostupnost L-argininu
- poškozená signalizace v endotelialních buňkách Gi protein závislých signálů
- snížená dostupnost kofaktorů pro syntézu NO (Ca2+, calmodulin, tetrahydrobiopterin, NADPH)
Endogenní inhibice NO syntázy (asymetrický dimetylarginin (ADMA))
Zvýšená inaktivace NO a/nebo jeho odbourávání
- vzestup neenzymatických glykačních molekul (AGE)
- aktivace polylové dráhy
- aktivace na diacylglycerolu (DAG) závislé protein kinázy C (PKC)
- uncoupling eNOS
Zvýšená tvorba endotelem tvořených kontrakčních faktorů a vasokonstrikčních prostanoidů
Závěr
Dle přehledu literatury, který zde předkládáme, se zdá nyní již jasné, že NO hraje důležitou roli v metabolických dějích spojených s obezitou a metabolickým syndromem. I když jeho efekt není dosud zcela jasný, zdá se, že kromě vazodilatačního efektu ovlivnění NOS vede k mnoha dalším dějům zahrnujícím změny redox potenciálu a změnu oxidativní fosforylace v mitochondrii. Zbývá nám ještě dlouhá cesta k odhalení všech funkcí NO v obezitě a metabolickém syndromu, přestože již hodně práce na tomto poli, jak ukazuje tento článek, bylo vykonáno.
Zkratky
ATP – adenosin trifosfát
BAT – hnědá tuková tkáň
BH4 – tetrahydro-biopterin
BMI – body mass index
CcO – cytochrom C-oxidáza
cGMP – cyklický guanosin monofosfát
CNS – centrální nervový systém
COX-2 – cyclooxygenáza 2
FADH2 – redukovaný flavin-adenosin-dinukleotid
HDL – high density lipoprotein
IRS-1 – insulin receptor-substrate-1
LPS – lipopolysacharid
L-NAME – L-arginine-metyl ester
NADPH/NADP+ – redukovaná/oxidovaná forma nikotin-adenosin-dinukleotid-fosfátu
NO – oxid dusnatý
PI3K-Akt – fosfo-inositol-trifosfát-Akt
PPARα – peroxisome proliferator-activated receptor α
SNP – nitroprussid sodný
TNFα – tumor necrosis faktor alfa
TxA2 – tromboxan A2
UCP – uncoupling protein
Ostatní zkratky jsou vysvětleny v textu.
Práce vznikla za podpory IGA MZ NR/9379-3/2007, VZ MSM 0021620807.
Adresa pro korespondenci:
MUDr. Mgr. Jiří Hodis
Farmakologický ústav 1. LF UK
Albertov 4,
120 00 Praha 2
fax: +420 266 038 218,
e-mail: hodis@atlas.cz
Sources
1. Knowles RG, Moncada S. Nitric oxide synthases in mammals. Biochem J 1994; 298 : 249–258.
2. Alderton WK, Cooper CC, Knowles RG. Nitric oxide synthases: structure, function and inhibition. Biochem J 2001; 357 : 593–615.
3. Ribiere C, Jaubert AM, Gaudiot N, et al. White Adipose Tissue Nitric Oxide Synthase: A Potential Source for NO Production. Biochemical and Biophysical Research Communications 1996; 222 : 706–712.
4. Farghali HA, Kameníková L. Oxid dusnatý-NO. In Lincová D, Farghali H, et al. Základní a aplikovaná farmakologie. Praha: Galen et Karolinum 2005; 301–307.
5. Hunsaker DM, Hunsaker JC. Obesity Epidemic in the United States (A Cause of Morbidity and Premature Death); in Forensic Pathology Reviews, Vol. 2. Humana Press Inc., DOI10. 1385/1592598722 ISBN 978-1-59259-872-4 (Online) 2007.
6. Hainer V. Základy klinické obezitologie. Praha: Grada Publishing a.s. 2004; 36.
7. Morley JE, Flood JF. Effect of competitive antagonism of NO synthetase on weight and food intake in obese and diabetic mice. Am J Physiol 1994; 266 (1Pt2): R164–R168.
8. Tsuchiya K, Sakai H, Suzuki N, et al. Chronic blockade of nitric oxide synthesis reduces adiposity and improves insulin resistance in high-fat-induced obese mice. Endocrinology 2007; 21: doi:10.1210/en.2006-1371.
9. Giulivi C, Kato K, Cooper CE. Nitric oxide regulation for oxygen consumption I: cellular physiology. Review. Am J Physiol Cell Physiol 2006; 291(6): C1225-31. Epub 2006 Aug 2.
10. Cleeter MW, Cooper JM, Darley-Usmar VM, et al. Reversible inhibition of cytochrome c oxidase, the terminal enzyme of the mitochondrial respiratory chain, by nitric oxide. Implications for neurodegenerative diseases. FEBS Lett 1994; 345 : 50–54.
11. Brown GC, Cooper CE. Nanomolar concentrations of nitric oxide reversibly inhibit synaptosomal respiration by competing with oxygen at cytochrome oxidase. FEBS Letters 1994; 356 : 295–298.
12. Torres J, Cooper CE, Sharpe M, Wilson MT. Reactivity of Nitric Oxide with Cytochrome c Oxidase:Interactions with the Binuclear Centre and Mechanism of Inhibition. Journal of Bioenergetics and Biomembranes 1998; 30.
13. Davies N, Trikkas C, Cooper CE. Does carbon monoxide inhibit cytochrome oxidase in vivo? Biochem SocTrans 1997; 25 : 406S.
14. Schweizer M, Richter C. Nitric oxide potently and reversibly deenergizes mitochondria at low oxygen pension. Biochemical and Biophysical research Communications 1994; 204(1): 169–175.
15. Clerc P, Rigoulet M, Leverve X, Fontaine E. Nitric oxide increases oxidative phosphorylation efficiency. J Bioenerg Biomembr 2007; 39 : 158–166 DOI 10.1007/s10863-007-9074-1.
16. Nisoli E, Clementi E, Carruba MO, Moncada, S. Defective Mitochondrial Biogenesis: A Hallmark of the High Cardiovascular Risk in the Metabolic Syndrome? Circ Res 2007; 100 : 795–806.
17. Bossy-Wetzel E, Lipton SA. Nitric oxide signaling regulates mitochondrial number and function. Cell Death and Differentiation 2003; 10 : 757–760. doi:10.1038/sj.cdd.4401244.
18. Nedergaard J, Ricquier D, Kozak LP. Uncoupling proteins: current status and therapeutic prospects – Meeting on Uncoupling Proteins. EMBO reports 2005; 6: No 10.
19. Mantena SK, King AL, Andringa KK, et al. Mitochondrial dysfunction and oxidative stress in the pathogenesis of alcohol – and obesity-induced fatty liver diseases. Free Radic Res 2008; doi:10.1016/j.freeradbiomed.2007.12.029.
20. Banks WA. The blood-brain barrier as a cause of obesity.Review. Curr Pharm Des 2008; 14 : 1606–1614.
21. ESC guidelines on Diabetes, Pre-diabetes and Cardiovascular Diseases. European Heart Journal 2007; 9(Suppl C): 1–74.
22. Leclercq IA, Morais ADS, Schroyen B, et al. Insulin resistance in hepatocytes and sinusoidal liver cells: Mechanisms and consequences – Review. Journal of Hepatology 2007; 47 : 142–156.
23. de Luca C, Olefsky JM. Inflammation and insulin resistance. FEBS Letters 2008; 582 : 97–105.
24. Martyn JAJ, Kaneki M, Yasuhara S. Obesity-induced Insulin Resistance and Hyperglycemia – Etiologic Factors and Molecular Mechanisms. Anesthesiology 2008; 109 : 37–48.
25. Hodis J, Kutinová-Canová N, Potmesil P, et al. The role of adrenergic agonists on glycogenolysis in rat hepatocyte cultures and possible involvement of NO. Physiol Res 2007; 56 : 419–425. Epub 2006 Aug 22.
26. Albuszies G, Vogt J, Wachter U, Thiemermann C, et al. The effect of iNOS deletion on hepatic gluconeogenesis in hyperdynamic murine septic shock. Intensive Care Med 2007; 33 : 1094–1101. DOI 10.1007/s00134-007-0638-7.
27. Guarino MP, Afonso RA, Raimundo N, et al. Hepatic glutathione and nitric oxide are critical for hepatic insulin-sensitizing substance action Am J Physiol Gastrointest Liver Physiol 2003; 284: G588–G594. First published December 2002; 4 : 10.1152/ajpgi.00423.2002.
28. Moore MC, DiCostanzo CA, Smith MS, et al. Hepatic portal venous delivery of a nitric oxide synthase inhibitor enhances net hepatic glucose uptake Am J Physiol Endocrinol Metab 2008; 294: E768–E777. First published January 2008; 22: doi:10.1152/ajpendo.00184.2007.
29. Kahn BB, Flier JS. Obesity and insulin resistance. J Clin Invest 2000; 106 : 473–481.
30. Thomas MS, Zhang WR, Jordan PM, et al. Signaling pathways mediating a selective induction of nitric oxide synthase II by tumor necrosis factor alpha in nerve growth factor-responsive cells. Journal of Neuroinflammation 2005; 19: doi:10.1186/1742-2094-2-19.
31. Yang WS, Lee WJ, Funahashi T, et al. Weight reduction increases plasma levels of an adipose-derived anti-infl ammatory protein, adiponectin. J Clin Endocrinol Metab 2001; 86 : 3815–3819.
32. Chen H, Montagnani M, Funahashi T, et al. Adiponectin stimulates production of nitric oxide in vascular endothelial cells. J Biol Chem 2003; 278 : 45021–45026.
33. Vos TA, Van Goor H, Tuyt L, et al. Expression of Inducible Nitric Oxide Synthase in Endotoxemic Rat Hepatocytes Is Dependent on the Cellular Glutathione Status. Hepatology 1999; 29 : 421–426.
34. Nishimura M, Izumiya Y, Higuchi A, et al. Adiponectin Prevents Cerebral Ischemic Injury Through Endothelial Nitric Oxide Synthase–Dependent Mechanisms. Circulation 2008; 117 : 216–223.
35. Ellger B, Langouche L, Richir M, et al. Modulation of regional nitric oxide metabolism: Blood glucose control or insulin?Intensive Care Med 2008; 34 : 1525–1533 DOI 10.1007/s00134-008-1118-4.
36. Creager MA, Lüscher TF, Cosentino F, Beckman JA. Diabetes and Vascular Disease: Pathophysiology, Clinical Consequences, and Medical Therapy: Part I. Circulation 2003; 108 : 1527-1532 DOI: 10.1161/01.CIR.0000091257.27563.32.
Labels
Addictology Allergology and clinical immunology Angiology Audiology Clinical biochemistry Dermatology & STDs Paediatric gastroenterology Paediatric surgery Paediatric cardiology Paediatric neurology Paediatric ENT Paediatric psychiatry Paediatric rheumatology Diabetology Pharmacy Vascular surgery Pain management Dental HygienistArticle was published in
Journal of Czech Physicians

- Advances in the Treatment of Myasthenia Gravis on the Horizon
- Possibilities of Using Metamizole in the Treatment of Acute Primary Headaches
- Metamizole at a Glance and in Practice – Effective Non-Opioid Analgesic for All Ages
- Metamizole vs. Tramadol in Postoperative Analgesia
- Spasmolytic Effect of Metamizole
Most read in this issue
- Fatty acids – 1. Occurrence and biological significance
- Osteoporosis: Whom, when and how to treat?
- Coffee, its legend, history, and influence on human health
- Nitric oxide in patients with obesity and metabolic syndrome