
Neurogenní plicní edém
Šedý J. Neurogenic pulmonary oedema
Neurogenic pulmonary oedema is a complication of severe central nervous system injury. The centre of neurogenic pulmonary oedema is assumed to be a group of dorsal ventrolateral medulla nuclei, which are activated by a combination of afferent pathway hyperactivity and a sudden increase of intracranial pressure. The sympathetic system plays a crucial role in the pathogenesis of neurogenic pulmonary oedema by activating a rapid cascade of processes, leading to interstitial and intraalveolar oedema, together with important haemorrhage. For the diagnosis of neurogenic pulmonary oedema, physical examination and chest X-ray are crucial. The differential diagnosis is not easy, but the chances of proper diagnosis are increased when the relation between the central nervous system injury and the pulmonary problems is considered. Targeted curative treatment of neurogenic pulmonary oedema does not exist yet; thus, the treatment options are mainly supportive and symptomatic. The most important ones are continuous patient monitoring, posture and ventilation and oxygenation support. There are several experimental models that can be used for studying the etiopathogenesis or treatment of neurogenic pulmonary oedema. The main goal of experimental studies is to elucidate a preventive and therapeutic approach that is able to prevent or treat neurogenic pulmonary oedema. In this context, the most promising agent is atropine.
Key words:
neurogenic pulmonary oedema, brain injury, spinal cord injury, sympathetic nervous system.
:
Jiří Šedý
:
Ústav experimentální medicíny AV ČR, v. v. i., Praha
:
Čas. Lék. čes. 2011; 150: 147-155
:
Review Article
Neurogenní plicní edém je komplikace závažného poškození centrálního nervového systému. Centrem vzniku neurogenního plicního edému je s největší pravděpodobností skupina jader rostrální ventrolaterální prodloužené míchy, které jej spouští na podkladě kombinace hyperaktivace jejich aferentních drah a náhle zvýšeného intrakraniálního tlaku. V patogenezi neurogenního plicního edému hraje zásadní roli sympatický nervový systém, který spouští rychlou kaskádu dějů, vedoucích k intersticiálnímu a intraalveolárnímu edému, který doplňuje výrazná hemoragická složka. V diagnostice neurogenního plicního edému má zásadní význam fyzikální vyšetření a RTG hrudníku. Diferenciálně diagnostická úvaha není snadná, avšak šance na správnou diagnózu výrazně rostou, pokud lékař dá do souvislosti poškození centrálního nervového systému a plicní potíže. Cílená kurativní léčba neurogenního plicního edému dosud neexistuje, proto je smyslem terapeutických snah zejména léčba podpůrná a symptomatická. Jako nejdůležitější opatření se jeví kontinuální monitorace stavu pacienta, polohování pacienta a podpora ventilace a oxygenace. V současné době existuje několik experimentálních modelů, které je možné využít pro studium etiopatogeneze i léčby neurogenního plicního edému. Hlavním cílem experimentálních snah je získání preventivního a terapeutického přístupu, který umožní zabránit a včas léčit neurogenní plicní edém. Z tohoto hlediska je zatím nejvíce slibný atropin.
Klíčová slova:
neurogenní plicní edém, poranění mozku, poranění míchy, sympatický nervový systém.
Úvod
Normální funkce plicní tkáně vyžaduje co nejtenčí bariéru pro výměnu plynů mezi krevním řečištěm a vzduchem v plicích. Za normálních okolností je pohyb tekutiny přes stěnu cév přísně regulován. Pokud dojde k poruše tohoto mechanismu z nejrůznějších příčin a relativní nebo absolutnímu zmnožení tekutiny v oblasti mimo cévní řečiště na úkor tekutiny uvnitř cév, vzniká otok tkáně – edém. Plicní edém může mít celou řadu příčin, může probíhat různě závažně, s dočasnými nebo trvalými důsledky (1). Pro klinickou praxi je důležité, že plicní edém poměrně často zásadně komplikuje zdravotní stav pacienta a může dokonce vést navzdory intenzivní léčbě k jeho smrti.
Neurogenní plicní edém (NPE) je definován jako akutně vzniklý intersticiální a/nebo intraalveolární edém plicní tkáně, který vzniká na základě předchozího poškození tkáně centrálního nervového systému (CNS) (2). Jako první popsal NPE Shanahan již v roce 1908 jako komplikaci opakovaných epileptických záchvatů u jedenácti pacientů a byl to právě on, kdo spojil těžké poškození CNS se vznikem plicního edému, a zároveň první, kdo použil pro tento edém název „neurogenní“ (3). Neurogenní plicní edém vzniká řádově v minutách až hodinách po poranění CNS. Vzhledem k jeho „neurogenní“ povaze jej řadíme k nekardiogenním (extrakardiálním) plicním edémům. Z hlediska mechanismu vzniku se však jedná o poměrně unikátní klinickou jednotku, neboť se zde synergicky uplatňují oba základní mechanismy vzniku plicního edému, jmenovitě vysoký tlak intravaskulární a intersticiální tekutiny společně se zvýšenou permeabilitou součástí plicního parenchymu, které se u jiných typů edému vyskytují izolovaně (4). Kromě toho je zde hemoragická složka vyjádřena výrazně více, než u kteréhokoliv jiného edému. Podle dostupných statistických údajů by se mohlo zdát, že se jedná o velice vzácnou klinickou jednotku. Velice rozporuplná data však naznačují, že je tato jednotka spíše řazena mezi ostatní plicní edémy a není kladen důraz na její neurogenní původ. Neurogenní plicní edém bohužel klinický obraz ostatních plicních edémů pouze připomíná, jeho nebezpečí tkví ve výše zmíněných patofyziologických odlišnostech, v jejichž důsledku je často klinický průběh výrazně proměnný, a tedy i hůře předvídatelný, což může vést k dramatickým situacím.
Epidemiologie neurogenního plicního edému
Není snad na světě klinická jednotka, jejíž epidemiologická data by vykazovala tak rozporuplné výsledky jako právě NPE. Podle různých klinických studií se NPE vyskytuje v 1 až 71 % případů poškození CNS. Většina studií naštěstí naznačuje, že pravda bude zřejmě někde uprostřed, okolo 20–30 % pacientů s těžkým, náhle vzniklým poškozením CNS. Nižší čísla jsou pravděpodobně zaviněna skutečností, že NPE je u celé řady pacientů s poškozením CNS maskován jinými faktory, nebo je jeho význam v dané chvíli považován za druhořadý, vzhledem k často velmi těžkému celkovému klinickému stavu a nutnosti řešení důležitějších, akutně život ohrožujících stavů (akutní tepenné krvácení, počínající známky herniace mozkové tkáně apod.). Zvýšená čísla jsou pak pravděpodobně způsobena označením jakéhokoliv plicního edému za neurogenní, což je opačný extrém. Mortalita NPE se pohybuje okolo 10 % (5).
Obecná etiopatogeneze plicního edému
Plicní edém obecně vzniká na podkladě porušené rovnováhy intravaskulární a intersticiální tekutiny. Může vznikat z celé řady příčin, které vedou k jednomu nebo více z následujících dějů: 1. zvýšení kapilárního hydrostatického tlaku představuje nejčastější příčinu. Vzniká při přetlaku v žilním systému plic, který může být způsoben jak vlastními plicními cévami, tak poruchou přečerpávání krve levým srdcem. Dalším důvodem může být zvýšení objemu cirkulujících tekutin buď v celém organismu, nebo významná centralizace oběhu do oblasti životně důležitých orgánů, způsobená nejrůznějšími příčinami; 2. zvýšení propustnosti kapilární stěny – tento mechanismus vzniká na podkladě přímého nebo humorálně podmíněného rozšíření prostoru mezi jednotlivými endotelovými buňkami, případně jejich poškozením. Vede k nárůstu extravazace tekutiny a krevních elementů do intersticia a odtud eventuálně do intraalveolárního prostoru; 3. zvýšení povrchového napětí vnitřní plochy alveolů – vzniká v okamžiku, kdy v důsledku poškození nebo změny struktury surfaktantu, které vede ke změně vlastností na hranici vzduch-tekutina, kolapsu příslušných částí plic a úniku tekutiny do alveolů; 4. snížení onkotického tlaku krevní plazmy – vzniká při relativním nebo absolutním poklesu množství bílkovin krevní plazmy, zejména albuminu. Výsledkem je změna osmotického gradientu a únik tekutiny z intravaskulárního prostoru do intersticia a 5. snížení lymfatické drenáže plicního parenchymu – vzniká na podkladě zánětu nebo metastatického rozsevu (6, 7). Může být rovněž vrozená. Vede k nedostatečnému odvádění lymfy plicní tkání a jejímu hromadění v plicním parenchymu. Předstupněm plicního edému je překrvení plicního parenchymu – kongesce plic. Vlastní plicní edém se projevuje zvýšením tekutiny nejdříve v mezibuněčné hmotě plicního parenchymu – intersticiální edém, následně únikem tekutin do oblasti plicních sklípků – intraalveolární edém a dále i do dalších úseků dýchacích cest. Neurogenní plicní edém je charakterizován významným stupněm dilatace plicních kapilár, kongescí kapilár krvinkami, intraalveolárním krvácením a intersticiálním (perivaskulárním) a intraalveolárním edémem, který je tvořen na proteiny bohatým tkáňovým mokem – exsudátem, jež obsahuje více než 70 % plazmatické hladiny proteinů.
Etiopatogeneze neurogenního plicního edému
Ačkoliv je v etiologii NPE podezřívána celá řada mechanismů, není přes všechny snahy etiopatogeneze NPE dodnes spolehlivě vysvětlena. Etiologie NPE je dnes nejčastěji definována jako neuro-humorálně-stresová. Předpokládaný mechanismus rozvoje NPE ilustruje obrázek 1. Patofyziologicky představuje vznik NPE nerovnováhu Starlingových sil, odvislých od permeability kapilárního endotelu, cévního povrchu a hydrostatického a osmotického tlaku (4, 6–8). Roli Starlingových sil při vzniku NPE vymezuje Starlingova rovnice (9). Podle této rovnice je míra exfluxu tekutiny přímo úměrná hydrostatickému tlaku v kapiláře a nepřímo úměrná onkotickému tlaku v kapiláře. Zatímco za normálních okolností je většina této tekutiny odváděna systémem lymfatických cév zpět do žilního řečiště, v případě zvýšeného úniku tekutiny extravaskulárně tyto mechanismy brzy nedokáží tento děj dostatečně kompenzovat a rozvíjí se nejprve intersticiální a následně intraalveolární edém plic. Při extrémně náhlých objemově-tlakových změnách v plicním kapilárním řečišti navíc dochází k mechanickému poškození kapilárních stěn a extravazaci krevních elementů, což je typické právě pro NPE.
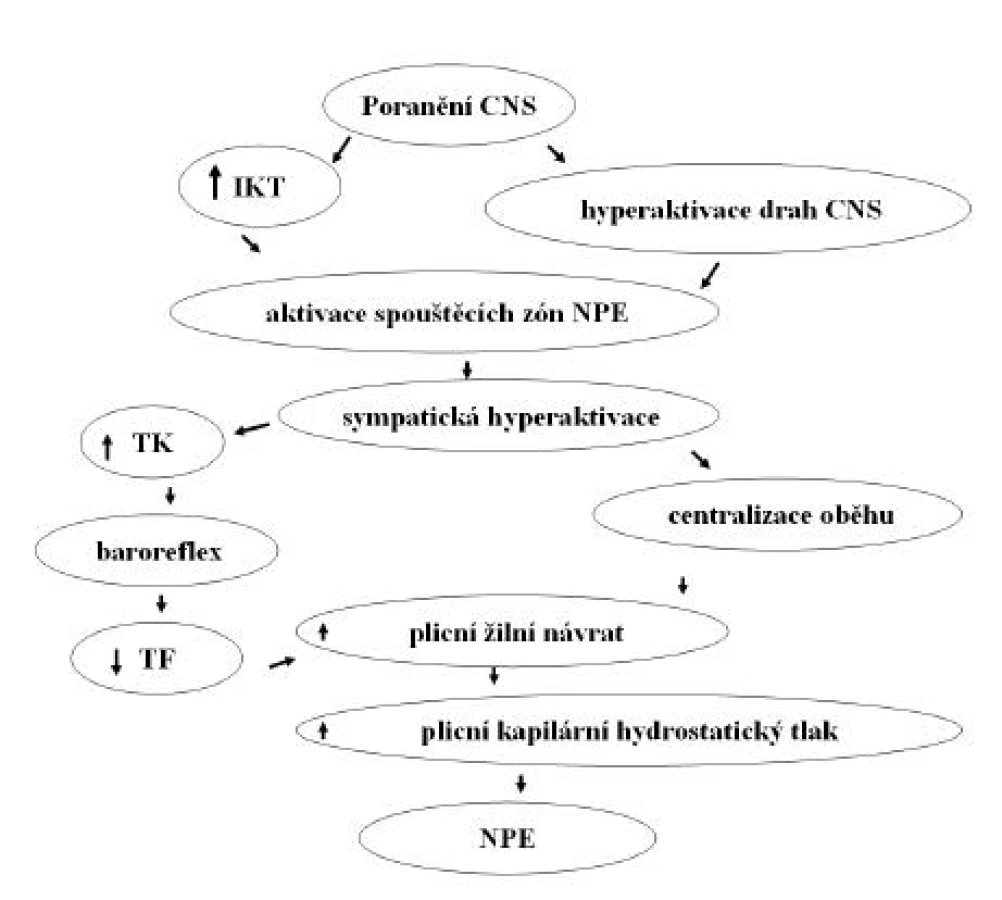
Role poranění nervového systému
Neurogenní plicní edém se rozvíjí nejčastěji v řádu hodin po poškození CNS nebo akutní exacerbaci chronického onemocnění mozku nebo míchy. Velmi rychle se rozvíjející NPE se může vyvinout i v řádu minut až desítek minut, tento typ je však spíše vzácný. Nejčastěji se rozvíjí u těžkých traumat CNS, u pacientů s epilepsií a v případech subarachnoidálního krvácení. Méně často se vyskytuje u pacientů se subdurálním krvácením, intracerebrálním nebo intramedulárním krvácením, hyponatremické encefalopatie, meningitidy, intrakraniálních a intraspinálních tumorů spojených s rozvojem hydrocefalu, u lupus erythematodes a u záchvatu roztroušené sklerózy (8). Kromě toho může NPE vzniknout velmi vzácně i iatrogenně při neurochirurgických zákrocích. Podobný mechanismus vzniku jako v případě NPE je podezříván i u plicního edému vznikajícího na podkladě feochromocytomu a u hand, foot and mouth disease (10, 11). Relativně sporné je označení NPE u toxických poškození tkáně CNS exogenními substancemi, jako jsou salicyláty (12), heroin (13), metadon (14). Efekty těchto látek jsou někdy přirovnávány k experimentálnímu modelu NPE, který je indukován vpravením veratrinu do cisterna magna. Neurogenní plicní edém byl popsán až u 20 % těžkých poranění mozku, charakterizovaných Glasgow coma scale < 8 (15). Studie Rogerse et al. (16) popisuje výskyt NPE ve 32 % případech poranění mozku s následkem smrti, přičemž 50 % pacientů s NPE zemřelo do 3 dnů. Výskyt NPE bývá v případech poranění mozku nejčastěji sdružen s náhlým vzestupem intrakraniálního tlaku (17). Byly však popsány i případy, kde prokazatelně ke vzestupu intrakraniálního tlaku nedošlo a přesto se edém rozvinul (18). Poškození plic v důsledku NPE je vedle infekce nejčastější příčinou kontraindikace jejich použití pro transplantaci u pacientů s diagnostikovanou mozkovou smrtí. Trulock (19) udává, že NPE je jednou z příčin toho, že pouze 20 % multiorgánových dárců má plíce ve stavu vhodném k transplantaci. Subarachnoidální krvácení, vznikající zejména na podkladě ruptury aneuryzmatu mozkové tepny, je jednou z nejčastějších příčin NPE. Velmi významná data udává studie Weira (20) popisující výskyt NPE u 71 % případů fatálního subarachnoidálního krvácení. Z této studie však vyplývá, že pouze 31 % pacientů mělo klinické známky NPE v období před úmrtím. V rozsáhlé retrospektivní studii na 477 pacientech nalezli Muroi et al. (21) NPE u 8 % pacientů. U všech pacientů se jednalo o rozsáhlý plicní edém s výraznou hemoragickou složkou. Edém byl signifikantně častější u pacientů, u kterých se příčina nacházela v dorzální části Willisova tepenného okruhu. Zvýšený intrakraniální tlak byl nalezen u 67 % pacientů s edémem. I když byly u 83 % pacientů nalezeny zvýšené srdeční markery, žádný z pacientů neměl v anamnéze ani v průběhu hospitalizace zjištěnu závažnou srdeční vadu. Neurogenní plicní edém měl zásadní dopad na návrat neurologických funkcí; Glasgow coma scale nižší než 4 mělo více než 77 % pacientů s NPE ve srovnání s 25 % pacientů bez NPE. V případě subarachnoidálního krvácení byl opakovaně popsán pozdní nástup rozvoje NPE, který se vyvíjel i několik dní po nejvýznamnější atace krvácení. Kromě toho se zpočátku mírný edém po různém období latence nebo zdánlivé rezoluce opět rozvíjí ve výrazně těžší až smrtelný stav (22). Epileptický záchvat je jednou z nejčastějších netraumatických příčin NPE. Nejčastější příčinou je záchvat typu grand mal. Neurogenní plicní edém se zde vždy rozvíjí v období po vlastním epileptickém záchvatu, u jednoho jedince i vícekrát za život (4, 23, 24). Ve studii Mulroye et al. (23) bylo zjištěno, že NPE ohrožuje zejména dětské pacienty s epilepsií. Celá řada experimentálních dat a klinických zkušeností ukazuje, že NPE může rovněž způsobit náhlá hyperaktivace většího množství drah, které vedou do center NPE. Typicky nastává tato situace u subarachnoidálního krvácení (2) nebo kompresní léze v hrudní části míchy (25–28). Experimentální data ukazují, že anestezie, aplikovaná epidurálně nebo intrathekálně do místa léze může rozvoji NPE zabránit, což lze velmi dobře sledovat i ve vymizení hemodynamické odpovědi (6, 29). Experimenty na ventilovaných zvířatech ukázaly, že NPE můžeme zabránit, pokud provedeme transekci krční míchy nad úrovní nebo přímo v úrovni míšní C7 (30, 31). V této oblasti se totiž nacházejí dráhy sympatiku, které převádějí tyto autonomní signály do oblasti plic. Vzato z opačného pohledu, rozvoj NPE můžeme potencovat, pokud míšní dráhy v této úrovni stimulujeme.
Role intrakraniálního tlaku
Epidemiologická data ukazují, že NPE vzniká zejména při rozsáhlejších poškozeních CNS, s výrazným podílem případů se zvýšeným intrakraniálním tlakem. V experimentu bylo prokázáno, že při náhlém zvýšení intrakraniálního tlaku dochází ke zvýšení tlaku systémového a mírnému zpomalení srdeční frekvence během 1 minuty. Zvýšení systémového a plicního tlaku spojené se zvýšeným žilním návratem, pozitivně inotropním účinkem na srdce a zvýšeným srdečním výdejem v závislosti na zvýšení tlaku intrakraniálního se označuje jako Cushingův reflex (5). Cílem tohoto reflexního mechanismu je za každou cenu perfundovat otékající tkáň CNS proti rostoucí periferní kapilární rezistenci; jeho důsledkem je však plicní edém. Cushing sám vysvětlil tento reflexní mechanismus na základě skutečnosti, že se v prodloužené míše hodnoty intrakraniálního tlaku přiblíží hodnotám tlaku systémového a mozkový perfuzní tlak klesne pod prahovou hodnotu, při které je mozková cirkulace ještě schopna autoregulace a zajištění 100 % nutričních potřeb (32). Při injekci plné krve nebo roztoku albuminu do cisterna magna umírá 50 % pokusných zvířat na komplikace spojené se vznikem NPE. Přitom množství vpravené tekutiny je mnohem důležitější než její složení. Někteří autoři prokázali, že role zvýšeného intrakraniálního tlaku při vzniku NPE je dána zejména tlakovou ischemií jader prodloužené míchy a deformace mozkového kmene, které vedou ke Cushingově odpovědi, zejména ve smyslu zvýšení krevního tlaku (sympatikus) a poklesu srdeční frekvence (parasympatikus) (33). Dnes se ukazuje, že mechanická deformace mozkového kmene má mnohem větší vliv na rozvoj systémové odpovědi, jejíž součástí je i NPE, než ischemická složka ve smyslu Cushingovy teorie. Jiní autoři prokázali, že při provedení bilaterální adrenalektomie před zákrokem na míše ke zvýšení systémového tlaku ani změnám srdeční frekvence nedochází. Na druhé straně, oboustranná vagotomie nemá žádný protektivní efekt při vzniku NPE, naopak rozsah edému ještě více zhoršuje. Zvýšený intrakraniální tlak prokazatelně poškozuje centra v prodloužené míše, která jsou podezřívána ze spuštění sympatické bouře. Klinické studie ukazují, že subarachnoideální krvácení z ruptury aneuryzmatu v povodí arteria vertebralis vede ke vzniku NPE častěji než ruptura aneuryzmatu v jiné lokalizaci. Nepřímé důkazy nasvědčují tomu, že ztráta krve může mít „protektivní“ vliv při rozvoji NPE, neboť se nemohou v dostatečné míře uplatnit významné tlakové změny, způsobené sympatickou bouří. Z doby války ve Vietnamu pochází kazuistika vojáka, který byl střelen nejdříve do břicha a poté do hlavy. Kulka, která zasáhla břišní dutinu, způsobila laceraci vena cava inferior a masivní vnitřní krvácení. Voják zemřel na následky poranění mozku, v plicích však nebyly nalezeny žádné známky plicního edému (5).
Hyperaktivace sympatického nervového systému
Na vzniku NPE se nejpravděpodobněji podílí náhle vzniklá systémová alfa-adrenergní stimulace sympatiku a uvolnění vazoaktivních substancí, označovaná jako katecholaminová bouře nebo méně často Cushingova odpověď. Výsledkem je generalizovaná vazokonstrikce a nárůst systémového a plicního tlaku, vedoucí k centralizaci oběhu. Následkem těchto změn je zvýšení plicního hydrostatického tlaku, poškození cévní a alveolární stěny, nárůst permeability alveolární stěny a uvolňování tekutiny a krevních buněk do intraalveolárního prostoru. Patofyziologický mechanismus, který se snaží vysvětlit vznik náhlých rozsáhlých hemoragií do plicního parenchymu na základě katecholaminové bouře, se označuje jako teorie výbuchu. Role sympatického systému při rozvoji NPE byla přímo i nepřímo prokázána celou řadou experimentů s využitím nejrůznějších experimentálních modelů. Příkladem je studie Novitzkyho et al. (34), kteří na opičím modelu zabránili rozvoji NPE, indukovaného nafouknutím balonku intrakraniálně, chirurgickou sympatektomií. Farmakologickým korelátem této studie je naše zjištění, že preventivní podání gangliového blokátoru pentolinia zcela zabrání rozvoji NPE u potkanů s poraněním míchy (25). Celkový pohled na tyto experimenty navíc ukazuje, že čím má pokusné zvíře více rozvinutý plicní autonomní systém, tím pravděpodobněji a tím těžší rozvine NPE. O náchylnosti k jeho rozvoji tedy vypovídá již samotný morfologický substrát (8). Někteří autoři, aby zdůraznili roli plicního řečiště a sympatiku při jeho vzniku, razili pro NPE termín neurohemodynamický plicní edém (8). Následky katecholaminové bouře jsou zvýšení systémového a plicního krevního tlaku, pokles tepové frekvence, centralizace krevního oběhu, zvýšený venózní návrat, zvýšení levostranného end-diastolického tlaku, zvýšení kapilárního hydrostatického tlaku a extravazace tekutiny, vedoucí k plicnímu edému.
Role krevního tlaku a tepové frekvence
V experimentech na zvířatech bylo opakovaně prokázáno, že rozvoji NPE předchází náhlé významné zvýšení systémového krevního tlaku. Blessing et al. (35) pozorovali zvýšení systémového tlaku o 40 mm u králíků, kterým experimentálně vyvolali NPE pomocí bilaterální destrukce A1 neuronů v medulla oblongata. Reis et al. (36) vyvolali NPE pomocí léze nucleus tractus solitarii na modelu laboratorního potkana a předpokládali, že smrt zvířete nastává na základě levostranného srdečního selhání v terénu těžké hypertenze. Blesssing et al. (35) pozorovali zvýšení cévní rezistence v abdominální aortě bezprostředně po inzultu, spouštějícím NPE o 350 %. Bradykardie je častým průvodním jevem při rozvoji NPE. Experimentálně bylo pozorováno, že bradykardie vzniká souběžně s elevací systémového tlaku (35). Podkladem bradykardie je pravděpodobně, podobně jako při aktivaci sympatiku, komprese a ischémie mozkového kmene. Bradykardie přímo následuje zvýšení intrakraniálního tlaku. Jádra mozkového kmene, odpovědná za tuto reakci, jsou pravděpodobně nucleus tractus solitarii a nucleus ambiguus. Experimentální data ukazují, že osu „mozkový kmen – srdeční frekvence“ není možné ovlivnit změnami systémového krevního tlaku. Zajímavé je, že experimentální intrakraniální hypertenze na levé straně mozkového kmene má výrazně větší vliv na rozvoj arytmií než na straně pravé. Pravděpodobně je to dáno asymetrickou autonomní periferní inervací srdce (37). Z těchto pokusů je zřejmé, že bradykardie pozorovaná u NPE může vznikat více na základě ovlivnění center v CNS než na podkladě periferního baroreflexu (27). Bradykardii u NPE je možné zvrátit pomocí bilaterální vagotomie nebo léze nucleus dorsalis nervi vagi.
Význam parasympatického nervového systému
Nervus vagus představuje jediný zdroj parasympatiku plic. Stimulace nervus vagus, například pomocí acetylcholinu, indukuje plicní vazodilataci a snížení kapilárního hydrostatického tlaku. Působení acetylcholinu je přitom dvojího typu – jednak působí na muskarinové receptory plicní hladké svaloviny cév, která takto ochabuje, jednak v plicním cévním řečišti inhibuje uvolňování katecholaminů (38). Naproti tomu, uvolnění vazoaktivního intestinálního polypeptidu z nervus vagus je příkladem působení alternativní, non-cholinergní, non-adrenergní cesty. V experimentech na morčeti, králíkovi a potkanovi bylo ukázáno, že oboustranné přetětí nervus vagus (bilaterální vagotomie) je schopné samo o sobě vyvolat plicní edém. Ačkoliv byl tomuto edému původně přiřazován stejný patofyziologický mechanismus jako edému neurogennímu – tj. excesivní aktivace sympatiku na základě vyřazení parasympatiku, tedy ono úplné odstranění závaží z jedné misky vah a úplné převážení misky druhé (39), další experimenty tyto úvahy nepotvrdily. Ukázalo se totiž, že bilaterální vagotomie pod odstupem nervus laryngeus recurrens plicní edém nevyvolává, a byly tak potvrzeny úvahy skeptiků, kteří tvrdili, že tento druh edému je způsobem obstrukcí dýchacích cest v důsledku laryngeálního a bronchiálního spazmu. Obstrukce dýchacích cest vyústí ve zvýšené nasávání a hromadění vzduchu v postižených plicních segmentech. V nepostižených plicních segmentech vzniká reflexním mechanismem negativní intersticiální tlak, který vyústí ve zvýšení transkapilárního filtračního tlaku v převzdušněných částech plic a dochází k rozvoji plicního edému. Nakonec se ukázalo, že bilaterální vagotomie pod odstupem nervus laryngeus recurrens nemá ani stupňující ani protektivní vliv na rozvoj NPE (8). Hlavním nebezpečím NPE je zejména náhlý a velmi razantní rozvoj, který může během několika minut ukončit život pacienta (40, 41). Z války ve Vietnamu pochází kazuistika naprosto zdravého vojáka, který byl v průběhu bitvy střelen zezadu do hlavy a na místě padl mrtev. Jeho pitva prokázala masivní NPE, vzniklý na základě rozsáhlého poranění mozku a zároveň vyloučila jinou příčinu plicního edému (5). V jeho patogenezi hraje významnou úlohu hemoragická složka, která je nejvíce vyjádřena právě u tohoto typu plicního edému. V našich experimentech vedla masivní exsudace sangvinolentní tekutiny z plic k úmrtí více než 40 % pokusných zvířat s nízkou hladinou isofluranové anestezie ve velmi krátké době (25, 26). Vznik NPE může být navíc potencován plicní kapilární vazokonstrikcí nebo změnami ve stěnách plicních cév v důsledku systémové choroby, jak bylo popsáno v případě systémového lupus erythematodes. Z řady experimentů vyplývá, že zvýšený krevní tlak hraje sice velmi důležitou roli, není však jediným zdrojem rozvoje NPE. V roce 1981 provedli Hoff et al. pokus, ve kterém modelovému zvířeti pomocí pouštění žilou regulovali tlak na normální hodnotu v kritické době rozvoje NPE. Neurogenní plicní edém byl sice menšího rozsahu, jeho rozvoj byl však pravidlem (42). Podobnou zkušenost máme i u našich experimentálních zvířat – pokud zvíře během výkonu v rámci experimentálního poranění míchy více krvácí, existuje menší pravděpodobnost, že zemře na následky NPE; edém však vytvořen bude. Tato zjištění odpovídají hemodynamickým vlastnostem krevního řečiště (28).
Poškození myokardu
V důsledku závažných hemodynamických změn dochází v průběhu rozvoje NPE i k sekundárnímu poškození myokardu. Syndrom bývá často označován jako syndrom myokardiálního otřesu (angl. neurogenic stunned myocardium) nebo Tako-Tsubo kardiomyopatie. Projevuje se nejčastěji reverzibilní generalizovanou hypokinezí srdečního svalu, doprovázeného sníženou ejekční frakcí. Vznik syndromu myokardiálního otřesu je nejpravděpodobněji způsoben náhlým vzestupem sérových katecholaminů, který je jedním z mechanismů, spouštějících NPE. Kromě toho se může podílet i katecholaminy vyvolaný nárůst systémové cévní rezistence. Syndrom je podmíněn generalizovaným výskytem mikronekróz v srdečním svalu (43). Mimořádně důležité je zjistění, že navzdory tomu, že syndrom myokardiálního otřesu vede v akutním stadiu často k výraznému zhoršení až smrti pacienta, při zvládnutí této situace nezanechává na myokardu téměř žádné funkčně-morfologické stopy a v průběhu 4–5 dnů se spontánně upraví. V těchto místech tedy spočívá velký potenciál pro budoucí výzkum, jehož cílem by mělo být terapeuticky ochránit srdeční sval proti působení negativního vlivu NPE a pomoci mu aktivně překlenout nejtěžší období, s výhledem vědomí, že v následujícím období bude schopen spontánní restituce ad integrum. Diferenciálně diagnosticky může být někdy obtížné odlišit syndrom myokardiálního otřesu od infarktu myokardu (44).
Látky ovlivňující vznik a rozvoj neurogenního plicního edému
Rozvoj a stupeň NPE ovlivňuje celá řada farmak a neurohumorálních působků. Mezi nejvýznamnější patří adrenalin, noradrenalin, neuropeptid Y, pentolinium, atropin a oxid dusnatý (28, 45). Adrenalin a noradrenalin hrají prokazatelně roli v průběhu katecholaminové bouře, infuze adrenalinu a noradrenalinu však per se NPE nezpůsobuje (46). Neuropeptid Y je neurohumorální působek, který má celou řadu farmakologických účinků, jako např. antinociceptivní, anxiolytický a orexigenní (zvyšující chuť k jídlu) a roli v modulaci cirkadiánního rytmu (47). Pokud je experimentálně do plic vpraven neuropeptid Y, který je mimojiné společně s noradrenalinem vylučován sympatickými nervy, a zvyšuje tak stupeň plicní vazodilatace a plicní vaskulární permeabilitu, reaguje organismus pokusného zvířete zvýšením plicní vaskulární permeability a vznikem NPE. Neuropeptid Y přitom působí přímo na endotelové buňky. Neuropeptid Y je schopen působením na GABA-ergní, glutamátergní a dopaminergní nervové dráhy ovlivňovat hloubku anestezie na úrovni CNS (47). Různé druhy anestetik, jako např. pentobarbital nebo ketamin, jsou zpětně schopny ovlivňovat receptory pro neuropeptid Y, a v důsledku pak i modulovat vznik NPE (40,41). Naše práce prokázaly, že podání pentolinia nebo atropinu dokáže zvrátit rozvoj NPE (25, 28, 45). Experimentální údaje ukazují, že endogenní opioidy (např. endorfiny) jsou zodpovědné za zvýšení plicní vaskulární permeability a objem extravaskulární tekutiny na podkladě zvýšení nitrolebního tlaku (48). Tato zjištění jsou založena na základě pozorovaného protektivního vlivu naloxonu (antagonista opioidních receptorů) při rozvoji NPE. Bylo prokázáno, že podání fentolaminu je schopno zabránit rozvoji NPE (49–51). Experimentální i klinická data ukazují, že beta-adrenergní složka je při rozvoji NPE méně významná. Colgan et al. (52) ukázal na psím modelu hypertenze, že propranolol redukuje plicní intravaskulární tlak a rozsah plicního zkratu. Při použití našeho modelu NPE jsme prokázali, že propranolol má mírně protektivní účinky na jeho rozvoj, které jsou však nesrovnatelně méně výrazné ve srovnání s alfa-adrenergní blokádou (27).
Anamnéza, klinický obraz a vyšetření
Z anamnestických údajů jsou z hlediska NPE důležité zejména parametry související se současným onemocněním CNS. Obecně řečeno jsou příznaky NPE poměrně různorodé a často nespecifické. Diagnostika proto není často snadná. Velmi důležité je dát tento klinický stav do souvislosti s akutně vzniklým poškozením CNS. Pokud je však již jednou pomyšleno na NPE, potvrzení diagnózy nečiní obvykle potíže. Klíčovým vyšetřením pro diagnostiku NPE je fyzikální vyšetření (zejména vědomí, srdeční a dechová frekvence, krevní tlak, typ dýchání, poklep a poslech plic).
Subjektivní příznaky
Mezi subjektivní příznaky NPE patří:
- náhle vzniklá dušnost (dyspnoe) – vzniká zejména v důsledku zvýšené dechové práce a větších tlakových změn a napětí v hrudním koši a v plicním parenchymu;
- bolesti na hrudi – vznikají ze stejných příčin jako dušnost, nemají charakter stenokardií;
- zhoršené odkašlávání – je způsobeno přetížením dýchacího svalstva předchozí námahou a nemožností kašlem odstranit edematózní tekutinu v alveolárních částech plic;
- bolesti hlavy, nauzea a zvracení – jsou způsobeny hypoxií. Často jsou projevem poškození CNS, zejména zvýšeného intrakraniálního tlaku.
- celkové oslabení – projevuje se pocity slabosti, schváceností, úzkostí až obavami o vlastní život, které vznikají v důsledku nemožnosti se dostatečně nadechnout a bolestí při dýchání. Pacient se výrazně potí.
Objektivní příznaky
Mezi objektivní příznaky NPE patří:
- vazba na poškození CNS – pro diagnostiku je klíčová. Může se jednat jak o poranění mozku nebo míchy, tak o náhle vzniklý stav poškození parenchymu CNS, způsobený krvácením, tlakem útvarů různé etiologie nebo náhlou exacerbací celkové chronické choroby.
- povrchnější dýchání – jedná se o obrannou reakci organismu na edémem způsobené zvýšení tlakových změn, potřebných pro ventilaci. Pacient více zapojuje pomocné dýchací svaly. Může být přítomno vpadávání supraklavikulárních jamek a prodloužení exspiria se spastickými bronchitickými fenomény.
- tachypnoe – důsledek hypoxie organismu. Vzhledem k tomu, že organismus nemůže zvyšovat dechovou práci zvětšením dechového objemu, musí takto činit zvýšením dechové frekvence.
- tachykardie – rovněž důsledek hypoxie. Vzniká reflexně, snahou o lepší zajištění krevního zásobení organismu.
- poslechový nález – typické jsou polopřízvučné až nepřízvučné vrzoty a chrůpky malých bublin, začínající při bázích plic a později se šířící na celý plicní parenchym. Chrůpky bývají typicky nad oběma plícemi, mohou však začínat na jedné plíci a později se šířit i nad druhou plíci.
- suchý dráždivý kašel – ke kašli dráždí tekutina přítomná v plicích;
- narůžovělé sputum až hemoptýza – jsou typické pro NPE v důsledku jeho hemoragické složky;
- hypoxemie – vzniká v důsledku nedostatečného okysličování krve kyslíkem, která se projevuje nízkým parciálním tlakem O2, sníženou saturací hemoglobinu kyslíkem, chladnými a bledými akry až cyanózou, vystupňovanou dušností, zapojováním pomocného dýchacího svalstva a řadou subjektivních nespecifických příznaků typu únavy a malátnosti. Hyperkapnie u plicního edému nebývá přítomna, neboť zvýšené ventilační úsilí v naprosté většině případů kompenzuje difuzní a ventilační poruchy. Častěji nacházíme hypokapnii, vznikající v důsledku zvýšeného ventilačního úsilí, která vede k rozvoji respirační alkalózy. Tento stav snižuje stupeň ionizace kalcia, a vede tak ke zvýšené neuromuskulární dráždivosti až křečím kosterního svalstva. Kromě toho může hypokapnie způsobovat vazokonstrikci mozkových cév, a tedy projevy sníženého zásobení krve kyslíkem v podobě točení hlavy, závratí až poruch vědomí.
- zvýšený intrakraniální tlak – je důsledkem nebo příčinou poškození CNS. Neurogenní plicní edém jej nevyvolává.
- poruchy vědomí – vznikají buď v důsledku hypokapnie, nebo častěji v důsledku generalizované hypoxie;
- spíše mírnější zvýšení teploty – NPE je nezánětlivý, proto významné zvýšení teploty odráží spíše jiný nebo nasedající zánětlivý stav;
- smrtelný chropot (angl. death rattle) – doprovází terminální stadium NPE v okamžiku, kdy není edematózní tekutina dostatečně odstraňována;
- minimální nebo žádné známky zánětu – důležitý diferenciálně diagnostický znak.
Vyšetření
Pro diagnostiku plicního edému je z pomocných vyšetření nejdůležitějším RTG hrudníku. Nacházíme zde obraz plicní žilní hyperemie a difuzní zastření plicního parenchymu v důsledku intersticiální edému, které v pozdějších stadiích přechází v husté zastření, odpovídající alveolárnímu edému. V případě NPE bývá zpočátku maximum zastření v perihilární oblasti, později se rozšiřuje na ostatní části plic. Pro diferenciální diagnostiku je důležité, že v případě kardiálních příčin doprovází RTG obraz zvětšení levostranného srdečního stínu. V případě prodělaného infarktu myokardu můžeme pozorovat i vyklenující se poinfarktové aneuryzma levé srdeční komory. V 90 % případů je možné pozorovat různý stupeň difuzního zastření obou plicních polí, zejména v hilové oblasti, nástřik plicních cév a normální velikost srdečního stínu. Monitoring plicního tlaku v zaklínění může poskytnout důležité diferenciálně diagnostické údaje v rámci odlišení kardiálního plicního edému, jeho nevýhodou je však invazivita. Kromě toho v případě plicního edému poměrně nepřesně odráží hodnoty kapilárního hydrostatického tlaku, a může proto často vykazovat normální hodnoty i v případě velmi vysokého kapilárního hydrostatického tlaku, prohlubujícího plicní edém (53). Biochemické testy jsou zde spíše druhořadé, časté monitorování pomocí ASTRUP může být přínosem. V periferní krvi je patrná významná metabolická acidóza se zvýšeným deficitem bazí. Vyšetření krevních plynů odhalí známky hypoxie a snížení parciálního tlaku oxidu uhličitého. Elektrokardiografie a echokardiografie pomohou vyloučit kardiogenní příčinu plicního edému. Echokardiografie pomůže odhalit syndrom myokardiálního otřesu, projevující se generalizovanou hypokinezí srdečního svalu, doprovázeného sníženou ejekční frakcí. Vyšetření srdečních enzymů pomůže diferenciálně diagnosticky odhalit infarkt myokardu a další příčiny kardiálního plicního edému. Pro diagnostiku syndromu myokardiálního otřesu je nejvýznamnější stanovení hladiny troponinu, která je u těchto pacientů nižší než 2,8 ng/ml a vyskytuje se konkomitantně s ejekční frakcí menší než 40 % a echokardiografickými abnormitami, při absenci jakýchkoliv změn na EKG (54). Zvýšené hodnoty mozkového natriuretického peptidu (BNP) naznačují, že příčinou plicního edému je levostranné srdeční selhání. Histopatologicky můžeme makroskopicky pozorovat zvětšené prosáklé plíce, na kterých jsou patrné subpleurální sufuze. Z plic vytéká narůžovělá zpěněná tekutina. Mikroskopicky je suverénním barvení hematoxylin-eosin. Na řezu můžeme pozorovat ztluštělé alveolární stěny a alveoly, vyplněné eozinofilním materiálem, mezi nímž jsou četné krevní elementy s převahou erytrocytů. Časté jsou atelektázy.
Diferenciální diagnostika neurogenního plicního edému
Diferenciální diagnostika plicního edému (tab. 1) záleží zejména v 1. nalezení nebo vyloučení kardiální příčiny a 2. rozlišení mezi edémem s převahou hydrostatického a permeabilního mechanismu vzniku. Odlišení nemusí být vždy jednoduché, neboť jednotlivé formy se mohou překrývat. Například, primárně hydrostatický mechanismus u kardiálního plicního edému může v případě náhlého nárůstu kapilárního hydrostatického tlaku způsobit tak rozsáhlou extravazaci intravaskulární tekutiny, že může připomínat permeabilní mechanismus vzniku.
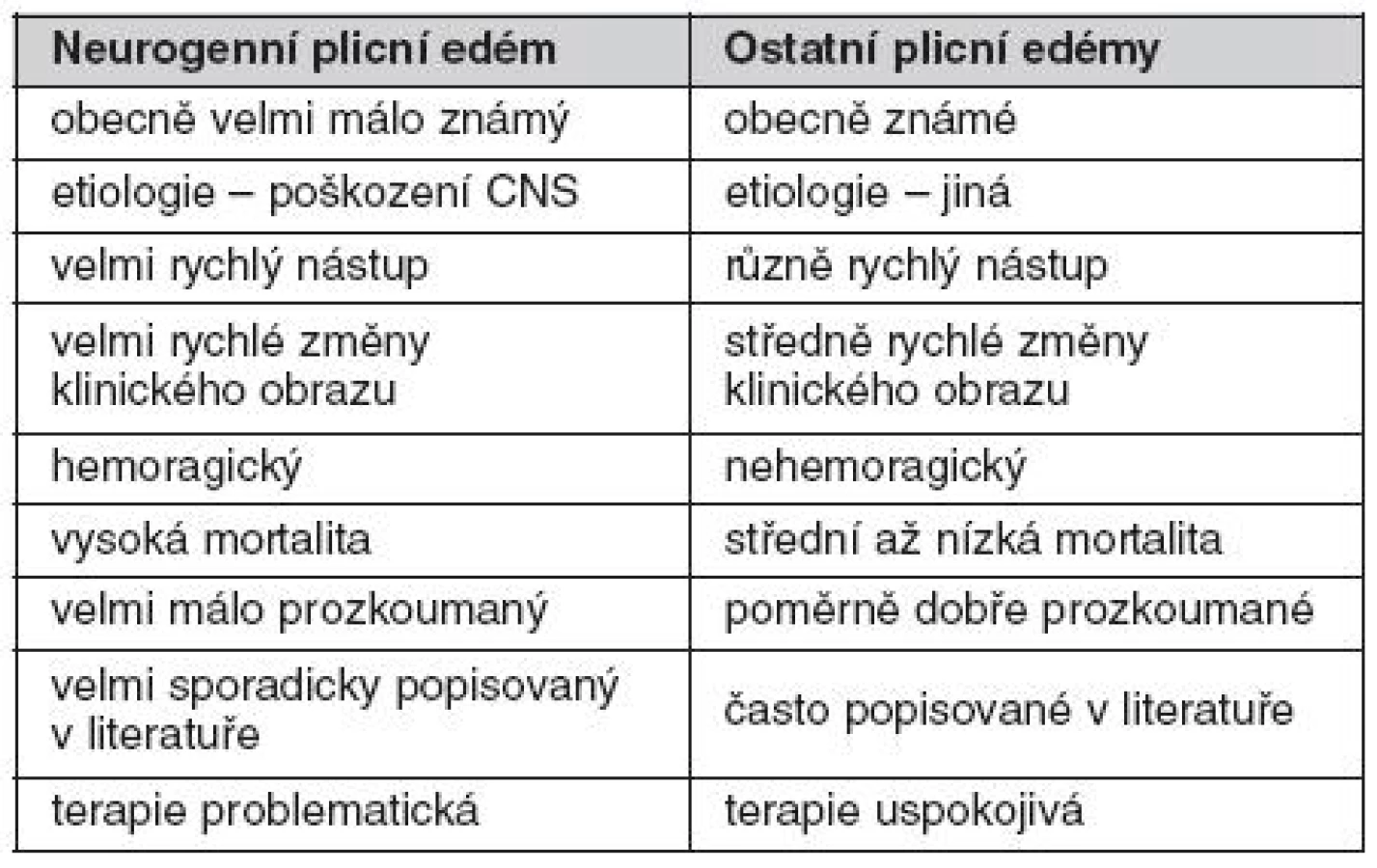
Základní diferenciálně diagnostická rozvaha je vedena na podkladě anamnézy, fyzikálního vyšetření a laboratorních hodnot. U nekardiálních edémů není obvykle v anamnéze žádná srdeční příhoda ani chronické srdeční onemocnění. U kardiálních edémů nacházíme při fyzikálním vyšetření studená cyanotická akra, zrychlený tep, kardiomegalii, distenzi krčních žil a vlhké chropy na plicích. Pacient s nekardiogenním edémem má obvykle teplá akra bez zásadních poruch tepové frekvence, bez distenze krčních žil a suché chropy na plicích. Z pomocných vyšetření nacházíme u kardiálních plicních edémů známky ischémie na EKG, zvýšené kardiální enzymy a perihilární distribuci vaskulární kongesce na RTG hrudníku. Plicní kapilární tlak obvykle převyšuje 18 mm Hg a poměr množství proteinů v edematózní tekutině ku množství proteinů v krevní plazmě nepřevyšuje 0,5. Naproti tomu u nekardiálních plicních edémů jsou EKG a kardiální enzymy normální, RTG hrudníku vykazuje vesměs periferní distribuci edematózní tekutiny, plicní kapilární tlak je obvykle nižší než 18 mm Hg a poměr množství proteinů v edematózní tekutině k množství proteinů v krevní plazmě je vyšší než 0,7. RTG nálezy se obvykle objeví relativně později, průměrně okolo 12 hodin po začátku kardiopulmonálních symptomů. V těžkých případech je na podkladě RTG vyšetření obtížné odlišit, zda se jedná o kardiogenní či nekardiogenní plicní edém. Studie však dokazují, že je možné v iniciálních fázích mezi těmito dvěma typy plicního edému najít odlišnosti, pokud se při hodnocení RTG snímku zaměříme na určité specifické rysy; pak je možné kardiogenní od nekardiogenního plicního edému odlišit s přesností na 91 % (55–57). Důležitá kritéria zahrnují:
- u nekardiogenního plicního edému je místem iniciální akumulace tekutiny plicní intersticium včetně peribronchiální oblasti a septálních linií. Poměrně rychle se však plicní edém mění v intraalveolární, kdy jsou alveolární prostory zcela vyplněny na proteiny bohatou tekutinou. Naproti tomu u kardiálního plicního edému dochází k zaplnění nitra alveolů tekutinou až v okamžiku, kdy již jsou překročeny reabsorpční kapacity intersticia.
- Kerleyho linie jsou typické pro kardiogenní plicní edém, zatímco u nekardiogenního plicního edému, vznikajícího na principu zvýšené permeability alveolo-kapilární membrány tyto linie přítomny nejsou. Lze je tedy považovat za znak typický pro kardiální plicní edém.
- periferní distribuce edematózní tekutiny v plicích a pleurální efuze jsou typické pro nekardiální plicní edém, zatímco u kardiálního se v iniciálních stadiích nevyskytují.
- u nekardiálního plicního edému jsou normální velikost a morfologie srdečního stínu a stíny velkých cév. Naproti tomu u kardiálního plicního edému jsou srdeční stín v oblasti levého srdce a šířka cév vycházejících ze srdce, zvětšeny. Neurogenní plicní edém může imitovat celá řada stavů, nejpravděpodobnější jsou však akutní plicní selhání (ALI – acute lung injury) a syndrom akutní dechové tísně (ARDS – adult respiratory distress syndrome). Tyto syndromy totiž vznikají na zánětlivém podkladě vedoucímu k difuznímu poškození alveolů (DAD – diffuse alveolar damage) a současně bez akutní vazby na poškození CNS. Mortalita ARDS a ALI je vyšší než mortalita NPE. Klinický obraz aspirační pneumonie je podobný NPE (pacient v těžkém stavu, dechová nedostatečnost, restriktivní porucha dýchání, tachykardie, tachypnoe). Aspirační pneumonie však nevzniká tak dramaticky rychle jako NPE a je u ní přítomna horečka. Je třeba dát pozor na stavy, kdy je horečka způsobena poškozením vlastního CNS (zejména oblasti ventrálního hypothalamu) – to však není na vrub NPE, tento nemá zánětlivou složku. Odeznění příznaků aspirační pneumonie navíc trvá déle, okolo 2 týdnů. Klinický obraz kardiálního plicního edému nejrůznější etiologie, např. v důsledku levostranného srdečního selhání, může rovněž imitovat NPE. Zde jsou nejdůležitější anamnestické údaje a neurologické a kardiologické vyšetření.
Terapie neurogenního plicního edému
Základem terapie je odstranění, minimalizace nebo stabilizace příčiny v CNS. Vzhledem k tomu, že cílená kurativní léčba NPE dosud neexistuje, je smyslem terapeutických snah zejména léčba podpůrná a symptomatická. Jako nejdůležitější opatření se jeví kontinuální monitorace stavu pacienta, polohování pacienta a podpora ventilace a oxygenace. Neurogenní plicní edém se u většiny pacientů podaří zvládnout do 48–72 hodin. Ačkoliv je léčbu možné považovat spíše za podpůrnou než kauzální, tedy jinými slovy, že poskytneme organismu možnost, aby se plicního edému sám zbavil, je tato léčba naprosto klíčová a často život zachraňující. Základem terapeutických snah je kontinuální monitorace životních funkcí pacienta, zejména měření krevního tlaku, tepové frekvence, dechové frekvence, EKG a saturace. Provádíme opakovaná hematologická a biochemická vyšetření krve. Monitorace tlaku v zaklínění může být velkým přínosem v průběhu terapie, neboť umožní udržení nízkých hodnot tlaku na srdeční úrovni a zároveň může zabránit nadměrnému snížení srdečního výdeje a hypoperfuzi mozku (58).
Poloha těla
Pro pacienta s NPE je klíčová poloha těla. Základem je taková poloha, která zabrání zvýšenému žilnímu návratu. Nutná je zvýšená poloha hlavy. Nejvýhodnější je poloha vsedě s dolními končetinami spuštěnými z lůžka. Tuto polohu však nelze uplatnit u pacientů s hypotenzí. V rámci první pomoci je někdy doporučováno dočasné přiměřené zaškrcení dolních kočetin v oblasti stehen, které redukuje žilní návrat z této oblasti. Zaškrcení pochopitelně musí umožňovat cirkulaci, nesmí tedy omezovat proud krve v tepnách. V některých případech může být výhodné pacienta uložit do polohy na břiše, jak doporučuje Marshall a Nyquist (59) nebo Fletcher a Atkinson (60). Tato poloha se velmi osvědčila i u pacientů s ARDS, jak ukazuje randomizovaná klinická studie Guerina (61). Studie Mancebo et al. (62) jasně ukazuje i snížení mortality u pacientů s ARDS, u kterých byla použita poloha na břiše již od prvních projevů onemocnění. Pro NPE zatím systematická studie bohužel neexistuje. Mechanismus terapeutického úspěchu polohy na břiše není zcela jasný, předpokládá se však redistribuce výměny plynů do oblastí méně postižených plicním edémem a zároveň zmírnění poškození plic způsobené umělou plicní ventilací. Při indikaci polohy na břiše však musíme vzít v úvahu, že u některých neurologických onemocnění, jako je intracerebrální krvácení, subarachnoidální krvácení, tepenné uzávěry v oblasti arteria cerebri media nebo poranění mozku, může poloha na břiše přivodit zvýšení intrakraniálního tlaku a takto zhoršit prognózu pacienta.
Oxygenace
Základním opatřením při podpoře oxygenace a ventilace je potlačení hypoxie pomocí přímého podávání kyslíku nosní nebo obličejovou maskou, případně pomocí endotracheální intubace a přetlakové mechanické ventilace pomocí pozitivního endexspiračního přetlaku (PEEP – positive end expiratory pressure). Izolované dodávání kyslíku zvyšuje parciální tlak kyslíku v plicních alveolech, a usnadňuje tak výměnu plynů. Kontinuální nebo dvoustupňové dodání vysokotlakého kyslíku kromě toho umožňuje úlevu přetíženým dýchacím svalům. Přetlaková mechanická ventilace pomocí PEEP značí iatrogenně navozenou situaci, kdy je na konci exspiria v dýchacích cestách vyšší tlak než atmosférický. Rozeznáváme tři úrovně PEEP. Nízká úroveň značí úrovně PEEP do 5 mm H2O, střední úroveň značí PEEP v rozmezí 5–15 mm H2O, vysoká úroveň značí PEEP nad 15 mm H2O. Pro ventilační podporu pacientů s NPE používáme nejčastěji střední úroveň, výjimečně vysokou úroveň PEEP. Kontinuální pozitivní přetlak v dýchacích cestách (CPAP – continuous positive airway pressure) pak značí situaci, kdy je u spontánně dýchajícího nemocného v dýchacích cestách udržen tlak vyšší než atmosférický po celou dobu dechového cyklu, tedy i v době inspiria. Kromě dodávky kyslíku a úlevy dechových svalů přetlaková mechanická ventilace 1. snižuje preload a afterload a takto usnadňuje srdeční činnost, 2. napomáhá redistribuci tekutiny v plicích z intraalveolárních do extraalveolárních prostor, kde tolik neinterferují s výměnou dechových plynů a 3. zvětšuje plicní objem a napomáhá tak prevenci rozvoje plicní atelektázy. PEEP vede ke zlepšení oxygenace v důsledku snížení plicního zkratu a ke zlepšení eliminace oxidu uhličitého na podkladě zvýšení alveolární ventilace. Je však třeba dbát toho, aby nedošlo k hyperinflaci, která se projeví naopak snížením perfuze ventilovaných alveolů, zhoršením eliminace oxidu uhličitého a dále dokonce zhoršením oxygenace – tento efekt závisí na podílu redistribuce krevního průtoku do neventilovaných oblastí. Bylo prokázáno, že v určitých oblastech plic probíhá průtok krve plicními kapilárami pouze v době exspiria, kdy je plicní kapilární tlak vyšší než plicní alveolární tlak. Velikost perfuze těmito oblastmi odpovídající velikosti plicního zkratu tak závisí jednak na použité hodnotě PEEP, jednak na velikosti dechového objemu, poměru inspiria a exspiria a hodnotě plicního kapilárního tlaku. Bylo prokázáno, že po překročení optimální hodnoty středního tlaku v dýchacích cestách, která u konkrétního pacienta závisí na i na aktuální hodnotě plicního kapilárního tlaku, dochází tímto mechanismem nejen ke zhoršení eliminace oxidu uhličitého, ale i oxygenace. Provzdušnění tekutinou vyplněných alveolů a roztažení kolabovaných alveolů v terénu plicního edému pomocí PEEP se označuje v zahraniční literatuře jako recruitment. Za tento děj je zodpovědný vrcholový alveolární tlak, dosahovaný v průběhu inspiria. Kromě působení na respirační systém má PEEP i poměrně velký vliv na hemodynamické poměry v malém oběhu. PEEP 1. snižuje venózní návrat do oblasti levého srdce a tedy i preload, 2. snižuje transmurální tlakový gradient, který musí být překonáván v průběhu srdeční kontrakce, což vede ke snížení afterloadu a s tím souvisí i 3. snížená spotřeba kyslíku myokardem – tyto efekty jsou nejvíce vyjádřeny u pacientů s levostranným srdečním selháním. Naproti tomu u pacientů bez levostranného srdečního selhání dominuje u aplikace PEEP snížení žilního návratu, a tedy i preloadu. U hypovolemických pacientů pak ke snížení srdečního výdeje. Vliv PEEP na afterload pravé komory závisí na redistribuci plicní reperfuze po zařazení PEEP a na aktuální velikosti plicního objemu. Plicní vaskulární rezistence dosahuje při nízkém plicním objemu vysokých hodnot, což je důsledek hypoxické plicní vazokonstrikce. Se zvyšováním plicního objemu plicní vaskulární rezistence klesá a po překročení optimální hodnoty plicního objemu dochází k jejímu opětovnému vzestupu v důsledku komprese kapilárního řečiště. Efekt aplikace PEEP na funkci pravé komory je tedy výsledkem komplikované interakce mezi snížením preloadu a ovlivněním afterloadu. PEEP má rovněž vliv na nitrobřišní tlak, který po zařazení PEEP stoupá. U pacientů s výrazně sníženou poddajností dutiny břišní může vést použití nadměrně vysokých hodnot PEEP k vzestupu nitrobřišního tlaku a omezení perfuze nitrobřišních orgánů. Nastavení hodnot PEEP v konkrétních situacích je stále předmětem diskuzí.
Farmakologická léčba
V časném stadiu mohou být užitečné alfa-blokátory a kortikoidy, farmakologická terapie však obvykle mnoho nevyřeší. Zvýšení diurézy představuje základní opatření při snižování preloadu. Snižuje hemodynamickou zátěž na levé srdce a pozitivně tak ovlivňuje jak srdeční sval, tak plicní hemodynamiku. Většina kličkových diuretik typu furosemidu, bumetanidu nebo torsemidu představuje úspěšnou podpůrnou terapeutickou modalitu při léčbě plicního edému, i v terénu hypoalbuminémie, hyponatrémie nebo hypochlorémie. Lékem první volby je furosemid, vzhledem k jeho venodilatačnímu účinku, kterým snižuje preload ještě před vlastním počátkem diuretického působení. Počáteční dávky furosemidu jsou doporučovány spíše nižší (do 0,5 mg/kg), teprve při jejich nedostatečném účinku se zvyšují (1 mg/kg), zejména u pacientů již léčených diuretiky nebo pacientů s renální insuficiencí nebo tam, kde je primární příčinou plicního edému hypervolémie. Nitráty slouží k vazodilataci jak koronárních tepen, které zlepší srdeční funkci, tak ke snížení preloadu levého srdce vazodilatací plicních žil. Zatímco u kardiogenních plicních edémů je jejich účinek významný, u NPE je jejich efekt nižší. V akutním stadiu používáme zejména sublinguálně aplikovaný nitroglycerin (1,2 mg každých 5 minut). Metodou volby, používanou zejména při systolickém tlaku vyšším než 100 mm Hg, je intravenózní podávání nitroprussidu sodného v dávce 0,1–5 μg/kg za minutu. Jeho podávání vyžaduje kontinuální monitoraci jeho hemodynamických parametrů. Morfinové preparáty mají kromě anxiolytického a analgetického účinku, který je v případě NPE rovněž žádoucí, také venodilatační efekt, který snižuje preload. Kromě toho snižuje stupeň dušnosti na podkladě odstranění stresu, útlumu vylučování katecholaminů, odstranění tachykardie a komorového afterloadu. ACE inhibitory mají význam zejména u pacientů s konkomitantní hypertenzí nebo hypertenzní krizí. Zpočátku se podávají spíše nižší dávky, následované vyššími udržovacími dávkami. U izolovaného NPE nemají zásadní efekt. Intravenózní podávání rekombinantního mozkového natriuretického faktoru (BNF) je nová efektivní léčebná modalita, která působí poměrně efektivní vasodilataci a snižuje tak preload. Iniciální dávka je 2 μg/kg, následované kontinuální infuzí 0,01 μg/kg za minutu. Při edému mozku a zvýšeném intrakraniálním tlaku aplikujeme příslušnou léčbu, která však ovlivňuje základní onemocnění a nikoli NPE.
Závěr
Neurogenní plicní edém je relativně málo častá, avšak závažná klinická jednotka. Poprvé byl popsán Shanahanem v roce 1908 jako komplikace opakovaných epileptických záchvatů. Jeho rozvoj je vázán na závažné poškození CNS. Centrem vzniku NPE je s největší pravděpodobností skupina jader rostrální ventrolaterální prodloužené míchy, které jej spouští na podkladě kombinace hyperaktivace jejich aferentních drah a náhle zvýšeného intrakraniálního tlaku. V patogenezi NPE hraje zásadní roli sympatický nervový systém, který spouští rychlou kaskádu dějů, vedoucích k intersticiálnímu a intraalveolárnímu edému, který doplňuje výrazná hemoragická složka. V diagnostice NPE má zásadní význam fyzikální vyšetření a RTG hrudníku. Diferenciálně diagnostická úvaha není snadná, avšak šance na správnou diagnózu výrazně rostou, pokud lékař dá do souvislosti poškození CNS a plicní potíže. Cílená kurativní léčba NPE dosud neexistuje, proto je smyslem terapeutických snah zejména léčba podpůrná a symptomatická. Jako nejdůležitější opatření se jeví kontinuální monitorace stavu pacienta, polohování pacienta a podpora ventilace a oxygenace. V současné době existuje několik experimentálních modelů, které je možné využít pro studium etiopatogeneze i léčby NPE. Hlavním cílem experimentálních snah je získání preventivního a terapeutického přístupu, který umožní zabránit a včas léčit NPE. Z tohoto hlediska je zatím nejvíce slibný atropin. Detailnější informace o neurogenním plicním edému můře čtenář nalézt v mé recentní monografii na toto téma (63).
Zkratky
| ALI | – akutní plicní selhání (acute lung injury) |
| ARDS | – syndrom akutní dechové tísně (adult respiratory distress syndrome) |
| BNF | – mozkový natriuretický faktor |
| CNS | – centrální nervový systém |
| CPAP | – pozitivní přetlak v dýchacích cestách (continuous positive airway pressure) |
| DAD | – difuzní poškození alveolů (diffuse alveolar damage) |
| NPE | – neurogenní plicní edém |
| PEEP | – pozitivní endexspirační přetlak (positive end expiratory pressure) |
Autor pro korespondenci:
MDDr.
MUDr. Jiří Šedý, PhD.
Ústav
experimentální medicíny AV ČR, v.v.i.
Vídeňská
1083, 142 20 Praha 4
fax:
+420 241 062 783, e-mail: jirisedy@hotmail.com
Sources
1. Vinš P. Plicní edém. Interní medicína pro praxi 2003, 11 : 540–547.
2. Fontes RB, et al. Acute neurogenic pulmonary edema: case reports and literature review. J Neurosurg Anesthesiol 2003; 15 : 144–150.
3. Shanahan WT. Acute pulmonary edema as a complication of epileptic seizures. N Y Med J 1908; 37 : 54–56.
4. Colice GL. Neurogenic pulmonary edema. Am Rev Respir Dis 1984; 130 : 941–948.
5. Simmons RL, et al. Respiratory insufficiency in combat casualties. II. Pulmonary edema following head injury. Ann Surg 1969; 170 : 39–44.
6. Šedý J, et al. Mechanism of neurogenic pulmonary edema development. Physiol Res 2008; 57 : 499–506.
7. Šedý J, et al. Rapid but not slow spinal cord compression elicits neurogenic pulmonary edema in the rat. Physiol Res 2009; 58 : 269–277.
8. Malik AB. Mechanisms of neurogenic pulmonary edema. Circ Res 1985; 57 : 1–18.
9. Starling EG. The Linacre Lecture on the Law of the Heart. London: Longmans, Green and Company 1918.
10. Rassler B. The role of catecholamines in formation and resolution of pulmonary oedema. Cardiovasc Hematol Disord Drug Targ 2007; 7 : 27–35.
11. McMinn PC. An overview of the evolution of enterovirus 71 and its clinical and public health significance. FEMS Microbiol Rev 2002; 26 : 91–107.
12. Broderick TW, et al. Salicylate-induced pulmonary edema. AJR Am J Roentgenol 1976; 127 : 865–866.
13. Steinberg AD, Karliner JS. The clinical spectrum of heroin pulmonary edema. Arch Intern Med 1968; 122 : 122–127.
14. Frand UI, et al. Methadone-induced pulmonary edema. Ann Intern Med 1972; 76 : 975–979.
15. Bratton SL, Davis RL. Acute lung injury in isolated traumatic brain injury. Neurosurgery 1997; 40 : 707–712.
16. Rogers FB, et al. Neurogenic pulmonary edema in fatal and nonfatal head injuries. J Trauma 1995; 39 : 860–866.
17. Baumann A, et al. Neurogenic pulmonary edema. Acat Anaest scand 2007; 51 : 447–455.
18. Popp AJ, et al. Delayed pulmonary dysfunction in head–injured patients. J Neurosurg 1982; 57 : 784–790.
19. Trulock EP. Lung transplantation. Am J Respir Crit Care Med 1997; 155 : 789–818.
20. Weir BK. Pulmonary edema following fatal aneurysm rupture. J Neurosurg 1978; 49 : 502–507.
21. Muroi C, et al. Neurogenic pulmonary edema in patients with subarachnoid hemorrhage. J Neurosurg Anesthesiol 2008; 20 : 188–192.
22. Fisher A, Aboul-Nasr HT. Delayed nonfatal pulmonary edema following subarachnoid hemorrhage. Case report. J Neurosurg 1979; 51 : 856–859.
23. Mulroy JJ, et al. Postictal pulmonary edema in children. Neurology 1985; 35 : 403–405.
24. Darnell JC, Jay SJ. Recurrent postictal pulmonary edema: a case report and review of the literature. Epilepsia 1982; 23 : 71–83.
25. Šedý J, et al. Low concentration of isoflurane promotes the development of neurogenic pulmonary edema in spinal cord injured rats. J Neurotrauma 2007; 24 : 1487–1501.
26. Šedý J, et al. A new model of severe neurogenic pulmonary edema in spinal cord injured rats. Neurosci Lett 2007; 423 : 167–171.
27. Šedý J, et al. Low degree of anesthesia increases the risk of neurogenic pulmonary edema development. Med Hypotheses 2008; 70 : 308–313.
28. Šedý J, et al. The role of nitric oxide in the development of neurogenic pulmonary edema in spinal cord injured rats: the effect of preventive interventions. Am J Physiol Regul Integr Comp Physiol 2009; 297: R1111–R1117.
29. Hall SR, et al. Intrathecal lidocaine prevents cardiovascular collapse and neurogenic pulmonary edema in a rat model of acute intracranial hypertension. Anesth Analg 2002; 94 : 948–953.
30. Chen HI, et al. Pulmonary edema and hemorrhage resulting from cerebral compression. Am J Physiol 1973; 224 : 223–229.
31. Chen HI, Chai CY. Integration of the cardiovagal mechanism in the medulla oblongata of the cat. Am J Physiol 1976; 231 : 454–461.
32. Cushing H. Concerning a definitive regulatory mechanism of the vasdomotor center which controls blood pressure during cerebral compression. John Hopkins Hosp Bull 1901; 12 : 290.
33. Thompson RK, Malina S. Dynamic axial brain–stem distortion as a mechanism explaining the cardiorespiratory changes in increased intracranial pressure. J Neurosurg 1959; 16 : 664–675.
34. Novitzky D, et al. Prevention of myocardial injury during brain death by total cardiac sympathectomy in the Chacma baboon. Ann Thorac Surg 1986; 41 : 520–524.
35. Blessing WW, et al. Hypertension, bradycardia, and pulmonary edema in the conscious rabbit after brainstem lesions coinciding with the A1 group of catecholamine neurons. Circ Res 1981; 49 : 949–958.
36. Reis DJ, et al. Brain lesions and hypertension: chronic lability and elevation of arterial pressure produced by electrolytic lesions and 6–hydroxydopamine treatment of nucleus tractus solitarii (NTS) in rats and cats. Prog Brain Res 1977; 47 : 169–188.
37. Krasney JA, Koehler RC. Heart rate and rhythm and intracranial pressure. Am J Physiol 1976; 230 : 1695–1700.
38. Bergofsky EH. Humoral control of the pulmonary circulation. Annu Rev Physiol 1980; 42 : 221–233.
39. Schmitt GH, Meyers FH. Characterization of the acute pulmonary edema that follows vagal section in the guinea pig. Am J Physiol 1957; 190 : 89–92.
40. Leal Filho MB, et al. Hemodynamic parameters and neurogenic pulmonary edema following spinal cord injury: an experimental model. Arq Neuropsiquiatr 2005; 63 : 990–996.
41. Leal Filho MB, et al. Importance of anesthesia for the genesis of neurogenic pulmonary edema in spinal cord injury. Neurosci Lett 2005; 373 : 165–170.
42. Hoff JT, et al. Experimental neurogenic pulmonary edema. Part 1: The role of systemic hypertension. J Neurosurg 1981; 54 : 627–631.
43. Marshall SA, Nyquist P. A Change of Position for Neurogenic Pulmonary Edema. Neurocrit Care 2009; 10 : 213–217.
44. Seow VK, et al. Neurogenic pulmonary oedema misdiagnosed as acute myocardial infarction in a comatose patient. Ann Acad Med Singapore 2007; 36 : 684–686.
45. Šedý J, et al. Atropine prevents the neurogenic pulmonary edema development. Med Hypotheses 2009; 73 : 42–44.
46. Rosell S. Neuronal control of microvessels. Annu Rev Physiol 1980; 42 : 359.
47. Naveilhan P, et al. Distinct roles of the Y1 and Y2 receptors on neuropeptide Y-induced sensitization to sedation. J Neurochem 2001; 78 : 1201–1207.
48. Peterson BT, et al. Effect of naloxone on the pulmonary vascular responses to graded levels of intracranial hypertension in anesthetized sheep. Am Rev Respir Dis 1983; 128 : 1024–1029.
49. Maron MB, Dawson CA. Pulmonary venoconstriction caused by elevated cerebrospinal fluid pressure in the dog. J Appl Physiol. 1980; 49 : 73–78.
50. Maron MB. Dose-response relationship between plasma epinephrine concentration and alveolar liquid clearance in dogs. J Appl Physiol 1998; 85 : 1702–1707.
51. Hakim TS, et al. Effects of sympathetic nerve stimulation on lung fluid and protein exchange. J Appl Physiol 1979; 47 : 1025–1030.
52. Colgan FJ, et al. Protective effects of beta blockade on pulmonary function when intracranial pressure is elevated. Crit Care Med 1983; 11 : 368–372.
53. Ganter BG, et al. Pulmonary capillary pressure. A review. Minerva Anestesiol 2006; 72 : 21–36.
54. Schubert A. Cardiovascular therapy of neurosurgical patients. Best Pract Res Clin Anaesthesiol 2007; 21 : 483–496.
55. Kithreotis P. Noncardiogenic pulmonary edema, ARDS, acute respiratory failure. Hell Respir Soc Publ 2000; 1 : 452–458.
56. Milne EN, et al. The radiologic distinction of cardiogenic and noncardiogenic edema. Am J Roentgenol 1985; 144 : 879–894.
57. Aberle R, et al. Hydrostatic versus increased permeability pulmonary edema. Diagnosis based on radiographic criteria in ctritically ill patients. Radiology 1988; 168 : 73–75.
58. Lagerkranser M, et al. Neurogenic pulmonary oedema: a review of the pathophysiology and clinical and therapeutic implications. Acta Med Scand 1982; 212 : 267–271.
59. Fletcher SJ, Atkinson JD. Use of prone ventilation in neurogenic pulmonary oedema. Br J Anaesth 2003; 90 : 238–240.
61. Guerin C. Ventilation in the prone position in patients with acute lung injury/acute respiratory distress syndrome. Curr Opin Crit Care 2006; 12 : 50–54.
62. Mancebo J, et al. A multicenter trial of prolonged prone ventilation in severe acute respiratory distress syndrome. Am J Respir Crit Care Med 2006; 173 : 1233–1239.
63. Šedý J. Neurogenní plicní edém, 1. vydání. Bratislava: Spišské vydavatelstvo 2011.
Labels
Addictology Allergology and clinical immunology Angiology Audiology Clinical biochemistry Dermatology & STDs Paediatric gastroenterology Paediatric surgery Paediatric cardiology Paediatric neurology Paediatric ENT Paediatric psychiatry Paediatric rheumatology Diabetology Pharmacy Vascular surgery Pain management Dental HygienistArticle was published in
Journal of Czech Physicians

- Advances in the Treatment of Myasthenia Gravis on the Horizon
- Possibilities of Using Metamizole in the Treatment of Acute Primary Headaches
- Metamizole vs. Tramadol in Postoperative Analgesia
- Spasmolytic Effect of Metamizole
- Metamizole at a Glance and in Practice – Effective Non-Opioid Analgesic for All Ages
Most read in this issue
- Tvarůžková Z, Pavlová Š, Doubek M, Mayer J, Pospíšilová Š. Lymphoproliferative disease in patients with autoimmune and inflammatory diseases: significance of antigenic stimulation and inflammatory processes
- Šedý J. Neurogenic pulmonary oedema
- Sobotka R, Hanuš T, Zemanová M. Principal value of surgical therapy in the renal cell cancer, chances of the biological treatment
- Hlaváčková L. Colleagues of Prof. J. Thomayer as in his notices from 1905–1918