
Kolorektální karcinom a kanonická Wnt signalizace
Colorectal cancer and canonical Wnt signalling pathway
Colorectal cancer is among the most frequent malignancies in the economically developed part of the world including the Czech Republic. From the molecular biological point of view, the development of colorectal cancer is in most cases linked to the pathologically activated canonical Wnt signalling pathway. The aim of this review is to present this intercellular signalling pathway, its role in the colorectal carcinogenesis and possible therapeutic implications according to our current knowledge.
Key words:
colorectal cancer, Wnt signalling pathway, tumorigenesis.
:
Martin Moravec
:
Univerzita Karlova v Praze, 3. lékařská fakulta, II. interní klinika FNKV, Fyziologický ústav AV ČR, Praha
:
Čas. Lék. čes. 2012; 151: 335-342
:
Review Article
Kolorektální karcinom je jednou z nejčastějších malignit v hospodářsky vyspělé části světa včetně České republiky. Na molekulárně biologické úrovni je ve většině případů jeho vznik spojený s patologicky aktivovanou kanonickou Wnt signalizací. Tato rešeršní práce si klade za cíl představit čtenářům tuto mezibuněčnou signalizační dráhu, její roli v kolorektální karcinogenezi i možná terapeutická východiska vyplývající z našich aktuálních znalostí.
Klíčová slova:
kolorektální karcinom, Wnt signalizace, nádorová transformace buňky.
Úvod do problematiky
Kolorektální karcinom (colorectal cancer, CRC) je jednou z nejčastějších malignit v hospodářsky vyspělé části světa a Česká republika patří k zemím s jeho nejvyšší incidencí (1, 2). Pochopení mechanismů účastnících se jeho vzniku a rozvoje může pomoci jeho účinnější léčbě. Objevy posledních více než 20 let ukazují, že hlavní roli v tomto procesu hraje patologicky zvýšená aktivita tzv. kanonické Wnt signalizační dráhy (3, 4).
Jedná se o mezibuněčnou molekulární signalizační dráhu objevenou v roce 1982 u octomilky obecné (Drosophila melanogaster), která ale plní zásadní funkce napříč celou živočišnou říší, a to i u člověka. Podporuje a reguluje proliferaci a diferenciaci buněk, nese informaci o jejich poloze a prostorové orientaci a ovlivňuje jejich migraci. Největší její význam u živočichů i u člověka je při časné ontogenezi, v průběhu formace tkání a orgánů. V dospělosti její význam klesá, ale nemizí. U člověka se účastní trvalé obměny některých tkání (kostní, epitelu gastrointestinálního traktu včetně kolorektální sliznice a dalších), mimo jiné udržováním zásoby tkáňových kmenových buněk (5–8). V humánní medicíně se setkáváme s řadou chorob spojených s její nedostatečnou (např. osteoporóza) či naopak nadměrnou aktivitou (mnoho nádorů, např. Wilmsův tumor, maligní melanom, karcinomy mammy, ovaria, jícnu, žaludku, jater, pankreatu či kolorekta) (9–12). Dle aktuálních znalostí mění expresi mnoha set genů a ovlivňuje asi čtyřicet jiných signalizačních drah (13).
Kanonická Wnt dráha je formou mezibuněčné regulace, kdy molekuly produkované jednou buňkou mění chování a genovou expresi buňky druhé. Těmito molekulami jsou Wnt glykolipoproteiny (Wnt). Ty se vážou na membránové receptory cílových buněk, a tak aktivují řadu intracelulárních pochodů vedoucích k nárůstu hladiny transkripčního faktoru ß-kateninu v cytoplazmě, k jeho přestupu do jádra a k jím zprostředkovaným změnám genové exprese. Protein ß-katenin je ústřední molekulou a efektorem celé kanonické Wnt dráhy (5–8).
Kromě kanonické existují i nekanonické Wnt signalizace. Jsou též iniciovány vazbou molekul Wnt na membránové receptory cílových buněk, mechanismy jejich působení ale nezahrnují aktivaci ß-kateninu a jím zprostředkované genové exprese. Řada extracelulárních, membránových i intracelulárních komponent kanonické i nekanonických drah jsou společné, což svědčí o jejich úzké provázanosti. O tom, která z nich bude spuštěna, rozhoduje řada více či méně známých faktorů. Patří mezi ně konkrétní typ Wnt a receptoru, resp. jejich kombinace, ale i vliv dalších membránových a cytoplazmatických regulačních molekul, přičemž původní koncept tzv. kanonických a nekanonických Wnt (dle signalizace, kterou mají aktivovat) se opouští. Výsledný efekt závisí tedy nejen na mezibuněčném signálu, ale do značné míry na samotné cílové buňce (7, 14–17). Rovněž nekanonické dráhy plní důležité role při tvorbě tkání a orgánů a jsou významnými regulátory cytoskeletu. V některých situacích mění aktivitu dráhy kanonické (pozitivně i negativně) a mohou mít význam i v kolorektální karcinogenezi (18–20). To je však oblast málo prozkoumaná. V tomto textu se budeme věnovat pouze dráze kanonické (dále již jen Wnt dráha), zájemce odkazujeme na jiné práce (21–24).
Kanonická Wnt dráha – komponenty, regulace, cílové geny
Wnt dráha začíná v buňkách produkujících signální Wnt. V tlustém střevě a konečníku jsou těmito buňkami myofibroblasty lokalizované subepiteliálně na dně slizničních krypt (invaginací zvětšujících povrch sliznice) (25). U člověka je známo devatenáct molekul Wnt. Obsahují glykosylové a lipidové zbytky, druhé jmenované (konkrétně jde o zbytky kyseliny palmitové a palmitolejové) jim propůjčují hydrofobní charakter a jsou nezbytné pro sekreci i funkci Wnt. Jejich vazbu zprostředkovává O-acyltransferáza Porcupine v endoplazmatickém retikulu buňky. Odtud jsou Wnt ve vazbě s proteinem Wntless (Wls) přenášeny vakuolárním systémem Golgiho aparátu k buněčné membráně a následně secernovány. Dle aktuálních znalostí je Wls spojen výhradně se sekrecí Wnt, která je tak oddělená od sekrece jiných molekul. Třetím důležitým aktérem počátku signalizace je proteinový komplex Retromer, jenž přenáší Wls z membrány zpět do Golgiho aparátu a brání jeho degradaci. Defekt každé z uvedených tří molekul Wnt dráhu narušuje a všechny jsou pravděpodobným místem její regulace (26–30) (obr. 1).
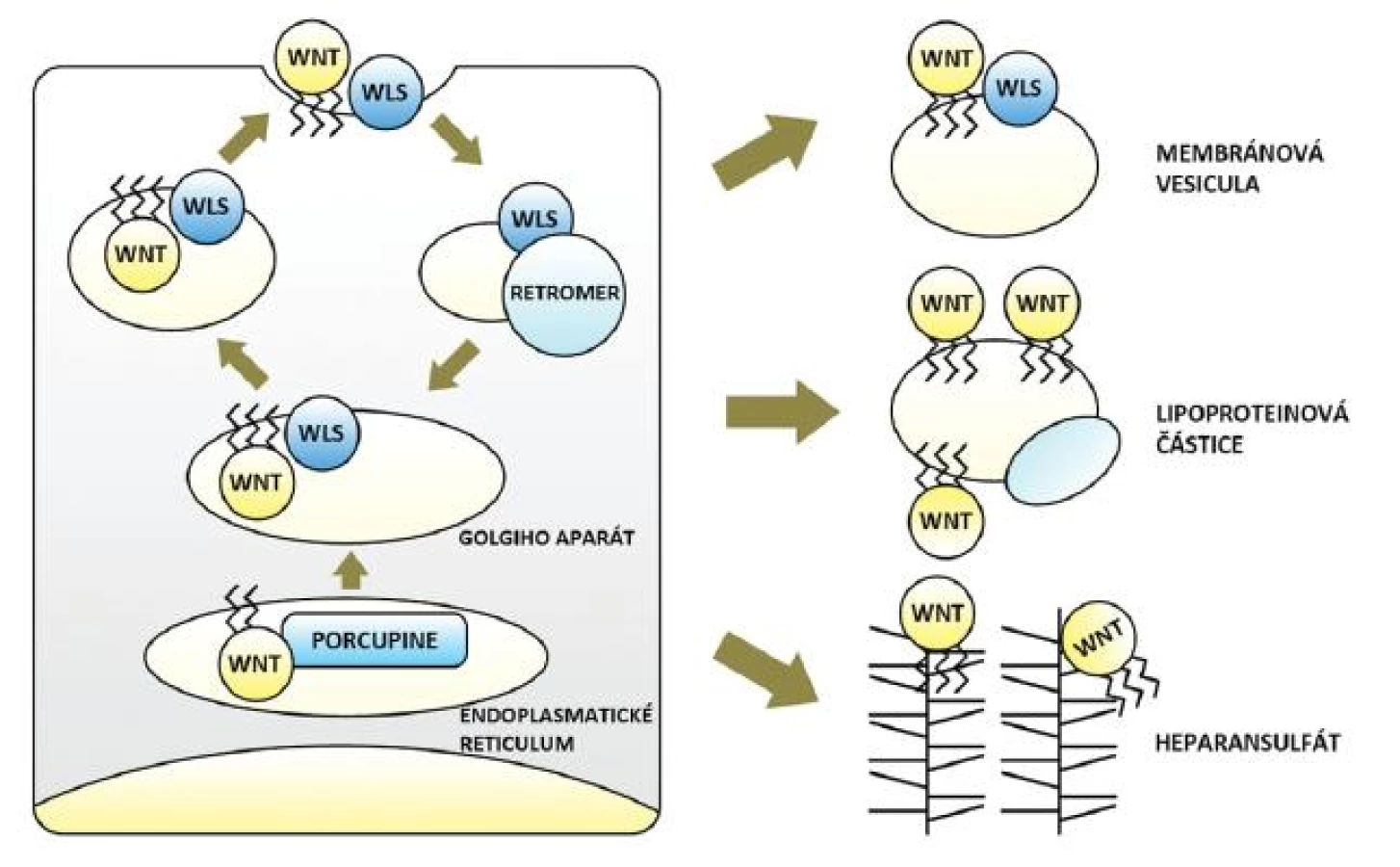
Transport hydrofobních Wnt ve vodném mezibuněčném prostředí vyžaduje určité pomocné mechanismy. Mohou být přenášeny vazbou na proteoglykany heparansulfáty (HSPG) nacházející se na membránách buněk i v extracelulární matrix. Wnt schopné difuze ve vodě jen na minimální vzdálenost překonávají díky síti HSPG vzdálenosti mnoha buněk. To potvrdil i elegantní experiment na octomilce obsahující klony buněk bez membránových HSPG. Wnt nebyly schopny tyto buňky překonat a signalizace se na jejich úrovni zastavila. Množství HSPG rovněž ovlivňoval gradient signalizace (31). Doložen je též přenos v komplexu s Wls ve vezikulách odloučených z membrány secernující buňky (32) a také přenos na větší vzdálenost ve vazbě na lipofilní částice (např. lipoproteiny včetně známého high-density lipoproteinu, HDL) (33, 34). Ač důkazy chybí, nelze vyloučit ani další mechanismy transportu pozorované u jiných signalizací (např. tvorba shluků Wnt ukrývající lipidové zbytky ve svém nitru). Účastnící se molekuly (HSPG, lipoproteinové částice a možná řada dalších) jsou opět významnými prvky regulace dráhy (6, 27–30) (obr. 1).
Mimo molekuly, které přenos Wnt umožňují, existuje mnoho dalších, které jej inhibují. Brání interakci Wnt s receptory na cílových buňkách, a to vazbou na jedny či druhé. Mezi hlavní patří Wnt inhibitory factor 1 (WIF1), solubilní Frizzled-related proteins 1-5 (sFRP 1-5) a Dickkopf 1 (DKK1) (5, 6). Mimo známou inhibiční funkci je ale možné, že svou vazbou Wnt do jisté míry stabilizují, a tak přenos ovlivňují i pozitivně (35).
V dalším kroku dochází k interakci Wnt s membránovými receptory na cílových buňkách. Hlavními z nich jsou Frizzled (FZD), jichž je u člověka známo deset a jež se účastní i nekanonických signalizací. Výhradně s kanonickou jsou spjaté low-density lipoprotein receptor-related protein 5 a 6 (LRP5, LRP6) fungující jako koreceptory FZD. DKK1 je specifickým inhibitorem LRP5/6, a tím i kanonické dráhy. Jsou známy i další receptory, ligandy a sekreční inhibitory/modulátory, které aktivují i inhibují Wnt signalizace včetně kanonické. Hrají ale důležité role jinde než v kolorektální sliznici, např. v kostní a nervové tkáni (14–16, 36).
FZD i LRP5/6 jsou molekuly transmembránové. Pro přenos signálu jsou podstatné konformační (tvarové) změny jejich intracelulárních domén, dané především jejich fosforylací. Při kanonické signalizaci je Wnt vázáno zároveň FZD i LRP5/6, čímž dochází k jejich seskupení a za účasti kinázy glykogensyntázy 3ß (GSK3ß) a kaseinkinázy 1 (CK1) k těmto fosforylačním změnám. Fosforylovaný LRP5/6 váže cytoplazmatický protein Axin, což je zásadní událost pro další přenos signálu. Fosforylačních změn i vazby Axinu se významně účastní další intracelulární protein Dishevelled (Dvl), jehož postižení vede k zástavě Wnt dráhy. Dvl je zároveň důležitou součástí i jiných buněčných signalizací (6, 8, 16, 36).
Ústřední molekulou kanonické dráhy je ß-katenin. Je nejen transkripčním faktorem, ale i významnou složkou cytoskeletu (účastní se mezibuněčné adheze) a při absenci kanonické signalizace plní především zde své funkce (37). Za této situace je jeho volná cytoplasmatická frakce významně snižována aktivitou tzv. ß-katenin degradačního komplexu. Jej tvoří proteiny Axin, GSK3, CK1 a APC (adenomatous polyposis coli), kde Axin tvoří kostru komplexu, GSK3 a CK1 v něm ß-katenin fosforylují a APC jej následně předkládá E3-ligáze ß-TrCP2, která na něj váže malý protein ubikvitin. Fosforylační změny a tato tzv. ubikvitinace slouží k rozpoznání ß-kateninu proteodegradačním aparátem buňky (proteazomem) a k jeho rozložení (6, 38).
Hladina Axinu je řádově nižší než zbylých tří proteinů a jeho množství je pro sestavení komplexu limitujícím faktorem. Navíc téměř neplní jiné funkce, a kanonickou signalizaci tak odděluje od jiných buněčných pochodů. Při aktivitě dráhy dochází k fosforylaci LRP, který pak na sebe Axin váže. Působí tím jeho relativní nedostatek a disociaci degradačního komplexu. Existují dvě rozdílné molekuly Axinu, které jsou z hlediska schopnosti degradovat ß-katenin rovnocenné, jejichž hladina je ale řízena odlišně. Zatímco exprese genu pro Axin2 je aktivována Wnt dráhou a je součástí její negativní zpětné vazby, exprese genu pro Axin1 je na Wnt dráze nezávislá (6, 39–41).
V případě, že volný cytoplazmatický ß-katenin není rozkládán, stoupá jeho hladina a dochází k jeho přestupu do jádra. Tento přestup je předmětem různých regulací, např. prostřednictvím dalších fosforylačně-defosforylačních změn, a je též ovlivňován jinými signalizačními drahami (viz níže) (5–8).
V jádře mění ß-katenin expresi mnoha genů. Sám není schopen vazby na DNA, ale účinkuje ve spojení s transkripčními faktory T-cell factors 1,3,4 (TCF1,3,4) a lymphoid enhancer factor 1 (LEF1), které tuto schopnost mají. Při nepřítomnosti ß-kateninu však TCF/LEF genovou transkripci inhibují, a to jak lokálně vazbou blokující promotory konkrétních genů, tak působením konformačních změn DNA na značnou vzdálenost (např. aktivací deacetyltransferáz či de - methyltransferáz histonů DNA či přímým vlivem na tvar DNA). Tak se mění přístupnost DNA pro transkripční aparát buňky, a tím výsledná genová exprese. Inhibiční účinek TCF/LEF často potencují další navázané molekuly (např. Groucho). ß-katenin je schopen je vyvázat, zaujmout jejich místo a zároveň poutat jiné molekuly působící aktivačně (např. BCL9, Pygopus). TCF/LEF se tak stávají transkripčními aktivátory a nadto opět dochází ke konformačním změnám DNA s výše popsanými důsledky (42) (obr. 2).

Efekt aktivované kanonické dráhy se značně liší podle druhu a stáří organismu, typu tkáně a buňky, její lokalizaci, funkčním statutu a nejspíše řady dalších determinant (5–8). To ukazuje na složitější regulace než jen pouhou přítomnost či absenci ß-kateninu v jádře. Jsou mimo jiné zprostředkované následujícími mechanismy. Lidské geny pro transkripční faktory TCF/LEF jsou čtyři, jejich produktů je ale daleko více (rozdílný splicing, posttranslační modifikace včetně fosforylace atd.) a ve svých účincích se liší. Některé působí aktivačně i bez ß-kateninu, jiné jej nejsou schopny vázat a jsou trvalými inhibitory (tzv. dominantly negative, dnTCF/LEF), další reagují s odlišnými promotory a ovlivňují jiné geny (43, 44). Širokou možnost modulace kanonické dráhy představuje zástup molekul vážící ß-katenin a TCF/LEF (viz výše). Některé jsou inhibitory díky schopnosti exportovat ß-katenin z jádra (např. Chibby a ICAT). Zásadně pak výsledný účinek Wnt dráhy ovlivňují konformační změny DNA, aktuálně exprimované geny a aktivita dalších signalizačních drah (5–8).
Škála přepisovaných genů tedy závisí na kontextu, ve kterém dráha probíhá. Ač jich jsou stovky, jen málo z nich je spouštěno univerzálně (např. axin2) (6). Pro kolorektální karcinogenezi jsou významné mimo jiné některé komponenty kanonické dráhy, jejichž ovlivnění lze chápat jako součást vlastní regulace (elevace DKK1, Axin2 a dnTCF/LEF a potlačení exprese LRP5/6 či některých FZD) (5, 6, 41, 44, 45). Dalšími jsou regulátory buněčného cyklu a proliferace (c-Myc a CyclinD1) a inhibitory apoptózy (Survivin) (5, 6, 46–48). Ještě jiné souvisí s migrací buněk a jejich vzájemným vztahem. Patří mezi ně enzym degradující mezibuněčné struktury matrix metalloproteináza-7, angiogenní růstový faktor endothelin-1 či skupina membránových receptorů a jejich ligandů EphB/EphrinB bránící volnému vmezeřování buněk. Buňka nesoucí ligand EphrinB inhibuje pohyb buňky s receptorem EphB – kanonická dráha snižuje expresi ligandu a zvyšuje expresi receptoru) (49–51).
Uvedená ukázka cílových genů Wnt dráhy je ve shodě s její primární úlohou při vývoji organismu i celoživotní obnově některých tkání (buněčná proliferace, diferenciace, migrace atd.), zároveň ale naznačuje její silný onkogenní potenciál a potřebu přesných regulací.
Kanonická Wnt dráha a kolorektální karcinogeneze
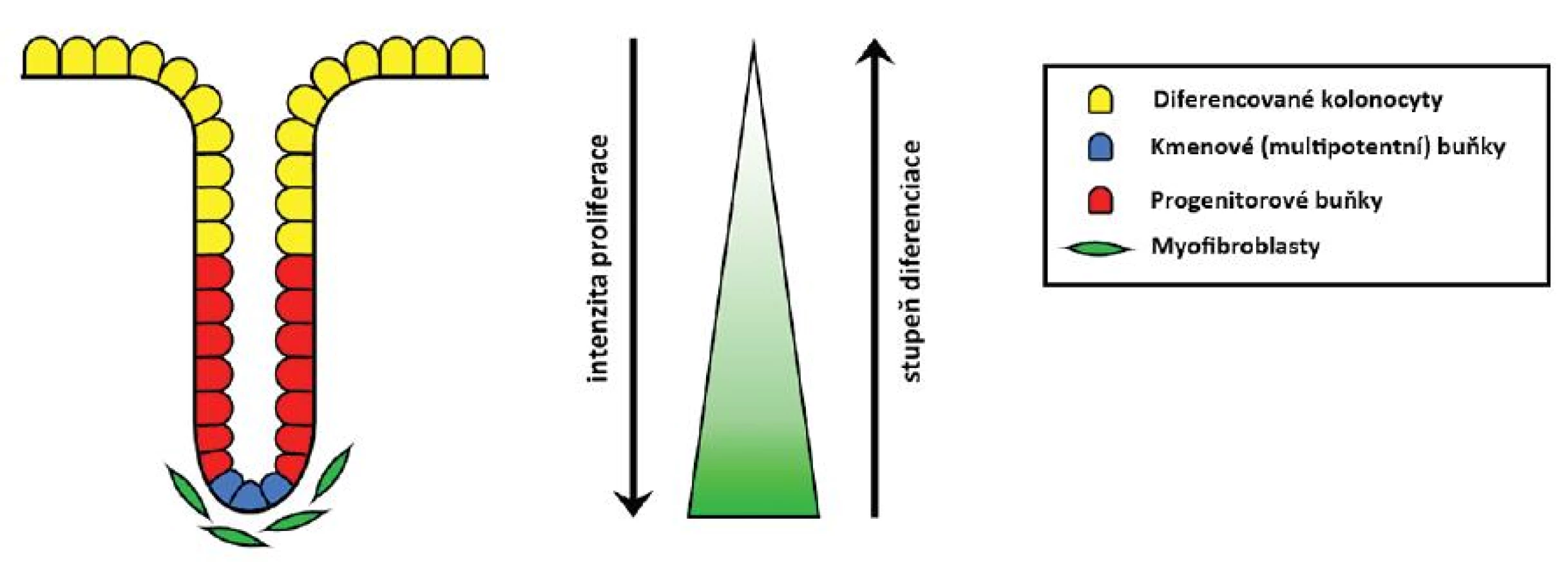
Epitel sliznice tlustého střeva a konečníku se obnovuje v pravidelných zhruba 3–6denních cyklech. To je možné díky multipotentním buňkám sídlícím na spodině slizničních krypt (též nazývaným tkáňové kmenové buňky). Ty zde sídlí celý život a mají schopnost neomezeného počtu dělení. Dávají vzniknout dceřiným progenitorovým buňkám, jež cestují apikálně, dále se dělí a diferencují ve zralé buňky sliznice – kolonocyty s absorpčními funkcemi, hlen secernující pohárkové buňky či buňky difuzního neuroendokrinního systému produkující různé hormonální působky. Zralé buňky pokrývají zhruba horní třetinu krypt. Cestují dále směrem ke střevnímu lumen, kde podstupují apoptózu, odlučují se a jsou nahrazovány novými (25, 38).
Celoživotní přežívání, nediferencovaný charakter, schopnost neomezeného počtu dělení i produkce různých typů dceřiných buněk není autonomní vlastností kmenových buněk. Do značné míry ji určuje Wnt signalizace, která je signalizací mezibuněčnou. Zdrojem Wnt glykolipoproteinů jsou myofibroblasty sídlící subepiteliálně na spodině krypt. Ony jsou nezbytné pro zachování místní zásoby kmenových buněk, a tím i celoživotní obměnu epitelu. Díky nim je aktivita dráhy vázána právě na tuto oblast. Směrem apikálním klesá, čímž je umožněn proces diferenciace a nakonec i apoptózy, ač samotné tyto pochody jsou řízeny ještě dalšími mechanismy (25, 52–54) (obr. 3).
O silném onkogenním potenciálu Wnt dráhy jsme se již zmínili a aktuální znalosti potvrzují její významnou úlohu v kolorektální karcinogenezi (4, 11).
Podle etiologie můžeme CRC dělit na hereditární (v užším smyslu při hereditárních syndromech – adenomatózní polypóze tračníku, Lynchově syndromu a dalších vzácnějších stavech – cca 10 %), na asociované s idiopatickými střevními záněty (cca 2–3%) a na sporadické (cca 85 %) (55–57). Při adenomatózní polypóze má postižený již vrozeně defekt jedné alely genu pro APC a při poruše druhé je zasažena funkce ß-katenin degradačního komplexu a trvale aktivována Wnt dráha (38, 57). U Lynchova syndromu (hereditary nonpolyposis colorectal cancer, HNPCC) jsou přítomny defekty reparačního aparátu DNA buňky (produkty tzv. mismatch repair genů) vedoucí ke kumulaci chyb genomu včetně chyb onkogenních (57–59). U idiopatických střevních zánětů souvisí riziko s délkou trvání a mírou aktivity onemocnění, kdy je DNA kolonocytů dlouhodobě poškozována noxami probíhajícího zánětu (např. oxidačním stresem při produkci kyslíkových radikálů) (56, 60–62). U sporadických CRC je přisuzována kombinaci vnějších rizikových faktorů (strava s nedostatkem vlákniny a nadbytkem živočišných tuků, obezita, kouření, stres atd.) a genetických dispozic, které však v současnosti nejsme schopni konkretizovat (63).
Histologický obraz karcinogeneze sporadického CRC zahrnuje většinou následující změny kolorektální sliznice. Prvním detekovatelným stadiem nádorových změn jsou tzv. aberrant crypt foci. Jde o izolované postižení pouze několika slizničních krypt, které může být charakteru buněčných atypií či atypické architektoniky krypt (či obou současně). Jsou následovány rozvojem klasického adenomu, většinou polypoidního, s narůstajícími dysplastickými změnami. Dalším stadiem je tzv. karcinom in situ, který již má semimaligní chování. Nepřerůstá sice bazální membránu epitelu, ale již infiltruje zdravé epiteliální buňky. Tzv. intramukózní karcinom je první stadium invazivního karcinomu. Již přechází přes bazální membránu do lamina propria sliznice, ale ponechává intaktní slizniční svalovou vrstvu. Má velmi nízké riziko generalizace (snad pro minimální množství lymfatických cév a krevních kapilár v lamina propria) a pro své chování je někdy klinicky označován jako „neinvazivní karcinom“ či dokonce „adenom s těžkou fokální atypií“. Nádor pronikající do slizniční svaloviny je již pokročilejší formou CRC, který nejen že prorůstá dále do hloubky střevní stěny i okolí, ale stoupá i riziko jeho lymfatického či hematogenního šíření. Metastazující karcinom představuje nejpokročilejší, generalizované stadium onemocnění (3, 25, 64).
Nadměrná aktivita Wnt dráhy je popisována již v adenomech (80–90 %) a obdobnou frekvenci (90 %) nacházíme i v CRC. Je spojována s iniciací celé karcinogeneze, avšak sama k rozvoji maligního stadia nestačí a musí následovat poruchy dalších buněčných regulací (3, 4, 57).
Také hereditární formy CRC jsou spojeny s tvorbou polypů (stovky až tisíce u adenomatózní polypózy, jednotlivé u HNPCC), které následně malignizují. Nadměrná aktivita Wnt dráhy stojí na počátku prakticky všech CRC při adenomatózní polypóze, u HNPCC o něco méně, cca v 65–90 % (38, 57, 65, 66). U idiopatických střevních zánětů (ulcerózní kolitidy a nověji potvrzeno i u Crohnovy choroby) dochází při dlouhodobém postižení tračníku zánětlivým procesem k dysplastickým změnám epitelu, na jejichž podkladě může dojít k nádorové přeměně. Postižení Wnt dráhy rovněž nacházíme, ale nemusí být spojeno s iniciací karcinogeneze, nýbrž s její progresí (61, 62, 67–69).
Téměř všechny CRC vykazují zvýšenou afinitu ke vzniku genetických chyb, tzv. genomovou nestabilitu. Dle cytogenetických metod, které umožňují mikroskopicky hodnotit chromozomové změny, se dělí na nestabilitu chromozomovou (CIN) a mikrosatelitní (MSI). První je spojena s hrubším postižením DNA (translokace či zlomy chromozomů, jejich ztráty, zmnožení apod.). V případě druhém jsou postižené tzv. mikrosatelitní sekvence, což jsou fragilnější a častěji poškozované úseky DNA, se kterými je však buňka schopna se za normálních okolností vypořádat. MSI působí změny postihující jednotlivé geny (jejich zkrácení, zmnožení, fúze atd.). CIN a MSI se v CRC prakticky společně nevyskytují (57–59, 61, 64–71).
Mechanismy, které způsobují genovou nestabilitu, jsou známy jen částečně. Téměř všechny HNPCC vykazují MSI, což souvisí s postižením genů reparačního aparátu DNA. Jejich poruchu nacházíme i u většiny sporadických CRC se známkami MSI (jich je cca 15 %). Většina sporadických CRC (85 %) je ale spojena s CIN, přičemž tyto mívají defekt proteinu APC. APC se účastní řady dějů souvisejících s cytoskeletem a jako součást dělícího vřeténka participuje i na segregaci chromozomů v mitóze. Příčinou CIN je nejspíše narušení této jeho funkce (38, 64). Aby se však mohly hrubé chromozomální poruchy udržet, musí být provázeny poruchou kontrolních mechanismů, které normálně v jejich přítomnosti spouští apoptózu (taková postižení nacházíme pravidelně v maligním stadiu onemocnění). Zajímavé však je, že N-konec proteinu APC (alespoň v jedné alele genu pro APC) je zachovaný v každém CRC a je zjevně pro přežití nádorové buňky nepostradatelný. To jistě souvisí s některou z řady funkcí, které tato molekula zastává. CIN a MSI odráží dvě rozdílné cesty kolorektální karcinogeneze (57–59, 61, 64–71).
Aberantní aktivace Wnt dráhy je dána především poruchou (mutací, delecí, útlumem genové exprese) tří jejích komponent – APC, ß-kateninu a Axinu. Defekty APC a Axinu narušují vznik a funkci ß-katenin degradačního komplexu, ty postihující ß-katenin jej činí odolným vůči degradaci. Důsledkem je vždy jeho stabilizace a nadměrná exprese genů Wnt dráhy. U sporadického CRC nacházíme postižení APC v cca 80–85 %, ß-kateninu do 10 % a Axinu vzácně (72–75). V podskupině nádorů s MSI (HNPCC i sporadických) je častý defekt ß-kateninu (cca 45 %) a Axinu (cca 20 %), méně APC (cca 20 %). Přitom postižení APC bývá mírnější, v 50 % doprovází defekt Axinu a není spojené s CIN (11, 57, 65, 66, 70).
Mimo klasická postižení Wnt dráhy (APC, ß-kateninu a Axinu) nacházíme v CRC změny i dalších jejích komponent. Patří k nim útlum genové exprese (tzv. silencing) jejích sekrečních inhibitorů mechanismem hypermetylace promotorů jejich genů (to brání jejich transkripci). Nachází se již v adenomech (sFRP1-2 cca 90 %, sFRP4-5 cca 15–30 %, WIF1 cca 75 %) a přetrvává i v karcinomech (76–78). Uvedená frekvence platí pro sFRP dokonce již ve stadiu aberrant crypt foci, které jsou definovány buď atypiemi architektoniky krypt (např. jejich zdvojováním), nebo buněčnými dysplaziemi (či kombinací obou). První typ s absencí dysplastických změn má zřetelně menší riziko maligní progrese a nachází se i fyziologicky (např. při hojení sliznice). Dysplastické změny bývají spojeny s klasickým postižením Wnt dráhy (APC, ß-katenin a Axin), které v prvním případě nejsou (mohou být ale jiné, např. K ras – viz dále) (25, 76, 79).
Nakolik změny exprese sFRP a WIF1 spojené s hypermetylací přispívají k rozvoji CRC není jasné. Je prokázáno, že zvyšují aktivitu Wnt dráhy i v přítomnosti klasických mutací (76). Někdy jsou přítomné i ve tkáni bez histologicky prokazatelných nádorových změn (77), často se vyskytují synchronně a jsou do určité míry reverzibilní (76). Stejné změny postihující geny komponent Wnt dráhy (sekreční inhibitory, ale i APC) často nacházíme v CRC asociovaných s idiopatickými střevními záněty a souvisí s délkou trvání a intenzitou střevních zánětů (67). Nadbytek živočišné potravy, který je rizikovým faktorem rozvoje CRC (63), rovněž působí útlum exprese sekrečních inhibitorů (je prokázáno pro WIF1), i když je nutné dodat, že mechanismus není nutně identický (80). Přemíra takové stravy poškozuje epitel kolon a vyžaduje jeho rychlejší regeneraci. Útlum WIF1 vede ke zvýšení aktivity Wnt dráhy a spolu s tím proliferace prekurzorových buněk (80). Jedním z důsledků může být vyšší riziko vzniku genetických chyb. Význam hypermetylačních změn a potlačení exprese těchto genů pro kolorektální karcinogenezi, stejně jako eventuální význam dietních faktorů pro tento proces nejsou ale dosud objasněny.
Tyto změny postihují i gen pro DKK1, sekreční inhibitor specifický pro kanonickou dráhu. Oproti předchozím však až v pokročilých stadiích CRC (cca 25 % karcinomů infiltrující okolí střeva či zakládající metastázy, v časnějších stadiích nebývá) (45).
Až v 50 % MSI CRC a velmi vzácně v ostatních, jsou pozorovány mutace TCF4 (člena TCF/LEF transkripčních faktorů). Pravidelně vedou k postižení části molekuly vážící inhibitory genové transkripce, a mohou tedy změnit jeho funkci a autonomně aktivovat část kanonické genové exprese (66, 81–84).
Z popisu Wnt dráhy vyplývá zásadní význam receptorů LRP5/6 a cytoplazmatického proteinu Dvl pro přenos signalizace. Dosud však v CRC nebylo nalezeno žádné primární postižení jejich genů (11). Jsou sice doklady o tom, že hladina molekuly Dvl2 je v CRC zvýšena, a může tak přispívat ke karcinogenezi. Jedná se ale nejspíše o změnu druhotnou v důsledku aktivity některých buněčných signalizací, snad i samotné Wnt dráhy, která její genovou transkripci zvyšuje (85).
Kromě alterace genomu vlastních neoplastických buněk se ukazuje důležitá úloha buněk v jejich okolí tvořících nádorové stroma. Wnt je signalizací mezibuněčnou a fyziologicky začíná v myofibroblastech spodiny slizničních krypt. V CRC vykazují jednotlivé buňky odlišnou míru aktivity Wnt dráhy, přičemž vysoká aktivita je spojena s nízkou diferenciací, vyšší proliferací a agresivnějším chováním (je v místech invaze nádoru do okolí), nízká naopak s diferencovaným charakterem a výrazně sníženou frekvencí dělení. Intenzita Wnt signalizace a s ní spojené biologické chování jednotlivých buněk není ale jejich autonomní vlastností. Je prokázáno, že nádorové stroma CRC produkuje molekuly, které jsou schopny tuto intenzitu měnit. I buňky diferencovanější získávají v případě jejího posílení vlastnosti oněch agresivnějších a naopak. Nádorové stroma tedy dokáže měnit chování samotných nádorových buněk. Oněmi molekulami nemusí být nutně Wnt glykolipoproteiny, stimulační efekt (vedoucí k malignějšímu chování) byl prokázán u hepatocytárního růstového faktoru (HGF) (52–54, 86).
Za geny aktivované Wnt dráhou a kódující proteiny zodpovědné za její onkogenní účinky připomeňme proliferačně působící c-Myc a Cyclin D1, antiapoptotický protein survivin, vaskulární růstový faktor endothelin-1 či matrix metalloproteinázu-7 degradující mezibuněčné adhezivní molekuly (viz výše).
Sama deregulace Wnt dráhy ke vzniku maligního onemocnění nestačí. Účastní se poruchy minimálně tři dalších signalizací – karcinogenezi inhibující dráha transforming growth factor beta (TGFß; účastní se receptory TGFßR1 a TGFßR2 a transkripční faktory SMAD2 a SMAD4), proliferaci stimulující signalizace epidermálního růstového faktoru (EGF; s receptorem EGFR a kaskádou vzájemně se aktivujících kinas K-ras, B-raf a MAPK) a další proliferační dráha fosfatidylinositol-3-kinázy (PI3K; postižena bývá PI3K či její inhibitor, fosfatáza PTEN). Mimoto nacházíme poruchy mechanismů apoptózy (proteiny p53 a BAX), změny telomerázové aktivity a další. CRC s CIN a MSI se liší frekvencí postižení jednotlivých genů, což částečně odráží jejich rozdílnou citlivost k oběma mechanismům genomové nestability. V obou případech jsou ale postiženy obdobné buněčné regulace a signalizace a patologická aktivace Wnt dráhy hraje mezi nimi zásadní roli, většinou již v iniciaci karcinogeneze (3, 64–66, 68, 69, 87–92).
Kanonická Wnt dráha a terapie kolorektálního karcinomu
Dosud je kurativní léčbou CRC prakticky pouze jeho resekce a cílem je proto jeho včasná detekce, což se v České republice daří jen asi v polovině případů. Přesto jsou poslední léta ve znamení výrazného pokroku. Přesnější staging onemocnění a nové chirurgické metody umožňují účinnější (mnohdy kurativní) odstranění jaterních metastáz a pětileté přežití takto operovaných pacientů dosahuje cca 50 %. Zároveň do klinické praxe vstoupila nová účinnější léčiva včetně biologické léčby cíleně zasahující konkrétní buněčné signalizace. K 1. únoru 2012 jsou v České republice schváleny bevacizumab, monoklonální protilátka inhibující angiogenní růstový faktor VEGF, a tím novotvorbu cév vyživujících nádor. Dále pak cetuximab a panitumumab, monoklonální protilátky inhibující receptor pro epidermální růstový faktor EGFR. Tato léčba sama o sobě není kurativní, ale prodlužuje přežití nemocných včetně asymptomatického stadia v řádu měsíců až let. Zároveň i původně neresekabilní jaterní metastázy mohou být díky této léčbě zcela odstraněny (2, 93, 94).
Význam Wnt dráhy u CRC je nepochybný. Experimenty na buněčných liniích lidských kolorektálních karcinomů ukazují, že její inhibice má schopnost zastavit růst a způsobit apoptózu nádorových buněk i v přítomnosti mutací dalších signalizačních drah (52, 76, 95–97). Snaha o vyvinutí léků, které by přímo zasahovaly Wnt signalizaci, však navzdory značnému úsilí zůstává doposud úspěšná jen na úrovni laboratorních experimentů (92, 98).
Možností je přitom teoreticky řada: od ovlivnění tvorby a transportu Wnt, jejich vazby na membránové receptory či zásah cytoplazmatických nebo jaderných komponent dráhy. Čím blíže by byl takový cíl interakci ß-kateninu a TCF/LEF, tím specifičtější by byla inhibice. Narušení časnějších událostí signalizace vede k vyššímu riziku postižení i jiných buněčných pochodů. Zásah konkrétního genového produktu aktivované dráhy zase postihuje jen část jejích účinků. Nemožnost selektivně ovlivnit pouze nádorovou tkáň je jiným omezením, neboť Wnt dráha plní v organismu řadu zásadních fyziologických funkcí.
Látky přímo bránící vazbě ß-kateninu a TCF/LEF byly identifikovány, ale i když skutečně inhibují Wnt signalizaci i růst buněčných linií kolorektálního karcinomu, jejich použití v praxi je zatím nereálné. Vazebná místa ß-kateninu s TCF/LEF, APC i cytoskeletárními molekulami se totiž překrývají a jejich narušení má závažné důsledky, např. defekty mezibuněčné adheze (95, 99). Podobně inhibice proteinu Dvl, který je důležitým pro transmembránový přenos Wnt signálu, inhibuje spolu s ním i další buněčné pochody, z nichž některé samy o sobě Wnt dráhu antagonizují (17, 100). Protilátky proti Wnt (prokázáno např. pro Wnt1) sice inhibují růst kolorektálních nádorových buněčných linií, nejsou však schopny zasáhnout izolovaně nádorovou tkáň (97). Neselektivní působení takových léčiv, ať už ve smyslu zásahu samotné Wnt dráhy, či pouze maligních buněk, tedy představuje nejvážnější překážku pro jejich užití v praxi.
Přesto není situace beznadějná. Genová transkripce řízená ß-kateninem je zesilována současnou aktivitou EGFR signalizace (zvyšuje přestup ß-kateninu do jádra) a tedy úspěch jejích inhibitorů v terapii CRC (viz výše) lze částečně přičíst i tomuto jejich účinku (101). Obdobně působí i HGF, růstový faktor produkovaný v CRC okolní nenádorovou tkání (viz výše). V současné době probíhají klinické studie s několika farmaky, která inhibují tuto signalizaci (102).
Velmi diskutovanými léčivy z hlediska kolorektální karcinogeneze jsou nesteroidní protizánětlivé léky (NSAID). Ty inhibují enzym cyklooxygenázu (COX), a tím tvorbu prostaglandinů, látek řídících v místě svého vzniku množství biologických pochodů včetně imunitních a zánětlivých. COX se vyskytuje ve dvou izoformách, kdy COX1 plní řadu fyziologických úloh, zatímco výskyt COX2 je spojen převážně s patologickými stavy. Nadměrná exprese COX2 je přítomna v kolorektálních adenomech (40 %) i karcinomech (85 %) a vede k inhibici apoptózy, novotvorbě cév a obecně k progresi onemocnění. Rovněž prostaglandiny produkované COX zvyšují přestup ß kateninu do jádra a aktivitu Wnt signalizace (69). Řada experimentů ukazuje, že NSAID jsou skutečně schopny inhibovat růst a progresi kolorektálních adenomů, a to alespoň z části inhibicí Wnt dráhy. Nejvíce zkoumány byly kyselina acetylsalicylová (ASA), sulindac, indomethacin a selektivní COX2 inhibitor celecoxib (98).
V USA byl dokonce od roku 1999 celecoxib schválen pro léčbu premaligního stadia adenomatózní polypózy tračníku (k tzv. chemoprevenci). To bylo následováno klinickou prospektivní studií zkoumající vliv celecoxibu na prevenci rozvoje sporadické formy CRC. Ta však musela být předčasně ukončena, neboť, ač byl pozorován příznivý efekt na rozvoj adenomů, 2–3krát (v závislosti na dávce) stoupl výskyt cévních mozkových příhod a srdečních infarktů (103). Celecoxib (a nejspíše ani ostatní selektivní COX2 inhibitory) se tedy k dlouhodobému užívání a prevenci CRC nehodí.
V roce 2011 byly publikovány výsledky metaanalýzy osmi rozsáhlých a dlouhodobých prospektivních klinických studií (celkem s 25 570 účastníky) sledujících primárně účinnost ASA v prevenci kardiovaskulárních chorob. V nich bylo pozorováno snížení výskytu CRC v průměru o 7 % při jejím dlouhodobém užívání, zároveň nesignifikantně nižší celkový počet úmrtí bez ohledu na příčinu a absenci vyššího výskytu závažných gastrointestinálních krvácení (104). Cíleně však, na rozdíl od předchozích, vztah ASA a CRC zkoumala americká studie z let 1992–2004 (s 39 876 účastníky), která ale žádný protektivní účinek neprokázala (při relativně nízké dávce 100 mg ASA obden) (105). V současné době není ASA jako prevence CRC doporučena, ale nové studie (s vyšším dávkováním) probíhají (106).
Známým protektivním faktorem rozvoje CRC je dostatečný přísun vlákniny v potravě. Experimenty na buněčných liniích lidského CRC s butyrátem sodným, monokarboxylovou kyselinou vznikající ve střevě fermentací některých druhů vlákniny, ukazují ve většině případů příznivý efekt na tyto buňky ve smyslu zpomalení proliferace, podpory diferenciace a indukce apoptózy (u některých linií ale překvapivě opačný). Butyrát v nich přitom Wnt dráhu stimuluje a uvedené účinky jsou přímo úměrné stupni této stimulace. Z toho je zřejmé, že jeho působení je komplexní a vyžaduje další studie (107–109).
Ač se přímý terapeutický zásah Wnt dráhy zatím nedaří, příklady nové biologické léčby, NSAID a snad i efektu vlákniny ukazují, že úspěšné mohou být strategie ovlivňující tuto signalizaci nepřímo.
Závěr
Úloha Wnt kanonické dráhy v rozvoji CRC je nesporná a to většinou již v prvních fázích tohoto onemocnění. Uplynulých dvacet let intenzivních výzkumů přineslo mnoho poznatků o jejích komponentách, regulacích, cílových genech a funkcích. Ač stále zbývá řada nejasností, současné znalosti pomáhají chápat její klíčové aspekty a současně identifikovat potenciální místa pro terapeutické zásahy.
Klinicky použitelný způsob její přímé inhibice zatím neznáme. Důvodem je komplexnost signalizačních systémů buňky a jejich regulací, kdy ovlivnění jedné komponenty vede k obtížně předvídatelným a potenciálně velmi závažným následkům. Nadto plní Wnt dráha řadu životně důležitých funkcí a problémem (ale obecně onkologickým) je selektivní zásah nádorové tkáně.
Příklady starých léčiv (NSAID), dietních faktorů (vlákniny) i moderní léčby však ukazují, že nejde o nereálný cíl. Ovlivnění Wnt dráhy nepřímo prostřednictvím signalizací, které ji modulují, je možná nejreálnější cestou v tomto úsilí.
Zkratky
- APC – adenomatous polyposis coli
- ASA – acetylsalicylová kyselina
- CK1 – kasein kináza 1
- CIN – chromozomová nestabilita
- COX1,2 – cyklooxygenáza 1, 2
- CRC – kolorektální karcinom
- DKK1 – Dickkopf 1
- dnTCF/LEF – dominantly negative T-cell factors/lymphoid enhancer factor
- Dvl – Dishevelled
- EGF – epidermální růstový faktor
- EGFR – receptor epidermálního růstového faktoru
- FZD – Frizzled receptors
- GSK3ß – kináza glykogensyntázy 3ß
- HDL – high-density lipoprotein
- HGF – hepatocytární růstový faktor
- HNPCC – hereditary nonpolyposis colorectal cancer
- HSPG – proteoglykan heparansulfát
- LEF1 – lymphoid enhancer factor 1
- LRP5,6 – low-density lipoprotein receptor-related peptid 5, 6
- MSI – mikrosatelitní nestabilita
- NSAID – nesteroidní protizánětlivé léky
- PI3K – fosfatidylinositol-3-kináza
- sFRP1–5 – solubilní Frizzled-related protein 1–5
- TCF1,3,4 – T-cell factor 1, 2, 3
- TGFß – transforming growth factor ß
- TGFßR1,2 – receptor typu 1, 2 pro transforming growth factor ß
- WIF1 – Wnt inhibitory factor 1
- Wls – Wntless
- Wnt – Wnt glykolipoproteiny
Za pomoc při přípravě děkuji svým školitelům doktorandského studia doc. MUDr. Milanu Kmentovi, CSc. a prof. RNDr. Jiřímu Páchovi, DrSc., dále MUDr. Jiřímu Švecovi, PhD., a za vytvoření obrazové přílohy Bc. Petru Moravcovi.
Článek vznikl za finanční podpory GA UK č. 70310.
ADRESA PRO KORESPONDENCI:
MUDr. Martin Moravec
II. interní klinika 3. LF UK a FNKV
Šrobárova 50, 100 34 Praha 10
e-mail: martin.j.moravec@seznam.cz
Sources
1. International variations and trends AND Colon and rectum. In: World Cancer Research Fund, American Institute for Cancer Research. Food, nutrition and physical activity, and the prevention of cancer: a global perpective. Washington DC: AICR 2007; 20, 280–288.
2. Dušek L, et al. Populační odhady počtu nemocných s kolorektálním karcinomem v ČR – jeden z nástrojů hodnocení včasné diagnostiky časných stadií a rekurence onemocnění. Farmakoterapie 2009; 5 : 11–20.
3. Fearon ER, Vogelstein B. A genetic model for colorectal tumorigenesis. Cell 1990; 61 : 759–767.
4. Najdi R, et al. Wnt signaling and colon carcinogenesis: beyond APC. J Carcinog 2011; 10 : 5.
5. Logan CY, Nusse R. The Wnt signaling pathway in development and disease. Annu Rev Cell Dev Biol 2004; 20 : 781–810.
6. MacDonald BT, et al. Wnt/ß-catenin signaling: components, mechanisms, and diseases. Dev Cell 2009; 17 : 9–26.
7. van Amerongen R, Nusse R. Towards an integrated view of Wnt signaling in development. Development 2009; 136 : 3205–3214.
8. Verheyen EM, Gottardi CJ. Regulation of Wnt/ß-catenin signaling by protein kinases. Dev Dyn 2010; 239 : 34–44.
9. Doucas H, et al. Changes in the Wnt signalling pathway in gastrointestinal cancers and their prognostic significance. Eur J Cancer 2005; 41 : 365–379.
10. Nusse R. Cancer. Converging on beta-catenin in Wilms tumor. Science 2007; 316 : 988–989.
11. Polakis P. The many ways of Wnt in cancer. Curr Opin Genet Dev 2007; 17 : 45–51.
12. Gallagher JC, Sai AJ. Molecular biology of bone remodeling: implications for new therapeutic targets for osteoporosis. Maturitas 2010; 65 : 301–307.
13. Vlad A, et al. The first five years of the Wnt targetome. Cell Signal 2008; 20 : 795–802.
14. van Amerongen R, et al. Alternative Wnt signaling is initiated by distinct receptors. Sci Signal 2008; 1: re9.
15. Kikuchi A, et al. Multiplicity of interactions of Wnt proteins and their receptors. Cell Signal 2007; 19 : 659–671.
16. Angers S, Moon RT. Proximal events in Wnt signal transduction. Nat Rev Mol Cell Biol 2009; 10 : 468–477.
17. Gao C, Chen YG. Dishevelled: The hub of Wnt signaling. Cell Signal 2010; 22 : 717–727.
18. Caldwell GM, et al. Reorganisation of Wnt-response pathways in colorectal tumorigenesis. Br J Cancer 2008; 98 : 1437–1442.
19. Toualbi K, et al. Physical and functional cooperation between AP-1 and ß-catenin for the regulation of TCF-dependent genes. Oncogene 2007; 26 : 3492–3502.
20. Gordon MD, Nusse R. Wnt signaling: multiple pathways, multiple receptors and multiple transcription factors. J Biol Chem 2006; 281 : 22429–22433.
21. Kohn AD, Moon RT. Wnt and calcium signaling: ß-catenin-independent pathways. Cell Calcium 2005; 38 : 439–446.
22. Veeman MT, et al. A second canon: functions and mechanisms of ß-catenin-independent Wnt signaling. Dev Cell 2003; 5 : 367–377.
23. Seifert JR, Mlodzik M. Frizzled/PCP signalling: a conserved mechanism regulating cell polarity and directed motility. Nat Rev Genet 2007; 8 : 126–138.
24. Semenov MV, et al. SnapShot: noncanonical Wnt signaling pathways. Cell 2007; 131 : 1378.
25. van den Brink GR, Offerhaus GJ. The morphogenetic code and colon cancer development. Cancer Cell 2007; 11 : 109–117.
26. Steinhauer J, Treisman JE. Lipid-modified morphogens: functions of fats. Curr Opin Genet Dev 2009; 19 : 308–314.
27. van den Heuvel M, et al. Mutations in the segment polarity genes wingless and porcupine impair secretion of the wingless protein. EMBO J 1993; 12 : 5293–5302.
28. Leronowicz MJ, Korswagen HC. Sailing with the Wnt: charting the Wnt processing and secretion route. Exp Cell Res 2009; 315 : 2683–2689.
29. Port F, Basler K. Wnt trafficking: new insights into Wnt maturation, secretion and spreading. Traffic 2010; 11 : 1265–1271.
30. Mikels AJ, Nusse R. Wnts as ligands: processing, secretion and reception. Oncogene 2006; 25 : 7461–7468.
31. Takei Y, et al. Three Drosophila EXT genes shape morphogen gradients through synthesis of heparan sulfate proteoglycans. Development 2004; 131 : 73–82.
32. Korkut C, et al. Trans-synaptic transmission of vesicular Wnt signals through Evi/Wntless. Cell 209; 139 : 393–404.
33. Panáková D, et al. Lipoprotein particles are required for Hedgehog and Wingless signalling. Nature 2005; 435 : 58–65.
34. Neumann S, et al. Mammalian Wnt3a is released on lipoprotein particles. Traffic 2009; 10 : 334–343.
35. Bovolenta P, et al. Beyond Wnt inhibition: new functions of secreted Frizzled-related proteins in development and disease. J Cell Sci 2008; 121 : 737–746.
36. Cadigan KM, Liu YI. Wnt signaling: complexity at the surface. J Cell Sci 2006; 119 : 395–402.
37. Nelson WJ, Nusse R. Convergence of Wnt, beta-catenin, and cadherin pathways. Science 2004; 303 : 1483–1487.
38. Schneikert J, Behrens J. The canonical Wnt signalling pathway and its APC partner in colon cancer development. Gut 2007; 56 : 417–425.
39. Lee E, et al. The role of APC and Axin derived from experimental and theoretical analysis of the Wnt pathway. PLoS Biol 2003; 1: E10.
40. Chia IV, Constantini F. Mouse axin and axin2/conductin proteins are functionally equivalent in vivo. Mol Cell Biol 2005; 25 : 4371–4376.
41. Lustig B, et al. Negative feedback loop of Wnt signaling through upregulation of Conductin/Axin2 in colorectal and liver tumors. Mol Cell Biol 2002; 22 : 1184–1193.
42. Mosimann C, et al. Beta-catenin hits chromatin: regulation of Wnt target gene activation. Nat Rev Mol Cell Biol 2009; 10 : 276–286.
43. Arce L, et al. Diversity of LEF/TCF action in development and disease. Oncogene 2006; 25 : 7492–7504.
44. Roose J, et al. Synergy between tumor suppressor APC and the beta-catenin-TCF4 target TCF1. Science 1999; 285 : 1923–1926.
45. Aguilera O, et al. Epigenetic inactivation of the Wnt antagonist DICKKOPF-1 (DKK-1) gene in human colorectal cancer. Oncogene 2006; 25 : 4116–4121.
46. Tetsu O, McCormick F. Beta-catenin regulates expression of cyclin D1 in colon carcinoma cells. Nature 1999; 398 : 422–426.
47. Arber N, et al. Increased expression of Cyclin D1 is an early event in multistage colorectal carcinogenesis. Gastroenterology 1996; 110 : 669–674.
48. Zhang T, et al. Evidence that APC regulates survivin expression: possible mechanism contributing to the stem cell origin of colon cancer. Cancer Res 2001; 61 : 8664–8667.
49. Brabletz T, et al. Beta-catenin regulates the expression of the matrix metalloproteinase-7 in human colorectal cancer. Am J Pathol 155(1999): 1033–1038.
50. Kim TH, et al. Beta-catenin activates the growth factor endothelin-1 in colon cancer cells. Oncogene 2005; 24 : 597–604.
51. Batlle E, et al. Beta-catenin and TCF mediate cell positioning in the intestinal epithelium by controlling the expression of EphB/ephrinB. Cell 2002; 111 : 251–263.
52. van de Wetering M, et al. The ß-catenin/TFC4 complex imposes a crypt progenitor phenotype on colorectal cancer cells. Cell 2002; 111 : 241–250.
53. Vermeulen L, et al. Wnt activity defines colon cancer stem cells and is regulated by the microenvironment. Nat Cell Biol 2010; 12 : 468–476.
54. de Sousa EM, et al. Targeting Wnt signaling in colon cancer stem cells. Clin Cancer Res 2011; 17 : 647–653.
55. Munkholm P. Review article: the incidence and prevalence of colorectal cancer in inflammatory bowel disease. Aliment Pharmacol Ther 2001; 18 Suppl 2 : 1–5.
56. Eaden JA, et al. The risk of colorectal cancer in ulcerative colitis: a meta-analysis. Gut 2001; 48 : 526–535.
57. Kinzler KW, Vogelstein B. Lessons from hereditary colorectal cancer. Cell 1996; 87 : 159–170.
58. Heinen CD, et al. DNA repair and tumorigenesis: lessons from hereditary cancer syndromes. Cancer Biol Ther 2002; 1 : 477–485.
59. Komarova NL, et al. Dynamics of genetic instability in sporadic and familial colorectal cancer. Cancer Biol Ther 2002; 1 : 685–692.
60. Choi PM, Zelig MP. Similarity of colorectal cancer in Crohnęs disease and ulcerative colitis: implications for carcinogenesis and prevention. Gut 1994; 35 : 950–954.
61. O’Sullivan JN, et al. Chromosomal instability in ulcerative colitis is related to telomere shortening. Nat Genet 2002; 32 : 280–284.
62. Harpaz N, Polydorides AD. Colorectal dysplasia in chronic inflammatory bowel disease: pathology, clinical implications, and pathogenesis. Arch Pathol Lab Med 2010; 134 : 876–895.
63. Watson AJ, Collins PD. Colon cancer: a civilization disorder. Dig Dis 2011; 29 : 222–228.
64. Fodde R, et al. APC, signal transduction and genetic instability in colorectal cancer. Nat Rev Cancer 2001; 1 : 55–67.
65. Grady WM. Genomic instability and colon cancer. Cancer Metastasis Rev 2004; 23 : 11–27.
66. Thorstensen L, Lind GE, et al. Genetic and epigenetic changes of components affecting the Wnt pathway in colorectal carcinomas stratisfied by microsatellite instability. Neoplasia 2005; 7 : 99–108.
67. Dhir M, et al. Epigenetic regulation of WNT signaling pathways genes in inflammatory bowel disease (IBD) associated neoplasia. J Gastrointest Surg 2008; 12 : 1745–1753.
68. Rudolph KL, et al. Telomere dysfunction and DNA damage checkpoints in diseases and cancer of the gastrointestinal tract. Gastroenterology 2009; 137 : 754–762.
69. Watson AJ. Apoptosis and colorectal cancer. Gut 2004; 53 : 1701–1709.
70. Oving IM, Clevers HC. Molecular causes of colon cancer. Eur J Clin Invest 2002; 32 : 448–457.
71. Heinen CD, et al. Microsatellite instability in colorectal adenocarcinoma cell lines that have full-lenght adenomatous polyposis coli protein. Cancer Research 1995; 55 : 4797–4799.
72. Sparks AB, et al. Mutational analysis of the APC/ß-catenin/Tcf pathway in colorectal cancer. Cancer Res 1998; 58 : 1130–1134.
73. Miyaki M, et al. Frequent mutation of ß-catenin and APC genes in primary colorectal tumors from patients with hereditary nonpolyposis colorectal cancer. Cancer Res 1999; 59 : 4506–4509.
74. Lammi L, et al. Mutations in AXIN2 cause familial tooth agenesis and predispose to colorectal cancer. Am J Hum Genet 2004; 74 : 1043–1050.
75. Bienz M, Clevers H. Linking colorectal cancer to Wnt signaling. Cell 2000; 103 : 311–320.
76. Suzuki H, et al. Epigenetic inactivation of SFRP genes allows constitutive WNT signaling in colorectal cancer. Nat Genet 2004; 36 : 417–422.
77. Caldwell GM, et al. The Wnt antagonist sFRP1 in colorectal tumorigenesis. Cancer Res 2004; 64 : 883–888.
78. Taniguchi H, et al. Frequent epigenetic inactivation of Wnt inhibitory factor-1 in human gastrointestinal cancers. Oncogene 2005; 24 : 7946–7952.
79. Jen J, et al. Molecular determinants of dysplasia in colorectal lesions. Cancer Res 1994; 54 : 5523–5526.
80. Ijssennagger N, et al. Dietary haem stimulates epithelial cell turnover by downregulating feedback inhibitors of proliferation in murine colon. Gut 2011: (Epub ahead of print).
81. Duval A, et al. Frequent frameshift mutations of the TCF-4 gene in colorectal cancers with microsatellite instability. Cancer Res 1999; 59 : 4213–4215.
82. Shimizu Y, et al. Frequent alterations in the Wnt signaling pahtway in colorectal cancer with microsatellite instability. Genes Chromosomes Cancer 2002; 33 : 73–81.
83. Waterman ML. Lymphoid enhancer factor/T cell factor expression in colorectal cancer. Cancer Metastasis Rev 2004; 23 : 41–52.
84. Duval A, et al. The human T-cell transcription factor-4 gene: structure, extensive characterization of alternative splicings, and mutational analysis in colorectal cancer cell lines. Cancer Res 2000; 60 : 3872–3879.
85. Metcalfe C, Ibrahim AE, et al. Cancer Res 2010; 70 : 6629–6638.
86. Korkaya H, Wicha MS. Cancer stem cells: nature versus nurture. Nat Cell Biol 2010; 12 : 419–421.
87. Chan TL, et al. BRAF and KRAS mutations in colorectal hyperplastic polyps and serrated adenomas. Cancer Res 2003; 63 : 4878–4881.
88. Batlle E, et al. EphB receptor activity suppresses colorectal cancer progression. Nature 2005; 435 : 1126–1130.
89. Clevers H, Batlle E. EphB/EphrinB receptors and Wnt signaling in colorectal cancer. Cancer Res 2006; 66 : 2–5.
90. Yamamoto H, et al. Somatic frameshift mutation in DNA mismatch repair and proapoptosis genes in hereditary nonpolyposis colorectal cancer. Cancer Res 1998; 58 : 997–1003.
91. Grady WM, et al. Mutation of the type II transforming growth factor–beta receptor is coincident with the transformation of human colon adenomas to malignant carcinomas. Cancer Res 1998; 58 : 3101–3104.
92. Pritchard CC, Grady WM. Colorectal cancer molecular biology moves into clinical practice. Gut 2011; 60 : 116–129.
93. Ondřichová L. Metastazující kolorektální karcinom – nic není jako dřív. Medical Tribune 2011; 7(25): B2,B4.
94. Vyzula R, et al. Zhoubný novotvar kolorekta (C18–20). Zhoubný novotvar řiti a řitního kanálu (C21). In: Vyzula R, et al. Zásady cytostatické léčby maligních onkologických onemocnění. 14. vydání. Brno: KAPCZ 2012; 52–66.
95. Barker N, Clevers H. Mining the Wnt pathway for cancer therapeutics. Nat Rev Drug Discov 2006; 5 : 997–1014.
96. Gehrke I, et al. Targeting the Wnt/beta-catenin/TCF/LEF1 axis in solid and haematologecal cancers: Multiplicity of therapeutic options. Eur J Cancer 2009; 45 : 2759–2767.
97. He B, et al. Blockade of Wnt-1 signaling induces apoptosis in human colorectal cancer cells containing downstream mutations. Oncogene 2005; 24 : 3054–3058.
98. Dihlmann S, et al. Wnt/beta-catenin-pathway as a molecular target for future anti-cancer therapeutics. Int J Cancer 2005; 113 : 515–524.
99. Lepourcelet M, et al. Small-molecule antagonists of the oncogenic TCF/beta-catenin protein complex. Cancer Cell 2004; 5 : 91–102.
100. Shan J, et al. Identification of specific inhibitor of the Dishevelled PDZ domain. Biochemistry 2005; 44 : 15495–15503.
101. Muller T, et al. Regulation of epithelial cell migration and tumor formation by beta–catenin signaling. Exp Cell Res 2002; 280 : 119–133.
102. Wang MH, et al. Potential therapeutics specific to c-MET/RON receptor thyrosin kinases for molecular targeting in cancer therapy. Acta Pharmacol Sin 2010; 31 : 1181–1188.
103. Bertagnolli MM, et al. Five year efficacy and safety analysis of the adenoma prevention with celecoxib trial. Cancer Prev Res (Phila) 2009; 2 : 310–321.
104. Rothwell PM, et al. Effect of daily aspirin on long-term risk of death due to cancer: analysis of individual patient data from randomised trials. Lancet 2011; 377 : 31–41.
105. Cook NR, et al. Low-dose aspirin in the primary prevention of cancer: the Womenęs Health Study: a randomized controlled trial. JAMA 2005; 294 : 47–55.
106. Cuzick J, et al. Aspirin and non-steroidal anti-inflammatory drugs for cancer prevention: an international consensus statement. Lancet Oncol 2009; 10 : 501–507.
107. Bordonaro M, et al. Butyrate-induced apoptotis cascade in colonic carcinoma cells: modulation of the ß-Catenin-Tcf pathway and concordance with effects of sulindac and trichostatin A but not curcumin. Cell Growth Differ 1999; 10 : 713–720.
108. Bordonaro M, et al. Cell type – and promoter-dependent modulation of the Wnt signaling pathway by sodium butyrate. Int J Cancer 2002; 97 : 42–51.
109. Lazarova DL, et al. Linear relationship between Wnt activity levels and apoptosis in colorectal carcinoma cells exposed to butyrate. Int J Cancer 2004; 110 : 523–531.
Labels
Addictology Allergology and clinical immunology Angiology Audiology Clinical biochemistry Dermatology & STDs Paediatric gastroenterology Paediatric surgery Paediatric cardiology Paediatric neurology Paediatric ENT Paediatric psychiatry Paediatric rheumatology Diabetology Pharmacy Vascular surgery Pain management Dental HygienistArticle was published in
Journal of Czech Physicians

- Advances in the Treatment of Myasthenia Gravis on the Horizon
- Possibilities of Using Metamizole in the Treatment of Acute Primary Headaches
- Metamizole at a Glance and in Practice – Effective Non-Opioid Analgesic for All Ages
- Metamizole vs. Tramadol in Postoperative Analgesia
- Spasmolytic Effect of Metamizole
Most read in this issue
- Deep calf vein thrombosis
- Acute febrile neutrophilic dermatosis – Sweet syndrome
- Eutreated and maltreated girls – childhood, partnership, maternity – a longitudinal study
- Colorectal cancer and canonical Wnt signalling pathway