
Význam DNA vyšetření mutací C282Y, H63D a S65C v HFE genu
Authors:
Monika Drastíková; Martin Beránek; Jaroslava Hegerová; Daniela Putzová
Authors‘ workplace:
Univerzita Karlova v Praze, Lékařská fakulta Hradec Králové, Ústav klinické biochemie a diagnostiky FN
Published in:
Čas. Lék. čes. 2012; 151: 428-431
Category:
Original Article
Overview
Východisko.
Hereditární hemochromatóza je poměrně časté dědičné onemocnění, pro které je charakteristické zvýšené vstřebávání železa a jeho následné ukládání do důležitých orgánů těla. Cílem práce je určit výskyt mutací C282Y, H63D a S65C ve skupině pacientů s podezřením na hereditární hemochromatózu a získané výsledky porovnat se zdravou českou populací.
Metody.
Soubor pacientů se skládal z 95 mužů a 45 žen (medián věku 55 roků, rozsah 20–83 roků). Soubor zdravých osob (kontrolní skupina) tvořilo 167 dobrovolníků (65 mužů a 102 žen, medián věku 25 roků, rozsah 18–62 roků). Genetická analýza mutací v HFE genu byla provedena pomocí metody PCR/RFLP.
Výsledky. Frekvence rizikových alel v souboru pacientů byly:
18,2 % pro mutaci C282Y; 17,5 % pro H63D a 1,8 % pro S65C. Frekvence rizikových alel v kontrolním souboru byly: 5,7 % pro mutaci C282Y; 12,3 % pro H63D a 0,6 % pro S65C.
Závěry.
Výsledky svědčí o trojnásobně vyšším výskytu mutace C282Y ve skupině pacientů s podezřením na hereditární hemochromatózu oproti kontrolní skupině (18,2 % vs. 5,7 %). Výskyt mutací H63D a S65C se v obou porovnávaných souborech statisticky nelišil.
Klíčová slova:
hemochromatóza, HFE gen, C282Y, H63D, S65C, PCR/RFLP.
ÚVOD
Hereditární (vrozená) hemochromatóza (HH) je dědičné onemocnění charakterizované zvýšenou absorpcí železa v tenkém střevě a jeho následným ukládáním do jater, myokardu, pankreatu, hypofýzy, kloubů a pokožky. Nadměrná akumulace železa v játrech má vliv na aktivaci hvězdicovitých buněk (Ito buňky, hepatic stellate cells) a jejich přeměnu v buňky podobné myofibroblastům (myofibroblast – like cells), které ve zvýšené míře produkují kolagen I., III. a IV. typu (1, 2). Hypersekrece kolagenu může vést k jaterní fibróze. Při HH jsou rovněž ve zvýšené míře Fentonovou reakcí tvořeny hydroxylové radikály, které vlivem oxidačního stresu přispívají k nekróze hepatocytů a k rozvoji hepatocelulárního karcinomu (3–5).
Prevalence HH v kavkazské populaci je přibližně 1 : 200–500 (6, 7), což ji řadí mezi nejčastější geneticky podmíněná onemocnění, ale též mezi choroby výrazně „poddiagnostikované“. Hereditární hemochromatóza se dělí do pěti základních podtypů HH1–HH5. Podtyp HH1, který představuje nejběžnější formu hemochromatózy, je podmíněn mutacemi v HFE genu. Ostatní podtypy souvisejí s genetickými polymorfismy v genech kódujících hemojuvelin, hepcidin, transferinový receptor 2, feroportin, transferin, ceruloplazmin a H řetězce feritinu, avšak jejich frekvence ani klinické projevy nejsou v porovnání s HH1 tak významné (8, 9).
HFE gen, poprvé popsaný v roce 1996, se nachází na krátkém raménku 6. chromozomu (6p21.3). Patří do rodiny hlavního histokompatibilního komplexu s vysokou homologií ke genům HLA I. třídy. Kódovaný HFE protein má 343 aminokyselin. Je tvořen třemi extracelulárními doménami α1–α3. Doména α3 se u zdravých osob nekovalentně váže s ß2-mikroglobulinem a vzniklý heterodimer je transportován na povrch buněk, kde obsazuje receptor pro transferin (10). Transferin nesoucí železo může vytěsnit HFE protein z této vazby, čímž je zprostředkován signál o dostatečné saturaci organismu železem. Uvolněný HFE protein následně moduluje expresi hepcidinu, který degradací feroportinu snižuje přenos železa z enterocytů, hepatocytů a makrofágů do krevního oběhu (11).
Nejčastější příčinou HH1 v kavkazské populaci je mutace C282Y ve 4. exonu HFE genu. Substitucí cysteinu za tyrosin v pozici 282. aminokyseliny dochází ke ztrátě jednoho ze čtyř disulfidických můstků v HFE proteinu. Tato změna konformace ovlivňuje jeho afinitu k ß2-mikroglobulinu a transport na povrch buněk, a v důsledku toho i vyšší absorpci železa z gastrointestinálního traktu.
Druhou významnou genetickou změnou v HFE genu je mutace H63D vedoucí k substituci histidinu za aspartát. Tato mutace neovlivňuje tvorbu heterodimeru HFE s ß2-mikroglobulinem, proto jsou její klinické příznaky mírné nebo se nemusí projevit vůbec. Mechanismus, jakým dochází ke zvýšenému vstřebávání železa, není u této mutace dosud přesně popsán. Třetí nejčastější mutací v HFE genu je S65C podmiňující substituci serinu za cystein v HFE proteinu. Ani zde není role mutace v patogenezi HH zcela objasněna.
Autozomálně recesivní přenos HH znamená, že k jejím fenotypovým projevům může docházet jen v případě přítomnosti mutace na obou alelách. Postižené osoby lze molekulárně geneticky charakterizovat buď jako homozygoty pro jeden typ mutace (nejčastěji s genotypem 282Y/282Y) nebo smíšené heterozygoty (2–6 % případů HH) (12, 13).
Hemochromatóza patří k tzv. střádavým onemocněním. Ke kumulaci železa dochází celoživotně a první projevy HH se manifestují mezi 40. a 50. rokem života. Klinické příznaky choroby nejsou specifické, objevuje se únava, hepatomegalie, svalové a kloubní bolesti, v pokročilejších stadiích srdeční arytmie, hypogonadismus, diabetes mellitus, jaterní cirhóza a hepatocelulární karcinom. Projevy jsou mnohdy komplikovány jiným onemocněním (steatózou jater, alkoholismem, poruchou krvetvorby, metabolickým syndromem apod.).
Pro diagnostiku HH se provádí klinické vyšetření, fyzikální vyšetření a biochemická analýza zahrnující zejména určení saturace transferinu, stanovení koncentrace feritinu, sérového železa a železa v jaterní tkáni. Nedílnou součástí diagnostického procesu je také molekulárně genetická analýza jako prostředek pro vyhledání samotné příčiny choroby a odhalující další rizikové jedince v rodině probanda. Bez laboratorní diagnostiky mohou být nespecifické projevy HH snadno přehlédnuty a choroba může zůstat dlouhodobě neléčena.
Cílem této studie bylo zjistit, jaký je výskyt mutací C282Y, H63D a S65C u souboru pacientů hepatologických poraden s příznaky akumulace železa a porovnat jej s výskytem zmíněných mutací ve zdravé populaci.
SOUBOR NEMOCNÝCH A POUŽITÉ METODY
Do studie bylo zařazeno 140 pacientů (vyšetřovaný soubor) z 12 zdravotnických center zabývajících se chorobami jater a poruchami metabolismu železa. Pro molekulárně genetickou analýzu byly použity DNA vzorky 95 mužů a 45 žen s mediánem věku 55 roků (rozsah 20–83 roků). Soubor zdravých osob (kontrolní soubor) tvořilo 167 dobrovolníků (65 mužů, 102 žen, medián 25 roků, rozsah 18–62 roků), od nichž byl získán Informovaný souhlas k DNA analýze. Studie byla provedena se svolením Etické komise Fakultní nemocnice Hradec Králové.
Izolace DNA (QIAamp Blood Mini Kit, Qiagen, Německo) byla u pacientů provedena z 200 μl nesrážlivé krve (K3EDTA) a u dobrovolníků z bukálního stěru (FlogSwabs, Copan Flock Technologies, Itálie). Pro amplifikaci byla použita polymerázová řetězová reakce (PCR). Reakční směs pro amplifikaci exonu 2 HFE genu (25 μl) obsahovala 100 ng DNA, 10krát koncentrovaný PCR pufr s 15mM roztokem MgCl2 (TaKaRa, Japonsko), 200 μM dNTPs, 0,4 μM primerů – F: 5’-ACA TGG TTG AGG CCT GTT GC 3’ a R: 5’-GCC ACA TCT GGC TTG AAA TT-3’ (Generi Biotech, ČR) a 1,5 U HS Taq polymerázy (TaKaRa). Směs pro exon 4 se lišila pouze sekvencemi použitých primerů – F: 5’ - CTG GAT AAC CTT GGC TGT ACC CCC -3’ a R: 5’ - CAG ATC CTC ATC TCA CTG -3’. Teplotní profil PCR reakce pro oba úseky genu se skládal z 5 minutové úvodní denaturace při 95 °C, poté následovalo 30 cyklů PCR (denaturace 30 s při 94 °C, annealing 30 s při 50 °C a elongace 30 s při 72 °C) v termocykléru Veriti™ 96-Well Thermal Cycler, Applied Biosystems, USA.
Restrikční směs se skládala z 10 μl PCR produktu, 1 μl restrikčního pufru a 1 μl příslušného restrikčního enzymu (vše New England Biolabs, USA): RsaI pro detekci mutace C282Y, BclI pro H63D a HinfI pro S65C. Inkubace probíhala 16 hodin při 37 °C (RsaI a HinfI) nebo při 50 °C (BclI). Analýza restrikčních fragmentů (RFLP) byla provedena na 3% agarózovém gelu s ethidiumbromidem. Přítomnost wild-type alely C282 potvrzovaly restrikční fragmenty o velikostech 171 a 18 bp, zatímco pro mutovanou alelu 282Y svědčily fragmenty 142 bp, 29 bp a 18 bp. V případě mutace 63D docházelo k zániku rozpoznávacího místa pro BclI, a proto byl po restrikci na gelu viditelný jeden fragment o délce odpovídající PCR produktu (208 bp). Naopak, fragmenty o délkách 138 bp a 70 bp identifikovaly wild-type alelu H63. Obdobně při vyšetření mutace S65C svědčil fragment dlouhý 208 bp o přítomnosti mutantní alely 65C, fragmenty 147 bp a 61 bp identifikovaly wild-type alelu S65.
VÝSLEDKY
Ve vyšetřovaném souboru bylo nalezeno 18 homozygotů pro mutaci C282Y (12,9 %) a deset heterozygotů (7,1 %) pro tuto mutaci. U mutace H63D bylo detekováno sedm homozygotů (5,0 %) a 32 heterozygotů (22,9 %). Heterozygotní forma mutace S65C byla prokázána u tří pacientů (2,1 %) a nebyl nalezen žádný homozygot pro S65C. Smíšená heterozygozita byla zjištěna u pěti pacientů (3,5 %), z toho u tří osob (2,1 %) se jednalo kombinaci alel 282Y/63D a u dvou osob (1,4 %) o kombinaci 282Y/65C (tab. 1).
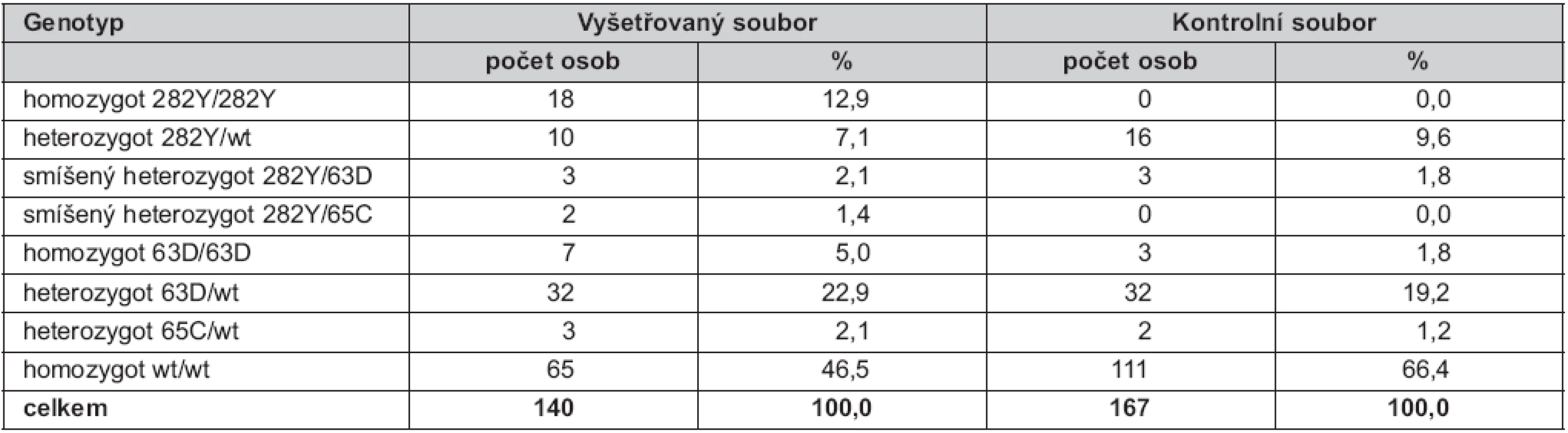
V kontrolním souboru jsme prokázali 16 heterozygotů pro mutaci C282Y (9,6 %), 32 heterozygotů pro H63D (19,2 %), dále tři homozygoty pro H63D (1,8 %), dva heterozygoty pro S65C (1,2 %). U tří dobrovolníků (1,8 %) byla zjištěna kombinace alel 282Y/63D (smíšená heterozygozita). V souboru nebyli nalezeni žádní homozygoti pro mutace C282Y ani S65C.
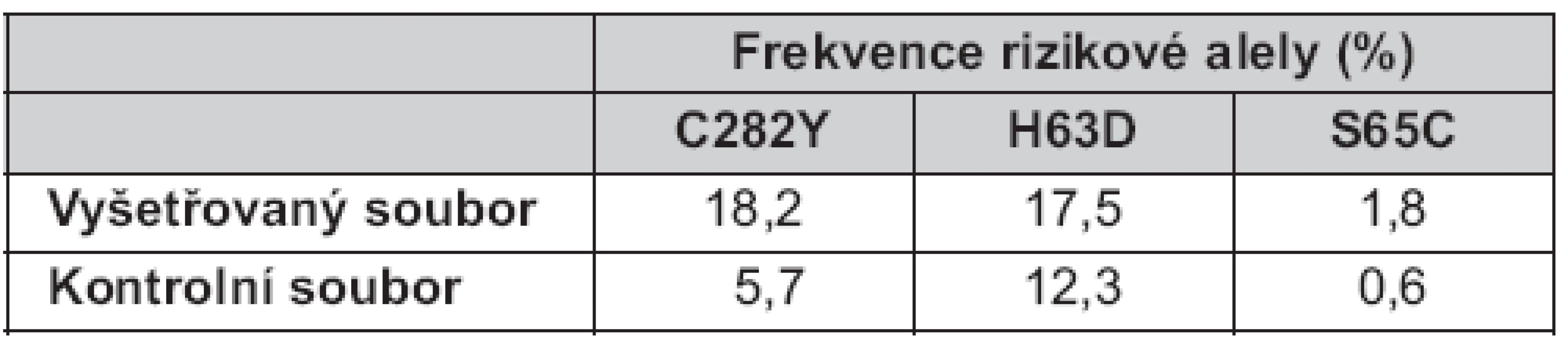
Celková frekvence všech rizikových alel ve vyšetřovaném souboru byla 37,5 % (18,2 % pro C282Y, 17,5 % pro H63D a 1,8 % pro S65C), tedy dvojnásobná než v kontrolním souboru: 18,6 % (5,7 % pro C282Y, 12,3 % pro H63D a 0,6 % pro S65C) (tab. 2).
Ve vyšetřovaném souboru bylo nalezeno celkem 30 osob s jedním z genotypů podmiňujících možné klinické příznaky HH (homozygoti 282Y/282Y, 63D/63D, smíšení heterozygoti 282Y/63D a 282Y/65C); v kontrolní skupině dosáhl jejich počet šesti. Predikce rizika HH ve vyšetřovaném souboru pacientů hepatologických ambulancí byla více než sedminásobná oproti souboru kontrolnímu (odds ratio 7,32; p < 0,001; 95% konfidenční interval: 3,27–16,39 %).
DISKUZE
Od roku 1996, kdy byl poprvé popsán HFE gen, byla publikována celá řada prací zabývajících se mapováním výskytu mutací v tomto genu. Jejich výsledky se shodují v konstatování, že majoritní podíl (80–90 %) na rozvoji HH má mutace C282Y, zejména je-li přítomna v homozygotní formě (14, 15). Na druhou stranu je třeba poznamenat, že penetrance C282Y není kompletní a choroba se manifestuje jen u části homozygotů (u 50 % mužů a 25 % žen) (16).
Z provedených populačních studií vyplývá, že nejvyšší frekvence rizikové alely 282Y se nacházejí v severozápadní Evropě, především v Irsku (14 %) a Velké Británii (8 %). V severní části Evropy (Norsko, Švédsko, Dánsko) se frekvence alely 282Y pohybují mezi 5,7–7,5 %. Není známo, zda byla tato mutace rozšířena do ostatních oblastí Evropy starověkými Kelty nebo skandinávskými mořeplavci, Vikingy (17, 18). Ve střední Evropě je frekvence 282Y 3,4–4,0 %. Ve státech jihovýchodní Evropy (Bosna a Hercegovina, Rumunsko, Srbsko, Makedonie) její výskyt klesá na 1,0–2,2 % (19). Mimo evropský kontinent se tato mutace objevuje vzácně.
V naší studii bylo zastoupení rizikové alely 282Y v kontrolní skupině vyšší než publikovali Čimburová a kol. (5,7 % vs. 3,4 %) v roce 2005 (20). Naše výsledky mohly být zatíženy chybou vyplývající z menšího počtu vyšetřených osob (n = 167).
Výsledky analýzy alely 282Y ve vyšetřovaném souboru (n = 140) prokázaly její trojnásobně vyšší výskyt u pacientů hepatologických ambulancí (18,2 %) oproti zdravé populaci. Celkem zde bylo nalezeno 18 homozygotů pro mutaci C282Y.
Jak již bylo zmíněno, mutace C282Y je považována za nejrizikovější mutaci z hlediska HH. Některé studie udávají, že podíl homozygotů ve skupině HH může být až 96% (21, 22). U heterozygotů pro C282Y dochází ke klinické manifestaci HH jen ojediněle. Spíše se ukazuje, že mutace může u heterozygotů akcelerovat orgánové poškození jiné primární etiologie (např. chronická hepatitida C, steatóza jater, hepatitida, alkoholismus apod.).
Frekvence mutace H63D se v evropské populaci pohybuje mezi 10 a 20 %. Nejvyšší výskyt byl popsán v Baskicku (30 %), Bulharsku, Španělsku a Portugalsku (> 20 %). U nás je její výskyt okolo 15 % (16). Frekvence mutace S65C se v kavkazské populaci pohybuje od 0,5 % (jihovýchodní Itálie) do 3 % (Švédsko, populace Saami) (23, 24). V České republice je frekvence S65C okolo 1 % (16). V našem kontrolním souboru jsme nalezli dva heterozygoty pro mutaci S65C a frekvence alely 65C byla 0,6 %.
Je známo, že u homozygotů pro H63D a smíšených heterozygotů 282Y/63D nebo 282Y/65C bývají příznaky HH mírnější. Výskyt mutací H63D (17,5 %) a S65C (1,8 %) v naší vyšetřované skupině se významně nelišil od frekvence zjištěné v kontrolním souboru i výše zmíněných populačních odhadů. Mezi heterozygoty pro S65C byla jedna žena ve věku 30 let s normálním biochemickým nálezem a 3 muži se zvýšenou sérovou koncentrací železa. Laboratorní nález však nemusí být způsoben přítomností mutace S65C.
U zbývajících 65 pacientů z vyšetřovaného souboru (46,5 %) nebyly prokázány mutace C282Y, H63D ani S65C v HFE genu. U těchto osob může být přetížení organismu železem způsobeno dalšími mutacemi genů, jejichž produkty se podílejí na absorpci, transportu nebo ukládání železa. K rozvoji příznaků hemochromatózy přispívají také nevhodné stravovací návyky, přítomnost toxinů v organismu, obezita, HCV infekce anebo dlouhodobě zvýšený příjem alkoholu (u mužů více než 60 g/den a u žen 40 g/den). Alkohol nejen inhibuje transkripci hepcidinu, ale v důsledku oxidačního stresu organismu také zvyšuje expresi transferinového receptoru 1 (5, 25).
Velké množství faktorů negenetického charakteru, které se objevuje v etiopatogenezi HH je jedním z důvodů, proč nelze zavést plošný populační screening mutací v HFE genu. Spíše je preferován tzv. selektivní screening osob v rizikových skupinách (16). Při diagnostice hemochromatózy je doporučováno kombinovat molekulárně genetická vyšetření s paletou biochemických testů zahrnujících určení saturace transferinu, volné vazebné kapacity séra pro železo, stanovení feritinu a železa v séru, případně v jaterní tkáni (26). Biochemické markery i přes svou nižší specifičnost a závislost na řadě faktorů (věk, dieta, stravovací návyky, menstruace a jiné ztráty krve) mohou také významně pomoci při monitorování účinnosti navržených terapeutických a dietních postupů.
ZÁVĚR
Hereditární hemochromatóza je značně rozšířeným genetickým onemocněním. K určení diagnózy HH se používají biochemická a molekulárně genetická vyšetření. Vzhledem k tomu, že se v našem státě neprovádí populační screening mutací v HFE genu, je nezbytné cíleně vyhledávat rizikové jedince pro toto onemocnění. Jednu z rizikových skupin představují pacienti hepatologických ambulancí se známkami přetížení organismu železem. Jak ukázala naše studie, u těchto osob se vyskytuje mutace C282Y několikanásobně častěji než ve zdravé populaci. Pro potvrzení nebo vyloučení HH u těchto osob (a jejich rodinných příslušníků) by mělo být vždy provedeno molekulárně genetické vyšetření HFE genu.
Zkratky
- BclI – Bacillus caldolyticus
- DNA – deoxyribonucleic acid (deoxyribonukleová kyselina)
- dNTPs – deoxynucleotide triphosphates (deoxynukleotid trifosfáty)
- HCV – hepatitis C virus (virus hepatitidy C)
- HH – hereditary hemochromatosis (vrozená (hereditární) hemochromatóza)
- HinfI – Haemophilus influenzae
- HLA – human leukocyte antigen (hlavní histokompatibilní komplex)
- HS Taq – hot start Thermus aquaticus
- K3EDTA – ethylene diamine tetraacetic acid trisodium salt
- PCR – polymerase chain reaction (polymerázová řetězová reakce)
- RFLP – restriction fragment length polymorphism (polymorfizmus délky restrikčních fragmentů)
- RsaI – Rhodopseudomonas sphaeroides
Studie byla provedena v rámci projektu Specifického vysokoškolského výzkumu č. 264902 (2012).
ADRESA PRO KORESPONDENCI:
Mgr. Monika Drastíková
Ústav klinické biochemie a diagnostiky LF a FN
Sokolská 581, 500 05 Hradec Králové
e-mail: monika.drastikova@fnhk.cz
Sources
1. Brůha R, Hůlek P, Petrtýl J. Jaterní cirhóza. In: Ehrmann J, Hůlek P, et al. Hepatologie. Praha: Grada Publishing 2010; 399–410.
2. Mormone E, George J, Nieto N. Molecular pathogenesis of hepatic fibrosis and current therapeutic approaches. Chem Biol Interact. 2011; 193 : 225–231.
3. Horák J. Genetická hemochromatóza. In: Ehrmann J, Hůlek P, et al. Hepatologie. Praha: Grada Publishing 2010; 339–445.
4. Kovář J. Biologický význam a fyziologické funkce železa. In: Horák J. Hemochromatóza. Praha: Grada Publishing 2010; 15–22.
5. Fargion S, Valenti L, Fracanzani A. L. Beyond hereditary hemochromatosis: new insights into the relationship between iron overload and chronic liver diseases. Dig Liver Dis 2011; 43 : 89–95.
6. Phatak PD, Sham RL, Raubertas RF, et al. Prevalence of hereditary hemochromatosis in 16031 primary care patients. Ann Intern Med 1998; 129 : 954–961.
7. Lyon E, Frank EL. Hereditary hemochromatosis since discovery of the HFE gene. Clin Chem 2001; 47 : 1147–1156.
8. Whittington CA, Kowdley KV. Review article: haemochromatosis. Aliment Pharmacol Ther 2002; 16 : 1963–1975.
9. Novotný J. Poruchy metabolismu železa II. Vnitř. Lék. 2005; 51 : 995–1006.
10. Feder JN, Penny DM, Irrinki A, et al. The hemochromatosis gene product complexes with the transferrin receptor and lowers its affinity for ligand binding. Proc Natl Acad Sci USA 1998; 95 : 1472–1477.
11. Deicher R, Hörl WH. New insights into the regulation of iron homeostasis. Eur J Clin Invest 2006; 36 : 301–309.
12. Mura C, Raguenes O, Férec C. HFE mutations analysis in 711 hemochromatosis probands: evidence for S65C implication in mild form of hemochromatosis. Blood 1999; 93 : 2502–2505.
13. Gurrin LC, Bertalli NA, Dalton GW, et al. HFE C282Y/H63D compound heterozygotes are at low risk of hemochromatosis-related morbidity. Hepatology 2009; 50 : 94–101.
14. Allen KJ, Gurrin LC, Constantine CC, et al. Iron-overload-related disease in HFE hereditary hemochromatosis. N Engl J Med 2008; 358 : 221–230.
15. Gómez-Llorente C, Miranda-León MT, Blanco S, et al. Frequency and clinical expression of HFE gene mutations in a Spanish population of subjects with abnormal iron metabolism. Ann Hematol 2005; 84 : 650–655.
16. Zlocha J, Kovács L, Požgayová S, et al. Molekulovo-genetická diagnostika a skríning hereditárnej hemochromatózy. Vnitř. Lék. 2006; 52 : 602–608.
17. Pedersen P, Melsen GV, Milman N. Frequencies of the haemochromatosis gene (HFE) variants C282Y, H63D and S65C in 6020 ethnic Danish men. Ann Hematol 2008; 87 : 735–740.
18. Olsson KS, Konar J, Dufva IH, et al. Was the C282Y mutation an Irish Gaelic mutation that the Vikings helped disseminate – Eur J Haematol 2011; 86 : 75–82.
19. Adler G, Clark JS, Łoniewska B, et al. Prevalence of 845G>A HFE mutation in Slavic populations: an east-west linear gradient in South Slavs. Croat Med J 2011; 52 : 351–357.
20. Čimburová M, Půtová I, Provazníková H, et al. S65C and other mutations in the haemochromatosis gene in the Czech Population. Folia Biol 2005; 51 : 172–176.
21. Hanson EH, Imperatore G, Burke, W. HFE gene and hereditary hemochromatosis: a HuGE review. Am J Epidemiol 2001; 154 : 193–206.
22. Cukjati M, Vaupotic T, Rupreht R, et al. Prevalence of H63D, S65C and C282Y hereditary hemochromatosis gene mutations in Slovenian population by an improved high-throughput genotyping assay. BMC Med Genet 2007; 69 : 1–9.
23. Pietrapertosa A, Vitucci A, Campanale D, et al. HFE gene mutations in Apulian population: allele frequencies. Eur J Epidemiol 2003; 18 : 685–589.
24. Beckman LE, Sjoberg K, Eriksson S, et al. Haemochromatosis gene mutations in Finns, Swedes, and Swedish Saamis. Hum Hered 2001; 52 : 110–112.
25. Pietrangelo A. Hemochromatosis: an endocrine liver disease. Hepatology 2007; 46 : 1291–1301.
26. Husová L, Dastych M, Votava M, et al. Hereditární hemochromatóza – opomíjená diagnóza. Čes a Slov Gastroent a Hepatol 2005; 59 : 188–194.
Labels
Addictology Allergology and clinical immunology Anaesthesiology, Resuscitation and Inten Angiology Audiology Clinical biochemistry Dermatology & STDs Paediatric dermatology & STDs Paediatric gastroenterology Paediatric gynaecology Paediatric surgery Paediatric cardiology Paediatric nephrology Paediatric neurology Paediatric clinical oncology Paediatric ENT Paediatric pneumology Paediatric psychiatry Paediatric radiology Paediatric rheumatology Paediatric urologist Diabetology Endocrinology Pharmacy Clinical pharmacology Physiotherapist, university degree Gastroenterology and hepatology Medical genetics Geriatrics Gynaecology and obstetrics Haematology Hygiene and epidemiology Hyperbaric medicine Vascular surgery Chest surgery Plastic surgery Surgery Medical virology Intensive Care Medicine Cardiac surgery Cardiology Clinical speech therapy Clinical microbiology Nephrology Neonatology Neurosurgery Neurology Nuclear medicine Nutritive therapist Obesitology Ophthalmology Clinical oncology Orthodontics Orthopaedics ENT (Otorhinolaryngology) Anatomical pathology Paediatrics Pneumology and ftiseology Burns medicine Medical assessment General practitioner for children and adolescents Orthopaedic prosthetics Clinical psychology Radiodiagnostics Radiotherapy Rehabilitation Reproduction medicine Rheumatology Nurse Sexuology Forensic medical examiner Dental medicine Sports medicine Toxicology Traumatology Trauma surgery Urology Laboratory Home nurse Phoniatrics Pain management Health Care Dental Hygienist Medical studentArticle was published in
Journal of Czech Physicians

- Advances in the Treatment of Myasthenia Gravis on the Horizon
- Memantine in Dementia Therapy – Current Findings and Possible Future Applications
- Possibilities of Using Metamizole in the Treatment of Acute Primary Headaches
Most read in this issue
- Aktuální pohled na fibromyalgii
- Význam DNA vyšetření mutací C282Y, H63D a S65C v HFE genu
- Fytoterapia kožných rán – prehľad experimentálnych a klinických štúdií v prvom decéniu 21. storočia
- Obecná psychopatologie – podklad pro objektivní psychiatrický nález*