
Rozštěpové vady
The clefts as inborn defects
The article refers usual facial clefts, which are not rare anomalies. Relation to other forms of so-called midline defects (limb clefts) is indicated. Syndromic and unusual clefts are rarer than isolated non-syndromic clefts. Clinical features, including minimal symptomatology, etiopathogenesis and population frequency are discussed. From the diagnostic point of view specific, prenatal, postnatal and differential diagnostic approaches are recognized. Preventive aspects, therapy and management of the disease (for cleft lip and palate defects, median cleft palate, broad spectrum of neural tube defects including anencephaly, limb clefts etc.) are important. We estimated the empiric risk of the recurrence and suggest methods for preconceptional preventive care.
Keywords:
cheilognathopalatoschizis – cleft palate – neural tube defects – limb clefts – etiopatogenesis – heredity – empiric risks and preconceptional care
Authors:
Miloslav Kuklík
Authors‘ workplace:
Genetické oddělení, Praha
3
Published in:
Čas. Lék. čes. 2013; 152: 185-191
Category:
Review Article
Overview
Článek shrnuje problematiku typických obličejových rozštěpů, o kterých je známo, že jsou relativně častými anomáliemi. Upozorňuje na jiné formy tzv. středočárových defektů spojených s rozštěpy, jako jsou např. rozštěpy končetin. Hovoří se o klinických projevech zahrnujících též minimální symptomatologii, dále o etiopatogenezi a populační frekvenci těchto vad, pokud je známa. Z diagnostického hlediska se článek zabývá specifickou, prenatální a postnatální a také diagnostikou diferenciální. Zdůrazněny jsou aspekty prevence, terapie a managementu jednotlivých chorob (CLP, CP, NTD a polygenních rozštěpů končetin). Jsou známa empirická rizika opakování těchto vad a existuje systém prekoncepční péče k jejich snížení.
Klíčová slova:
rozštěp rtu – patra a čelisti – mediánní rozštěp patra – rozštěp páteře – etiologie – dědičnost – empirická rizika a prekoncepční péče
ROZŠTĚP RTU, PATRA A ČELISTI (CHEILOGNATOPALATOSCHIZIS, CLP)
Rozštěpové vady včetně rozštěpu rtu, patra a čelisti se vyskytují v lidské populaci od nepaměti. Výskyt je znám např. v některých středověkých šlechtických populacích (Babenbergové v Rakousku), dále se vady CLP vyskytují četněji v některých izolovaných populacích ostrovních jako např. na Maltě. Péče o nemocné s touto vadou má v České republice dlouhou a dobrou tradici a je spojena v plastické chirurgii se jménem akademika prof. MUDr. Františka Buriana, DrSc., v prenatální fetoskopické diagnosticeprof. MUDr. Antonína Zwingera, DrSc., v lékařské genetice s prof. MUDr. Marií Tolarovou, DrSc. a dalšími následníky.
Etiologie a patogeneze
Tyto vady patří k onemocnění z nedostatku mezenchymálních struktur a nedostatečné proteosyntézy. Tím dochází k nespojení příslušných obličejových struktur. Vznik rozštěpu je determinován souhrou vnitřních faktorů (polygenní determinace z určitého počtu genů malého účinku s aditivním či multiplikačním působením). K objasnění příčiny vzniku vady u konkrétního pacienta je třeba pečlivý rozbor těhotenské anamnézy matky pacienta a zohlednění tzv. kritických a senzitivních period v 1. trimestru těhotenství (časová lokalizace případné teratogenní noxy v těhotenství do 35. dne po početí). Součástí polygenního systému pro vznik rozštěpu je také polymorfní gen MTHFR (metylentetrahydrofolátreduktáza), který má více funkcí.
Je snaha vyčlenit větší množství genů, které se podílejí na etiologii a patogenezi CLP za pomocí tzv. multigenomiky využívající tzv. genetických čipů. Praktický význam tyto studie však zatím nemají.
Experimentální studie využívající především ptačích modelů ukázaly určité analogie v etiopatogenezi u rozštěpů zobáků (experimentální kuřecí kmeny) a patra u savců a člověka. Vznik rozštěpu po experimentální expozici myšího hydrokortizonu, kdy se předpokládá útlum proteosyntézy, nelze jednoznačně přenášet na člověka – i v rámci jedné species totiž existuje značná farmakogenetická a farmakokinetická odlišnost.
Geneticka rizika a výskyt (obr. 1)
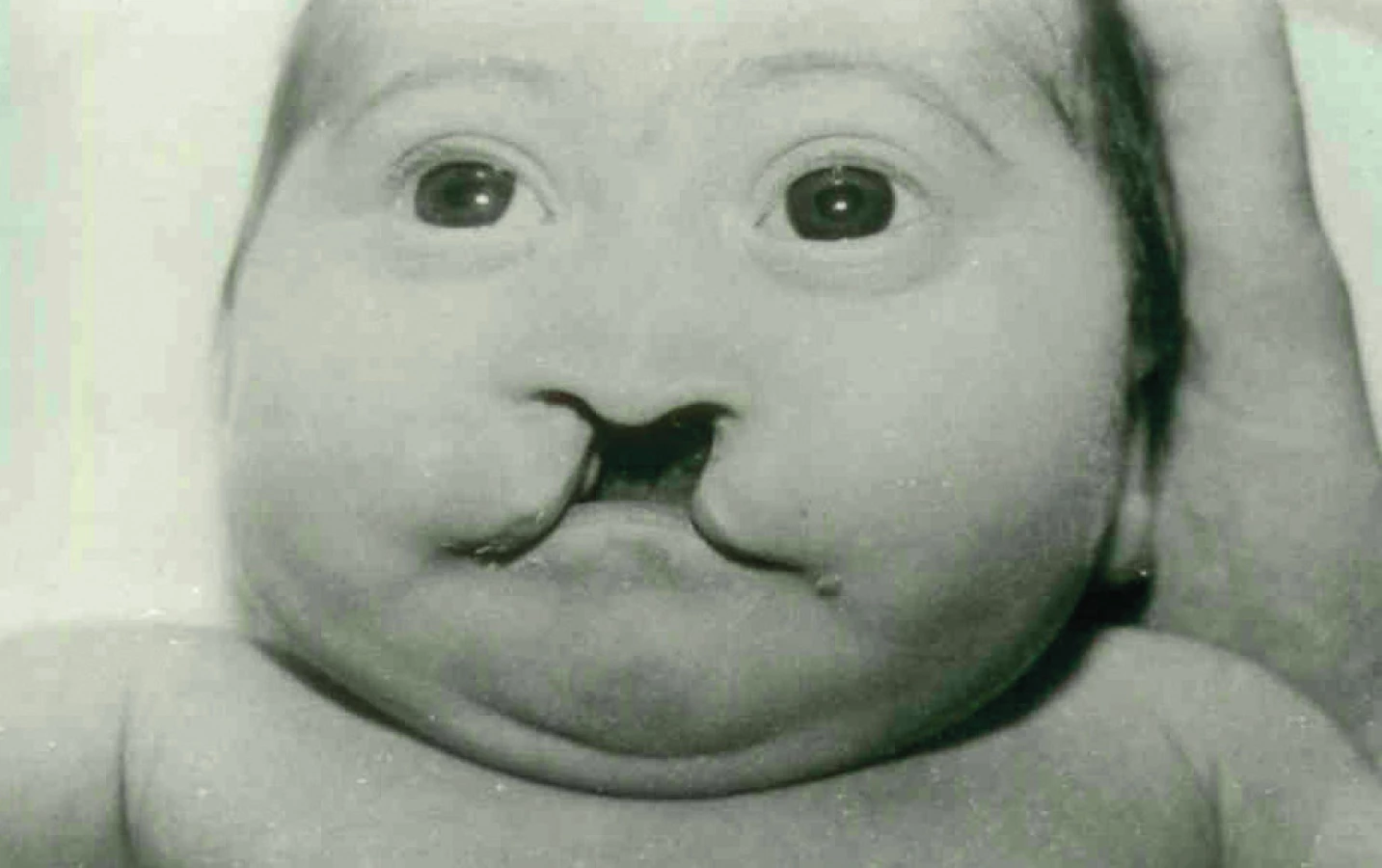
Frekvence rozštěpu rtu, patra a čelisti kolísají v různých populacích a také v čase. Nejedná se o řídké anomálie. Oboustranný rozštěp CLP je asi ve 20 % všech případů. Jednostranný rozštěp je na levé straně častější – 70 % (3). U oboustranných rozštěpů je ve více než 80 % zasaženo i patro, u jednostranných o něco méně (70 %) (1). V malé části případů je v rozštěpové linii přítomen kožní můstek (asi v 10 %) (1, 2). Rozštěp rtu, patra a čelisti se vyskytuje u všech lidských plemen a etnik.
Existuje pozorování nakupení vady v krátkém časovém období (clustering efekt) v některém geograficky omezeném území, což svědčí o případném zásahu teratogenní noxy. Rozštěpy se často vyskytují v sériích, epidemiích, tj. právě zmíněný cluster efekt. Často je to v návaznosti na některé infekce, především virová onemocnění. Tak tomu bylo např. v Čechách na Kladensku v roce 1975 v závislosti na proběhlou epidemii hepatitidy A (virus jako teratogenní noxa ?) a také v okolí Jindřichova Hradce v závislosti na předpokládané zvýšené přirozené radiaci (radiová emanace – radioaktivní radon z ložisek uranu).
Případný vliv zevního prostředí a zevních faktorů (peristatických, teratogenních) na vznik vady může objasnit pečlivý anamnestický rozbor těhotenství matky nemocných.
Frekvence CLP u nás je to přibližně 1 : 1500 jedinců ze všech novorozených. Izolovaný nesyndromologický rozštěp má rizika opakování v příbuzenstvu v souladu s pravidly polygenní dědičnosti, kdy u izolovaného případu (tj. ojedinělého v rodokmenu, v širší rodině) je riziko opakování vady pro příbuzného 1. stupně dáno Edwardsovým vzorcem druhé odmocniny z populačního výskytu. V naší populaci je to konkretně druhá odmocnina 1 z 1500, což je průměrný přibližný výskyt, když opomineme kolísání výskytu vady v čase, výsledně 4–5 %.
Na rozdíl od mendelovské dědičnosti riziko opakování má paměť a zvyšuje se s počtem nemocných v rodině. Vyšší empirická rizika jsou též pro vady (CLP) s oboustranným výskytem u jednoho člena rodokmenu (izolovaný výskyt), a to 8–10 %. Vždy se jedná o nesyndromologický rozštěp, který není součástí dalších vad. Pro opakovaný výskyt rozštěpů v rodině se riziko zvyšuje, v tomto smyslu existují tabulky empirických rizik, použitelné ovšem v konkrétní dané populaci. Nelze přenášet empirická rizika z jedné populace do druhé, protože v každé populaci je jiný výskyt dané vady. Na druhé straně minimální symptomatologie v podobě jednostranného frustního rozštěpu rtu znamená menší riziko než zmíněných 4–5 % pro příbuzného izolovaného případu 1. stupně. Vždy je nutné zohlednit stupeň vyjádření vady ve vztahu k určování stupně rizika.
Výše rizika vzniku rozštěpové vady může být příznivě ovlivněna a snížena systémem prekoncepční péče, kdy jsou režimovými opatřeními prokazatelně pozitivně eliminovány nepříznivé vlivy vedoucí k jejich vzniku. Je to především sanace zánětlivých fokusů budoucí matky, optimalizace jejího zdravotního stavu ještě před těhotenstvím a samozřejmě také v době těhotenství (zaléčení např. endokrinních a interních onemocnění apod.), nutrigenomická opatření. Výše rizika může být ovlivněna také opačně nepříznivými teratogenními vlivy nasedajícími na labilní geneticky nevyvážený terén daného jedince. Stejná teratogenní noxa působící v kritickou senzitivní periodu může u jednoho jedince vadu způsobit a u druhého ne.
Riziko opakování vady pro příbuzné pacienta 2. stupně klesá na 2 %. Pro polygenní dědičnost rozštěpů CLP je charakteristický prudký pokles rizika se stupněm příbuznosti, tj., u 3. stupně příbuznosti klesá prakticky na populační úroveň (resp. je jen teoreticky nepatrně vyšší). U onemocnění existují i minimální projevy (mikrosymptomy). Pro odhad opakování empirického rizika vzniku vady u potomka či sourozence v širším příbuzenstvu má velký význam výskyt případných mikrosymptomů v příbuzenstvu. Tyto mikrosymptomy patří často do okruhu antropogenetických znaků označovaných jako epigenetické znaky, v kriminalistice pak termínem zvláštní znamení. Tyto mikrosymptomy je pak z hlediska zvažování empirického rizika nutné pokládat za výskyt plně vyvinuté vady. Pro potomky samotných nosičů těchto mikrosymptomů bez výskytu vady v rodině (rodokmenu) není riziko zvýšeno. Mikrosymptomy ve smyslu gaussovského rozlišení kvantitativního znaku přecházejí plynule přes tzv. práh k vzniku vady (změna kvantity v novou kvalitu). Toto pojetí kvantitativního znaku lze aplikovat i na podíl faktorů způsobujících vadu (polyfaktoriální, polygenní determinace znaku přes kritický práh). Prekoncepční opatření zdánlivě ovlivní tzv. posun prahu vzniku vady; tento práh je však neměnný, jeho posun je zdánlivý, mění se pouze vyvolávající faktory, nikoliv genové dispozice.
Empirické riziko pracuje na rozdíl od mendelovské dědičnosti s pamětí. Empirické riziko se může v dané rodině změnit – např. při odhadu rizika pro potomka u izolovaného jednostranného rozštěpu počítáme pro potomka 4–5 %. Jestliže se však toto riziko v těhotenství a po porodu nového jedince realizuje, jde v rodině již o výskyt dvou vad a empirické riziko CLP se pro případné další těhotenství odhaduje na 9–10 %, což je fakt pro statisticky neorientované postižené rodiny problém a zdroj nedůvěry. Jde pouze o to, že předem nevíme, jaký je genetický podíl v konkrétní rodině na vzniku vady.
Součástí polygenního systému jsou také gonosomy. Jako u většiny polygenních vad je zde rozdílné zastoupení výskytu vady podle pohlaví, i když se nejedná o dědičnost vázanou na pohlaví. Z pěti nemocných jsou tři muži (sex ratio 3 : 2 ve prospěch mužů).
Rozdílný výskyt rozštěpů podle pohlaví má vliv i na odhad empirického rizika. Riziko lze modifikovat podle pohlaví: Empirické riziko pro potomka či sourozence u izolovaného výskytu je vyšší, dojde-li k výskytu u ženy, ne u muže, kde je nižší. V etiopatogenezi se předpokládá u nemocných žen větší podíl genetických faktorů než u mužů, kde v etiopatogenezi statisticky působí více faktorů peristatických, exogenních.
Pozornost byla věnována i případné stranové predilekci u výskytu jednostranných rozštěpů rtu, patra a čelisti, která však není výrazná a výsledky jednotlivých studií se mohou lišit podle rozsahu sledovaného souboru a populace. Obecně bývá udávána mírná pravostranná predilekce u nás, levostranná v zahraničí (USA) (1), podobně jako je tomu u končetinových vad. To by mohlo svědčit též pro účast homeotických genů, především MSX 1 a MSX 2 v etiopatogenezi rozštěpů.
Mikrosymptomy CL(P) (minimální projevy)
U rozštěpů rtu bez další symptomatologie může být přítomna vrozená jizva za situace, kdy původní malý rozštěp se ve fetálním období nakonec spojil. V místě rozštěpové linie může být u sourozenců či rodičů, vzácněji u vzdálenějších příbuzných chybění špičáku či ageneze horních postranních řezáků (2 + 2) v horní čelisti, popř. mikrodoncie či dystopie zubu, zejména horního špičáku v této oblasti.
Rodiče dětí s CL(P) mají častěji než ostatní populace progenii, oploštění obličeje. K dalším mikrosymptomům patří asymetrie patra, asymetrie nosních křídel je patrná již pouhou aspekcí. V oblasti molárů bývají u pacientů či příbuzných těchto nemocných ageneze zubů v této oblasti. V místě rozštěpové linie může být i hyperodoncie, a to nejen u zjevně nemocných, ale i u jejich příbuzných.
Při vyhledávání mikrosymptomů se může uplatnit antropometrické vyšetření (kraniometrie, kefalometrie, RTG lbi), které objektivizuje často subjektivní hodnocení (dojem) pozorovatele.
Příbuzenské a výběrové sňatky (vztahy)
Příbuzenské sňatky v rodinách a rodokmenech s CLP zvyšují empirické riziko vzniku vady u příbuzných (usnadňují manifestaci prahu vzniku choroby). Stejné situace mají výběrové sňatky dvou stejně nemocných jedinců se stejnou vadou (efekt aditivního účinku genů – polygenů se může doplňovat s multiplikací). Výběrové sňatky znamenají, že v lidské populaci neplatí zcela Hardyho - Weinbergův-Castleův zákon, a osoby stejné inteligence, zájmů, handicapu, zdravotního stavu mají častěji společné potomky. Tato skutečnost působí proti panmixii. Empirické riziko CLP pro potomky při sňatku (vztahu) dvou stejně nemocných jedinců s jednostrannou vadou činí až 36 %, je tedy podstatně zvýšeno proti jiným situacím. Příbuzenský sňatek v rodině (např. bratranecký, ale i jiný), kde se vyskytl CLP, má podobný efekt, ale ne s tak vysokým rizikem (bude záviset na stupni příbuznosti).
V některých zeměpisně izolovaných oblastech relativně malých populací (izoláty) může být výskyt rozštěpů vyšší než jinde, kde je panmixie relativně zachována (horské oblasti, ostrovní populace), v Evropě je zmiňována populace ostrova Malta.
Matematicko-statistické modely mohou být zajímavé, ale pro konkrétní rodiny nejsou tak důležité a významné jako konkrétní diagnostika již v prenatálním období. Matematické modely stanovení rizik využívají pravděpodobnostních modelů podle Bayese a jejich rozbor zde by byl již mimo rámec tohoto sdělení.
Prenatální diagnostika (obr. 2)
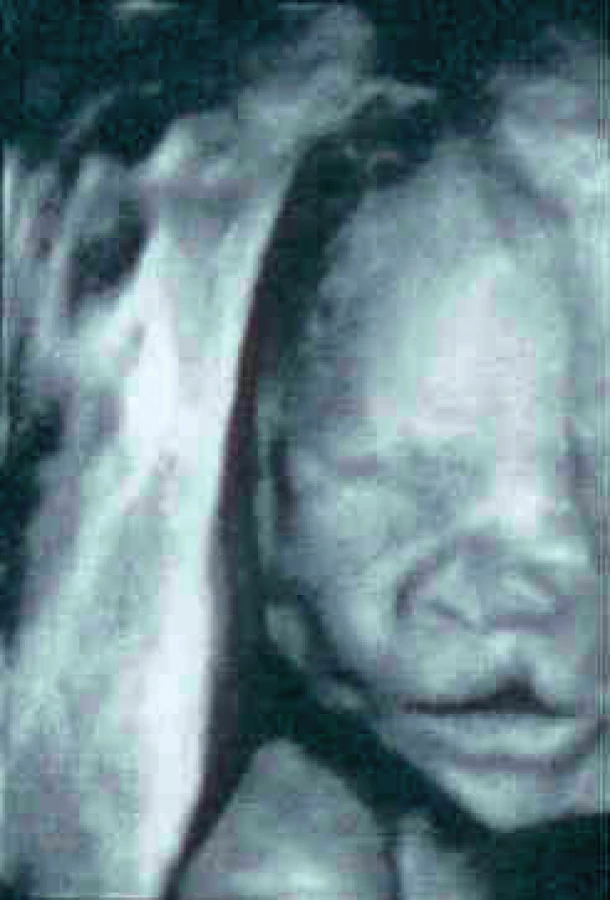
Prenatální diagnostika je založena v současnosti na zobrazovacích metodách s vyšším stupněm rozlišení, především ultrazvukových, jako je dvojdimenzionální a třídimenzionální ultrazvuk, a na NMR metodách (nukleární magnetická rezonance). Obě tyto neinvazivní zobrazovací metody prodělaly v posledních letech a desetiletích velký rozvoj a vytlačily invazivní fetoskopická sledování. Avšak i fetoskopická technika prodělala rozvoj.
Ultrazvuková vyšetření (UZ) v rozlišení struktur velmi pokročila – poskytuje však ještě stále hrubší, zrnitý obraz. Plastičtější, měkčí obraz poskytuje nukleární magnetická rezonance (NMR, MRI), která výhodně doplňuje vyšetření UZ, zejména v nejasných případech. Ke kvalitě tkání se může vyjádřit NMR lépe než ultrazvukové vyšetření, nastavení sledování fosfátového spektra může lépe objasnit materiálové vlastnosti tkání. U neinvazivních zobrazovacích metod je třeba vzít v úvahu možnost optických klamů daných nedokonalostí lidského zrakového analyzátoru a možnosti přehlédnutí vadné struktury, ale i možnosti falešné negativity nálezu. Diagnóza závisí na sonografických podmínkách konkrétního těhotenství (vizualizační podmínky dané obezitou těhotné, množstvím plodové vody, aktuální polohou plodu apod.). Třírozměrná ultrazvuková diagnostika je oblíbena u těhotných, neboť poskytuje obraz srozumitelný i laikovi. Je považována za nadstandardní vyšetření poskytující kromě třetího rozměru i poněkud plastičtější obraz a na mnohých pracovištích je pacienty proplácena. Pro ultrazvukového specialistu je však postačující dvourozměrný obraz.
Z historického hlediska je třeba připomenout invazivní fetoskopická vyšetřování pomocí vláknité optiky. Zavádění flexibilního fetoskopu do dělohy a do amniové dutiny představuje analogii endoskopických metod používaných v gastroenterologii, pneumologii a gynekologii.
V těhotenství je však využití fetoskopie spojeno s rizikem předčasného odtoku plodové vody a možností zanesení infekce, obojí s možností spontánního abortu.
Z hlediska předčasného odtoku plodové vody je přímá souvislost s průměrem fetoskopu, kdy došlo nyní k jeho minimalizaci. Pohled vláknitou optikou vybavenou osvětlením znamená pohled v úzkém zorném poli, jakoby klíčovou dírkou. Navíc je zde omezení podmíněné např. zkalením plodové vody vzniklé z různých důvodů (zánětlivá infekce, krvácení atd.). Tato metoda byla využívána zejména v osmdesátých a devadesátých letech 20. století, dnes je potřeba minimálně, i když fetoskop patří ke standardnímu vybavení gynekologicko-porodnických klinik.
Fetoskopie byla a je využívána zejména u oboustranných rozštěpů s vyšším empirickým rizikem opakování v potomstvu. Řada matek takto nemocných nechtěla, aby jejich potomek byl stejně stigmatizován a vystaven řadě plasticko-chirurgických zákroků, a při zjištění vady žádala přerušení těhotenství. Z etického hlediska je však sporné, zda takový přístup je přijatelný. Lékař odmítající vystavit indikaci přerušení těhotenství z důvodu zjištění jednostranného malého rozštěpu rtu se tak může dostat do konfliktu s těhotnou, která má pocit, že neprovedením interrupce je jejímu dítěti tzv. „zkažen život“.
Stejně tak se může rozvíjet mezioborový konflikt, kdy na stejnou situaci zjištění rozštěpu má jiný názor plastický nebo dětský chirurg provádějící reparační operace a gynekolog indikující interrupci z důvodů zjištění vrozené vady (nedostatečná legislativa neurčující přesně při indikaci přerušení těhotenství diagnózu a její stupeň). Podrobnější direktivy by tak usnadnily rozhodování. Tento poznatek má obecnější charakter a nevztahuje se pouze na diagnózu CLP.
Diferenciální diagnostika CLP
Rozštěpové vady typu CLP doprovázejí řadu mendelovských syndromů, kde se pak dědičnost vady řídí typem mendelovské dědičnosti syndromu (symptomatologie syndromu v další generaci však nemusí být vždy plně vyjádřena, rozdíl vad u rodičů a dětí a mezi sourozenci je v rámci intrafamiliární variability v syndromologii běžný, mezi jednotlivými symptomy včetně CLP je určitý stupeň volnosti).
CLP provází zejména vrozené chromozomální aberace, zejména trizomii 13, mendelovské choroby. Existuje však určitý kryptický syndrom oboustranného rozštěpu CLP spojený s píštělemi dolního rtu, tzv. syndrom van der Woudeové, kdy se vyskytují symetrické dvě píštěle dolního rtu zároveň se symetrickým oboustranným rozštěpem CLP v horní čelisti. Jeho dědičnost na rozdíl od polygenního rozštěpu CLP je autozomálně dominantní a očekávané riziko opakování je vyšší – 50 %. Diagnostika může být problematická, když jsou píštěle dolního rtu frustní, představující jen malé dolíčky. Pro diferenciální diagnostiku od prostého oboustranného rozštěpu CLP má význam cheiloskopie. Je to metoda analogická dermatoglyfice, tj. získávání otisků struktury papilárních linií z dlaní či plosek nohou. Zde se jedná o otisky rtů.
Cheiloskopie u CLP
Cheiloskopie je aspekce struktury rtu za pomoci otisků, analogicky jako u dermatoglyfiky. Otisky je nejlépe provádět za pomoci mastné rtěnky na křídový papír (možné formou hry i u malých dětí). Lze tak zobrazit u oboustranných rozštěpů rtu, patra a čelisti frustní formy píštělí dolního rtu, zařazující vadu symptomaticky do syndromu van der Woudeové s vyšším, autozomálně dominantním 50% rizikem.
Léčba a management CLP
Přístup k operativě rozštěpů by přesahoval rozsah tohoto sdělení, proto se jen stručně zmíníme o vývoji přístupu k této problematice. O první operativu a péči o nemocné se u nás zasloužil akademik Burian a za dobu posledních desetiletí přístup k chirurgickému řešení prodělal delší vývoj a má svá specifika odlišný od ostatní dětské chirurgie.
Zatímco v minulosti bylo chirurgické řešení CLP načasováno na pozdější vhodnou dobu, nyní je využíváno techniky fetálního hojení v novorozeneckém období, zejména pro CL menšího rozsahu. CLP není indikací k přerušení těhotenství, jde o vadu kosmetickou bez mentální retardace či závažného handicapu zamezujícího normální zařazení do společnosti. Úspěchy této metody jsou překvapující. U rozsáhlých komplikovanějších rozštěpů s technikou fetálního hojení nevystačíme. Při plasticko-chirurgické operaci bylo využíváno tzv. Z plastiky nezbytné pro nedostatek tkáňového materiálu.
Prekoncepční péče
Prekoncepční péče směřující k prevenci polygenní vady v disponovaných rodinách vyplývá z poznatků embryologie, některé z nich se datují již do čtyřicátých let 20. století. Nedostatek listové kyseliny (acidum folicum) či její zvýšenou spotřebu lze kompenzovat při proteosyntéze farmakologicky. Další je nezbytná dodávka vitaminů B skupiny (zejména B1, B6, B12), methioninu jako promotoru proteosyntézy. Někteří význam nutrigenomických opatření zpochybňují, argumentují nízkými výchozími empirickými riziky, je však jisté, že tato metodika má propracovaný teoretický podklad (kompenzace mutace MTHFR, stimulace proteosyntézy acidum folicum a methioninem jako promotorickou aminokyselinou, prokazatelně nedostatek tkáňového materiálu u této vady, analogicky, jako je tomu u jiných rozštěpových vad, zejména rozštěpů neurální trubice). Rozštěpy obličejové doprovází často jev siamských dvojčat s rachischizou, vyskytujících se častěji v některých geografických oblastech jihovýchodní Asie, což svědčí pro nutrigenomické vlivy dané nedostatkem některých stopových prvků v půdě a ve vodě a potřebných vitaminů ve stravě.
Důležité je, aby farmakologická substituce byla aplikována v kritických a senzitivních periodách pro vznik CLP charakteristických (do 35. dne vývoje).
Závěr
CLP představuje významnou polygenní vadu a její problematika musí být řešena komplexně za spoluúčasti genetika, pediatra, stomatologa – zejména ortodonta, plastického chirurga a dětského chirurga. Význačnou pomoc představují v diagnostice zobrazovací metody, naděje se vkládají do multigenomiky v poznání etiologie a genové příčiny této polygenní vady. Vzhledem k relativní četnosti vady je nutné této tématice věnovat patřičnou pozornost.
MEDIÁNNÍ ROZŠTĚP (CP) – CLEFT PALATE
Mediánní rozštěp je odlišný od předchozího CLP rozštěpu dobou svého vzniku a anatomickou lokalizací.
Výskyt mediánního (zadního) rozštěpu je nerovnoměrný dle pohlaví, ze tří pacientů jsou nemocné dvě ženy. Je to opačný trend ve vztahu k pohlaví, než je u CLP. Mediánní rozštěp je méně častý než rozštěp rtu, patra a čelisti (CLP), vyskytuje se buď jako izolovaný (polygenní rozštěp) nebo jako součást syndromů, kde jeho výskyt může být jen fakultativní. Výskyt je těžko přesně určitelný, je proměnlivý v místě a čase (v různých etnických skupinách).
Etiologie
Stejně jako rozštěp rtu, patra a čelisti je podmíněn vznik CP polygenně (polyfaktoriálně).
Nejedná se o rozštěp tkání, ale o nedostatečnou proteosyntézu, nedostatek mezenchymální tkáně. Defekt je lokalizován ve střední čáře a řadí se proto k tzv. středočárovým defektům (defekty střední čáry, midline defekty). Jde o linii určující bilaterální symetrii těla (organismu). Součástí polygenního systému je m.j. gen MTHFR, pravděpodobně i řada dalších genů včetně tzv. homeotických určujících uspořádání axiální osy a tkání podél ní (páteř), ale také končetin.
Polovina případů postihuje pouze patro měkké (1, 2).
Patogeneze
Nedostatek mezenchymální tkáně v kritických a senzitivních periodách v embryogenezi do 55. dne po oplození vajíček vede k nespojení středočárových struktur. Genetická dispozice k této patologii se kombinuje často s teratogenními vlivy, které jsou součástí polygenního a polyfaktoriálního systému. Mírnější formy rozštěpu se týkají jen patra měkkého, resp. uvuly nebo submukózních struktur. Submukózní rozštěp znamená neúplné svalové spojení s intaktním povrchem mukózy (1, 2). Podmiňuje huhňavou řeč, diagnostika musí vzít v úvahu i palpaci. Je-li rozštěp rozsáhlejší, týká se i patra tvrdého.
Empirická rizika a dědičnost
Empirická rizika u izolovaného rozštěpu CP se řídí pravidly polygenní dědičnosti dle tzv. Edwardsova vzorce – což je druhá odmocnina populačního výskytu pro příbuzného 1. stupně.
Empirické riziko je podstatné ještě pro příbuzného 2. stupně a je zhruba poloviční než pro příbuzného 1. stupně. Pro příbuzné 3. stupně se prakticky blíží populačním hodnotám výskytu rozštěpu. Jde tedy o rychlý pokles empirického rizika se vzdálenějším stupněm příbuznosti.
MIKROFORMY ROZŠTĚPU
Jsou to rozštěpy uvuly typu uvula bifida a uvula bipartita. Jejich výskyt v rodinách, kde již je plně vyvinutý případ vady, má význam pro určování rizika. Odhad empirického rizika (ERČ) včetně mikroforem se řídí tabulkami empirických rizik.
CP jako součást syndromů (obr. 3)
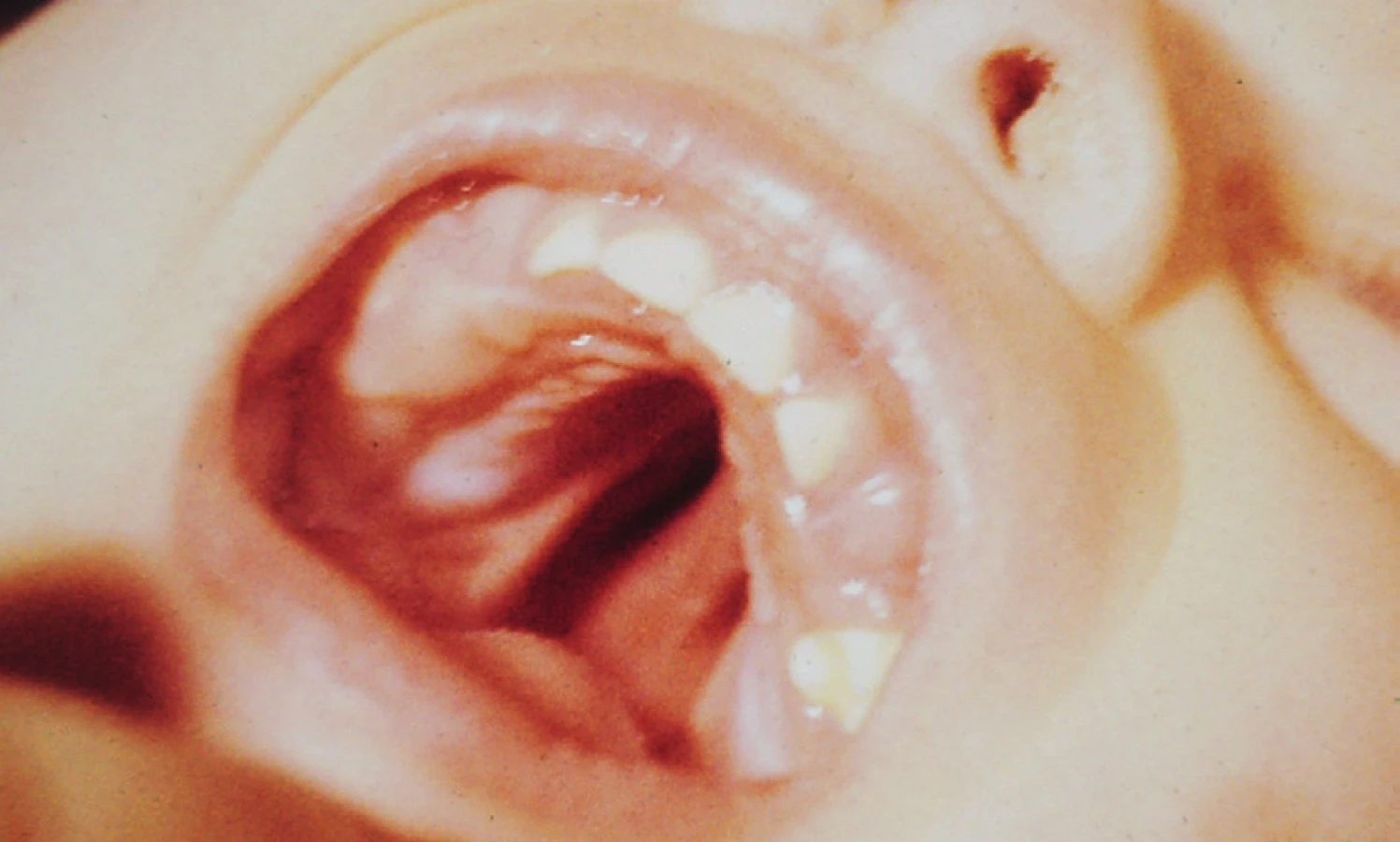
CP, které jsou součástí mendelovského syndromu (syndromologický rozštěp), mají podobně jako syndromologický rozštěp CLP dědičnost řídící se typem dědičnosti syndromu, jehož jsou součástí. CP je zde pouhým symptomem a jeho výskyt nemusí být vždy konstantní. Riziko mendelovské dědičnosti je pak konstantní, na rozdíl od polygenní dědičnosti nezávisí na počtu přítomných případů v rodokmenu.
Asociace rozštěpů
Defekty střední čáry se mohou asociovat, sdružovat. Existuje skutečně častá koincidence s defekty střední čáry. Existuje tzv. syndrom asociace rozštěpů CP + NTD, CP + rozštěpy končetin, poruchy septace (rozštěpy) v gynekologické oblasti (uterus septus, subseptus, bicornis, unicornis), uterus septus cum vagina septa, vagina duplex, rozštěp močové trubice, močového měchýře, stěny břišní. Duplicity spojené s rozštěpy a poruchami septace existují byť vzácně i u mužského pohlaví. Všechny tyto vady souvisejí s homeotickými geny. Od rozštěpů se prostřednictvím homeotických genů můžeme dostat i k problematice poruch končetin, rozštěpy končetin spojené s poruchami střední osy končetin. Homeotické geny určují, zda příslušné struktury jsou ve správný čas v embryogenezi na správném místě, a jsou tak společným jmenovatelem správného či nesprávného umístění struktur, tvarů a funkcí.
Poruchy septace souvisejí i s poruchami rotace orgánů a jejich zrcadlovým uspořádáním – situs viscerum inversus totalis, týkající se dutiny hrudní a břišní, či partialis týkající se pouze dutiny hrudní (dextrokardie s atypickou lobací plic), popř. pouze dutiny břišní (cave atypické umístění appendixu) či neúplná rotace známá u syndromu Ivemarkova a Kartagenerova. Bylo by jistě zajímavé sledovat vztah rozštěpů a končetinových vad u těchto poruch rotace orgánů. Končetinové vady u poruch rotace jsme popsali v roce 2004 (3).
Od mediánního rozštěpu k rozštěpům končetin (obr. 4a–c)


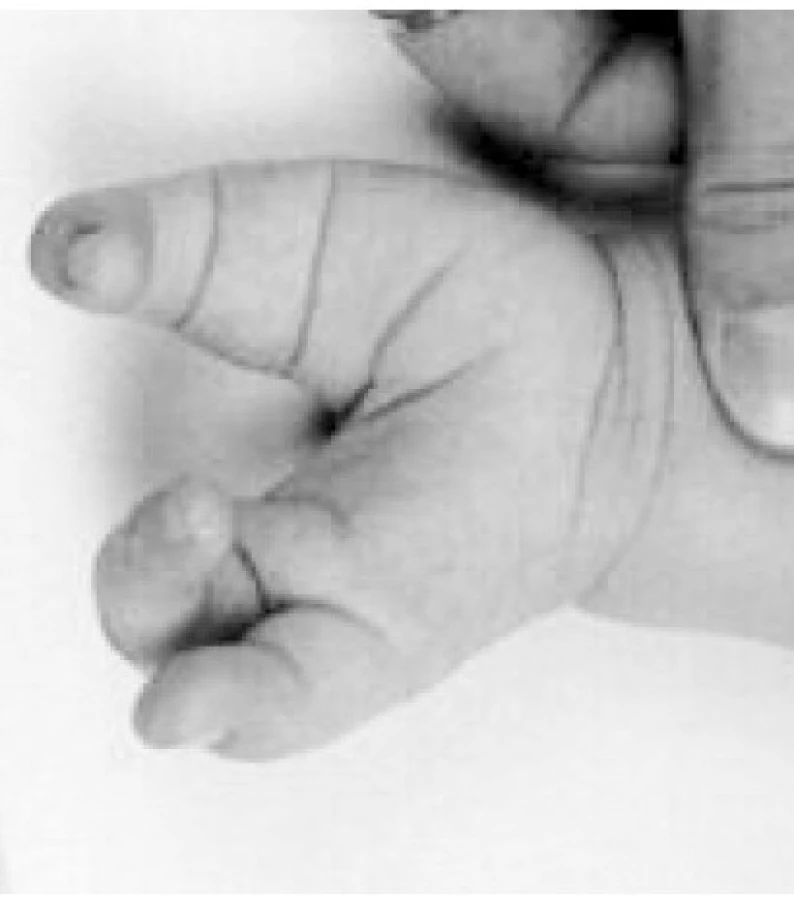
Jedná se o rozštěpy končetin v jejich uspořádání podél střední osy, souvislost je s homeotickými geny a rozštěpy páteře, tj. neurální trubice typu NTD. Rozštěpy končetin mají typickou redukci počtů paprsků podél střední osy končetiny, dle stranové predilekce jsou rozlišovány typ femur – tibie – radius s postižením uvedených oblastí a typ femur – fibula – ulna.
Vzácné typy obličejových rozštěpů
Patří k nim laterální obličejový rozštěp, začíná v koutku úst až k tragu boltce na stejné straně, jeho průběh může být do jisté míry variabilní (1, 2, 4). Dále je to šikmý obličejový rozštěp jako extrémně vzácný typ s dalšími variantami (5). Výklad těchto typů by byl již mimo rámec tohoto sdělení. Rozštěp mandibuly, rtu a jazyka ve střední čáře je extrémně řídký (1, 2).
Diferenciální diagnostika obličejových rozštěpů
Musí se vzít v úvahu velké množství syndromů, kdy jednotlivé typy rozštěpů jsou jejich pouhou součástí. Existuje asi 200 syndromů provázených rozštěpy (2) a jejich genetická prognostika a rizika rekurence jsou značně odlišné. Bizarní rozštěpy vyskytující se v nezvyklých variantách se nedají vždy vysvětlit embryologicky, ale jde o poruchy ektomezenchymu nejasného původu. Část se dá vysvětlit biomechanicky v souvislosti s amniovými pruhy (1, 2).
NEURAL TUBE DEFECT (NTD)
Rozštěpy páteře mají podobnou či stejnou etiopatogenezi jako mediánní rozštěpy. Rozdílné je načasování vzniku vady v embryogenezi z hlediska senzitivních a kritických period.
V lokalizaci rozštěpů, resp. neuzavírání neurální trubice, existuje tzv. kraniokaudální gradient, rozštěpy v kraniální oblasti a anencefalie vznikají časněji než ty, které jsou distálněji lokalizované.
Anencefalie
Nejtěžší a časnou formou NTD je anencefalie. Bývá často součástí blastopatie anencefalus + akardius – postihující dva zárodečné listy. Vady tohoto druhu jsou neslučitelné se životem a jsou součástí zamlklých abortů. Nejextrémnějším příkladem neurálního tubárního defektu je kraniorachischiza = totální dysrafismus, kdy mozek i páteřní kanál jsou kompletně otevřené. Kalva chybí, pouze bazální části frontální, parietální a okcipitální kosti jsou přítomny. Postižení CNS je variabilní – hemisféry a mozeček jsou rudimentární a mohou úplně chybět. Zbývající mozková tkáň je smíšená s angiomatózní tkání. Orbity jsou zmenšené, s protruzí bulbů. Poškození obličejového skeletu je variabilní a tvrdé patro je malformováno (2). Duplicita obličeje je přítomna v mnoha případech. Malformace břišní a hrudní stěny je přítomna velmi často, stejně tak anomálie gastrointestinálního traktu, ledvin a genitálií. Kombinace se spina bifida occulta, pilonidálními cystami a skoliózou v určitých případech je přítomna též. Sítnice je patologicky změněna (koloboma, angiomatózní proliferace) (2), optický nerv je neúplný a hypoplastický. Srdce, plíce, ledviny a nadledviny jsou hypoplastické. Přední lalok hypofýzy chybí v 25 % případů.
Výskyt
Vyskytují se ve všech etnických skupinách a rasách, často u tzv. siamských dvojčat (srostlice), jejichž výskyt je největší v jihovýchodní Asii. NTD je relativně častý (common disease) a zároveň je obtížné určit jeho skutečnou frekvenci, neboť se mnohdy nachází u spontánních abortů. Setkali jsme se s kazuistickými popisy, kdy u spontánně časně potracených plodů jsou popisovány meningomyelokély (MMC) velikosti hrášku v LS oblasti.
NTD je častou a závažnou vadou, avšak v dnešní době více než v minulosti chirurgicky léčitelnou v závislosti na tíži a stupni (rozsahu) vady.
Vznik NTD je proměnlivý v čase a prostoru, jsou výrazné geografické závislosti. Ve Spojených státech amerických je frekvence 1 : 1000 živě narozených (2, 6, 7), v izolovaných oblastech Irska a Walesu bývá incidence uváděna 5–7 : 1000 živě narozených.
Pro rozlišení teratogenně vzniklých rozštěpů je nutná pečlivá gynekologická anamnéza těhotenská s rozborem všech rizikových faktorů, důležitá je jejich časová lokalizace. Pátrání po možných příčinách se podobá často práci kriminalistiky.
Příbuzná afekce ve střední čáře skeletu kalvy – encefalokéla se vyskytuje vzácněji než meningomyelokéla. Encefalokéla anterior je nejčastější v Africe, Thajsku a Indii. Okcipitální encefalokéla se vyskytuje především u žen.
Etiopatogeneze
Polygenní determinace, nedostatečná saturace acidum folicum, přítomnost mutací MTHFR se zřetelně uplatňují, stejně tak hyperpyrexie v kritické a senzitivní periodě, časová lokalizace vzniku vady v době těhotenství. Vady vznikající časněji jsou lokalizovány kraniálně, vady vznikající v embryogenezi později jsou lokalizovány kaudálně. Kraniorachischiza se reprezentuje jako nejčasnější porucha neurální trubice a spadá do období mezi 17.–23. dnem po početí. Samotná anencefalie je etiopatogeneticky zařazena mezi 23. a 26. den embryonálního života jako porucha kraniálního neuroporu (2, 6, 7).
Etiopatogeneticky příbuzná, ale odlišná je středočárová porucha zvaná encefalokéla (2, 8–10). Může mít lokalizaci occipitální v 75 %, parietální v 10 % nebo přední v 15 %. Etiopatogeneze je méně známa než u NTD.
Empirické riziko a dědičnost NTD
Empirické riziko vychází z polygenní (polyfaktoriální) determinace a ze zmiňovaného Edwardsova vzorce.
Při určování empirického rizika nesmíme zapomínat na eventuální přítomnost minimálních projevů rozštěpu u rodičů v LS oblasti. Je to tzv. spina bifida occulta týkající se většinou L1, L2 obratlů – jednoho nebo dvou obratlů, popř. se jedná o canalis sacralis apertus kosti křížové. Tyto vady představují plynulý přechod od variant k patologii. L1, L2, rozštěpy bývají často náhodným nálezem u osob bez další genetické zátěže. Tento malý rozštěp sám o sobě představuje relativně neškodný epigenetický znak. Avšak v souvislosti s rozštěpy NTD musí být považován za minimální symptomatologiii zvyšující empirické riziko. Nositel spina bifida occulta kryté kůží může mít kožní korelát v podobě vlasového víru či vpadliny, nebo jiné kožní projevy (tzv. dimples) v inkriminované oblasti jako signální fenotyp ještě před RTG vyšetřením. Minimální symptomatologie NTD si všímá i biologicky orientovaná psychiatrie ve vztahu k enuréze (ve spojení s poruchami příslušného segmentu inervace močového měchýře). Osoby s poruchami minimální symptomatologie NTD trpí v dětství častěji enurézou než osoby bez této symptomatologie a častěji v dospělosti mají problémy se závislostmi na alkoholu, tabáku či jinými návykovými látkami spadajícími do problematiky tzv. AT ordinací (alkoholismus a tabakismus) – vlastní pozorování. Tyto minimální projevy jsou v populaci ve 4–8 % (více pro neuzavřený canalis sacralis apertus). Uzavírání oblouků křížové kosti nastává fyziologicky až po 10. roce života. Proto je při hodnocení těchto minimálních rozštěpů (dysrafismů) nutná opatrnost u mladších jedinců a vyčkat v hodnocení v příslušném věku.
U jednoho z rodičů či sourozenců dětí s NTD (příbuznost 1. stupně) minimální NTD = dysrafismus je přítomen ve více než 60–90 %. Tento jev je nutné zakalkulovat do výpočtu empirického rizika rekurence pro další těhotenství (potomka či sourozence). Vícečetný výskyt NTD v rodině znamená vyšší riziko rekurence a je nutné je zvažovat i pro širší příbuzenstvo, než je 1. a 2. stupeň – jak je tomu u ojedinělého (izolovaného) výskytu. Jestliže jsou v rodině dva postižení sourozenci, pak riziko opakování činí 20–25 % (2, 11).
Asociace rozštěpů a poruch septace již byla zmíněna (CP, NTD, rozštěp ruky či nohy). Rozsáhlý rozštěp páteře se nazývá rachischiza.
Dysrafismus souvisí s poruchami segmentace a poruchy tohoto typu s homeotickými geny, které určují, aby příslušné struktury v embryogenezi byly ve správný čas na správném místě.
Existují také syndromologické NTD (obr. 5) – u poruch segmentace typu Klippelova-Feilova syndromu, u syndromů mendelovského i polyfaktoriálního charakteru. Anencefalokela spojená s acardiem je blastopatií neslučitelnou se životem a projevuje se jako missed abortion.
Existují též syndromologické středočárové encefalokély v rámci syndromů, kdy riziko výskytu je pak dáno zpravidla mendelovskou dědičností syndromu, jehož je encefalokéla součástí.
Diagnostika
Diagnostika je možná prenatálně. Především zobrazovací ultrazvukové metody učinily velký pokrok. Zobrazovací metody nukleární magnetické rezonance a UZ odhalí na rozdíl od biochemické diagnostiky i NTD kryté kůží. Dnes již nedochází k selhání diagnózy UZ jako v jejích počátcích v sedmdesátých a osmdesátých letech 20. století. Dnes je již neuvěřitelné, že byly nerozpoznány a přehlédnuty diagnózy anencefalie zobrazovacími metodami včetně selhání diagnostiky pomocí alfa1-fetoproteinu (AFP). Selhání biochemie je možné i dnes a biochemická diagnostická metoda (6, 7) je vytlačována a doplňována zobrazovacími metodami. U syndromologické diagnostiky využíváme možnosti diagnostiky vrozených chromozomálních aberací či určení genového defektu základní diagnózy. Diagnostika encefalokély je obdobná. Setkali jsme se s relativně benigním případem okcipitální encefalokély u dítěte s normálním vývojem s defektem rozměru 2 × 2 cm, řešení bylo neurochirurgickou protetikou – arteficiálním krytem kalvy destičkou.
Terapie
U NTD pokročila zejména terapie chirurgická, úspěch závisí především na rozsahu vady. Od konce padesátých let 20. století dětská chirurgie učinila význačné pokroky v této oblasti a setkáváme se s řadou dospělých. Významným způsobem tak poklesl počet osob inkontinentních a odkázaných na invalidní vozík. Úspěch chirurgického zákroku závisí samozřejmě na rozsahu vady a prevenci infekčních komplikací. Je proto velmi důležité sterilní krytí rozštěpu fyziologickým roztokem po porodu a ošetřování ranné plochy.
Jsou zaznamenávány také snahy o fetální chirurgické zákroky v této oblasti, u plodů, kde je tato vada zjištěna. Předcházely jim také experimenty na zvířatech (primáti).
Prevence
Preventivní opatření lze analogicky jako u orofaciálních rozštěpů rozdělit na primární a sekundární. Primární prevence vychází z poznatků o etiopatogeneze a ze znalosti kritických vývojových period. Prevence spočívá opět v suplementaci listovou kyselinou, neboť na etiopatogenezi rozštěpů obecně se podílí v polygenním systému některé mutace genu pro metylentetrahydrofolát zvyšující spotřebu kyseliny listové v organismu. Nedostatečný přísun acidum folicum, methioninu a vitaminu B12 a dalších působků snižuje u disponovaných jedinců proteosyntézu a potencuje vznik rozštěpu.
Zkratky
AFP alfa1-fetoprotein
CLP rozštěp rtu, patra a čelisti (cheilognatopalatoschizis)
CL(P) mírná forma rozštěpu rtu a patra s minimálním podílemrozštěpu patra
CP mediánní rozštěp
ERČ empirické riziko
MMC meningomyelokéla
MTHFR metylentetrahydrofolátreduktáza
NMR, MRI nukleární magnetická rezonance
NTD neural tube defect
UZ ultrazvukové vyšetření
ADRESA PRO KORESPONDENCI:
MUDr. Miloslav Kuklík, CSc.
Genetické oddělení
Olšanská 7, 130 00 Praha 3
e-mail: honza.kuklik@volny.cz
Sources
1. Lynch HT, Kimberling WJ. Genetic counseling in cleft lip and cleft palate. Plast Reconstr Surg 1981; 68 : 800–815.
2. Goodman RM, Gorlin RJ. The malformed infant and child. Oxford: University Press 1983.
3. Kuklík M, Mařík, I., First, T. Syndromy spojené s poruchou rotace orgánů – situs viscerum inversus. Pohybové ústrojí 2004; 11 : 78–90.
4. Boo-Chai K. The transverse facial cleft: its repair. Br J Plast Surg 1969; 22 : 119–124.
5. Boo-Chai K. The oblique facial cleft? A report of two cases and a review of 41 cases. Br J Plast Surg 1970; 23 : 352–359.
6. Crandall BF, Lebherz TB, Freihube R. Neural tube defects: maternal serum and prenatal diagnosis. Pediatr Clin N Am 1978; 254 : 619–629.
7. Milunski A, Alpert E. Prenatal diagnosis of neural defects. Obstet Gynecol 1976; 48 : 1–5, 1976; 49 : 6–12, 1977; 49 : 532–536, 1980; 55 : 1980; 60–66.
8. Cohen MM, Jr, Lemire RJ. Syndromes with cephaloceles. Teratology 1982; 25 : 161–172.
9. Harverson G, Bailey IC, Kiryabwire JWM. The radiological diagnosis of anterior encephaloceles. Clin Radiol 1974; 25 : 317–322.
10. Mc Laurin RL. Parietal cephaloceles. Neurology (Minneap.) 1964; 14 : 764–772.
11. Pietrzyk JJ. Neural tube malformations: complex segregation analysis and recurrence risk. Am J Med Genet 1980; 7 : 293–300.
Labels
Addictology Allergology and clinical immunology Angiology Audiology Clinical biochemistry Dermatology & STDs Paediatric gastroenterology Paediatric surgery Paediatric cardiology Paediatric neurology Paediatric ENT Paediatric psychiatry Paediatric rheumatology Diabetology Pharmacy Vascular surgery Pain management Dental HygienistArticle was published in
Journal of Czech Physicians

- Advances in the Treatment of Myasthenia Gravis on the Horizon
- Possibilities of Using Metamizole in the Treatment of Acute Primary Headaches
- Metamizole at a Glance and in Practice – Effective Non-Opioid Analgesic for All Ages
- Metamizole vs. Tramadol in Postoperative Analgesia
- Spasmolytic Effect of Metamizole
Most read in this issue
- Rozštěpové vady
- Dědičnost ortodontických anomálií
- Studie na dvojčatech ve stomatologii
- Ortodontické anomálie a sekulární trend v utváření orofaciální oblasti