
Molekulární patologie cholangiocelulárních nádorů
Molecular pathology of cholangiocellular carcinomas
Cholangiocellular carcinoma is a relatively rare malignant tumor, originating from cholangiocytes, with poor prognosis and late diagnosis. It is a malignancy with a variable biological etiology, numerous genetic and epigenetic changes. Its incidence in the Czech Republic is about 1.4 per 100,000 people per year.
For good prognosis and long-term survival, early diagnosis with surgical treatment is important. In these cases, a 5-year survival rate is about 20-40 %. In the early diagnosis imaging methods and histopathological verification play an essential role, whereas laboratory oncomarkers are not yet sufficiently accurate. The same applies for genetic markers. This leads to the search of new molecular targets and the high effort in the introduction of cytological and molecular-biological methods with high specificity and sensitivity into routine practice. Current early diagnosis is based on the use of efficient imaging methods.
The use of genetic testing, and especially knowledge of the molecular basis of this disease, will be of a great benefit. The observation of the association between the genetic pathways, IDH1, RAS-MAPK etc., and genetic mutations of genes, such as TP53, KRAS, SMAD4, BRAF, IDH1/2, may be significant. From the molecular point of view, it is also interesting to monitor oncogenic potential in HBV/HCV infection.
Keywords:
cholangiocellular carcinoma – oncomarkers – mutation of genes – KRAS – p53
Authors:
Libor Staněk 1,2,3; Robert Gürlich 1; Jan Hajer 4; Martin Oliverius 1; Renata Soumarová 5 5
Authors‘ workplace:
Chirurgická klinika 3. LF UK a FN Královské Vinohrady, Praha
1; Ústav histologie a embryologie 1. LF UK v Praze
2; Vysoká škola zdravotnictví a sociální práce sv. Alžběty v Bratislavě
3; 2. interní klinika 3. LF UK a FN Královské Vinohrady, Praha
4; Onkologická klinika 3. LF UK a FN Královské Vinohrady, Praha
5
Published in:
Čas. Lék. čes. 2019; 158: 64-67
Category:
Review Article
Overview
Cholangiocelulární karcinom je poměrně vzácný maligní nádor vycházející z cholangiocytů, se špatnou prognózou a zpravidla pozdně stanovenou diagnózou. Jedná se o malignitu s různorodou biologií a řadou genetických i epigenetických změn. Jeho incidence v ČR činí asi 1,4 případu na 100 000 obyvatel za rok.
Pro příznivou prognózu a naději na delší přežití je důležitá časná diagnostika s chirurgickým řešením. V těchto případech dosahuje 5letého přežití asi 20–40 % pacientů. V časné diagnostice hrají zásadní roly zobrazovací metody a histopatologická verifikace; laboratorní onkomarkery zatím nejsou dostatečně vypovídající. Totéž platí také o genetických markerech, což vede k hledání nových molekulárních cílů a zavedení cytologických a molekulárně biologických metod s vysokou specificitou a senzitivitou do rutinní praxe. Současná racionální časná diagnostika spočívá v účelném využití zobrazovacích metod.
Využití genetického testování, a hlavně znalost molekulární podstaty tohoto onemocnění bude velkým přínosem. Jako významné se jeví sledování asociace s genetickými dráhami IDH1 a RAS-MAPK a dále sledování genetických mutací genů TP53, KRAS, SMAD4, BRAF, IDH1/2 a dalších. Z molekulárního hlediska nelze podcenit také onkovirový potenciál infekce HBV/HCV.
Klíčová slova:
cholangiocelulární karcinom – onkomarkery – mutace genů – KRAS – p53
EPIDEMIOLOGIE A DIAGNOSTIKA
Cholangiocelulární karcinom (CCC) je vzácné nádorové onemocnění, které nemá příliš dobrou prognózu. Histologicky vychází ze stěny žlučovodů, může vzniknout z malých žlučových kanálků, jež se nacházejí uvnitř jater, nebo z velkých žlučovodů mimo jaterní tkáň. Rozlišujeme dva podtypy, a to Klatskinův tumor, který vzniká v oblasti ductus choledochus a představuje více než 90 % CCC, a ampulom vznikající ze společného žlučovodu v oblasti ústí do tenkého střeva, tzv. Vaterově papile.
Mezi rizikové faktory pro vznik CCC patří genetické predispozice, životní styl (konzumace alkoholu, syrových sladkovodních ryb, užívání tabáku), expozice látkám z vnějšího prostření (dioxinu, vinylchloridu a dalším) a jejich interakce. Rizikovým faktorem může být infekce virem HBV/HCV, chronické biliární a jaterní onemocnění jako primární sklerotizující cholangitida, přítomnost žlučových kamenů, vrozené biliární malformace a cirhóza. Riziko vzniku CCC zvyšuje také diabetes mellitus. Rozmanitost těchto rizikových faktorů na všech výše uvedených úrovních činí z CCC velmi heterogenní onemocnění.
V diagnostice hrají zásadní roli zobrazovací metody a histopatologická verifikace a hledají se genetické markery. V tomto kontextu byla provedena retrospektivní studie na kohortě 66 pacientů s chirurgicky resekovaným CCC. Předoperační CT snímky byly hodnoceny radiology a porovnávány s genetickou analýzou a klinickými daty pacientů (1). Sledována byla asociace mezi výsledkem CT vyšetření a genetickými drahami (IDH1 a RAS-MAPK). Studie definovala tři kritéria, která jsou spojena s vyšším rizikem úmrtí: nekrózu (poměr rizik [HR] 2,95; 95% interval spolehlivosti [CI] 1,44–6,04; p = 0,029), satelitní uzly (HR 3,29; 95% CI 1,35–8,02; p = 0,029) a zvýšenou vaskularizaci (HR 2,63; 95% CI 1,28–5,41; p = 0,029). Ovšem mezi těmito kritérii a genovými změnami v drahách IDH1 a RAS-MAPK nebyly nalezeny žádné významné souvislosti (p = 0,63–0,84). Práce tedy naznačuje asociaci mezi CT zobrazením a celkovým přežitím, nebyla však zjištěna žádná souvislost mezi zobrazovacími metodami a definovanými genetickými drahami.
V histologické verifikaci je CCC charakterizovaný tubulárními strukturami (CK7 a CK19-pozitivními) a nápadnou fibroprodukcí, na rozdíl od klasického HCC (pozitivní Hep Par-1), který až na fibrolamelární variantu nemá vazivové stroma (2). CCC má nepříznivou biologickou povahu a obecně jeho diagnóza představuje ve většině center kontraindikaci k transplantaci jater.
TERAPIE
V případě CCC výsledky léčby stále nejsou uspokojivé. Jedinou potenciálně kurabilní metodou je chirurgická resekce, avšak pouze malá část nemocných je indikovaná k radikálnímu výkonu. Mikroskopicky negativní okraje po R0 resekci patří mezi nejdůležitější prognostické faktory dlouhodobého přežívání. Většina pacientů je ovšem odkázána na paliativní léčbu, jejímž cílem je zajištění celkové kvality života nemocných s CCC (3). Součástí komplexní léčby je adjuvantní chemoterapie a radioterapie. Zlepšení výsledků léčby a prognózy nemocných s CCC lze v budoucnu očekávat od molekulární diagnostiky a rozšíření povědomí o molekulárních mechanismech tohoto onemocnění s následným zavedením nových terapeutických monoklonálních protilátek či inhibitorů tyrosinkináz (4) (obr. 1).
(převzato z: American Association for Cancer Research, 2016)
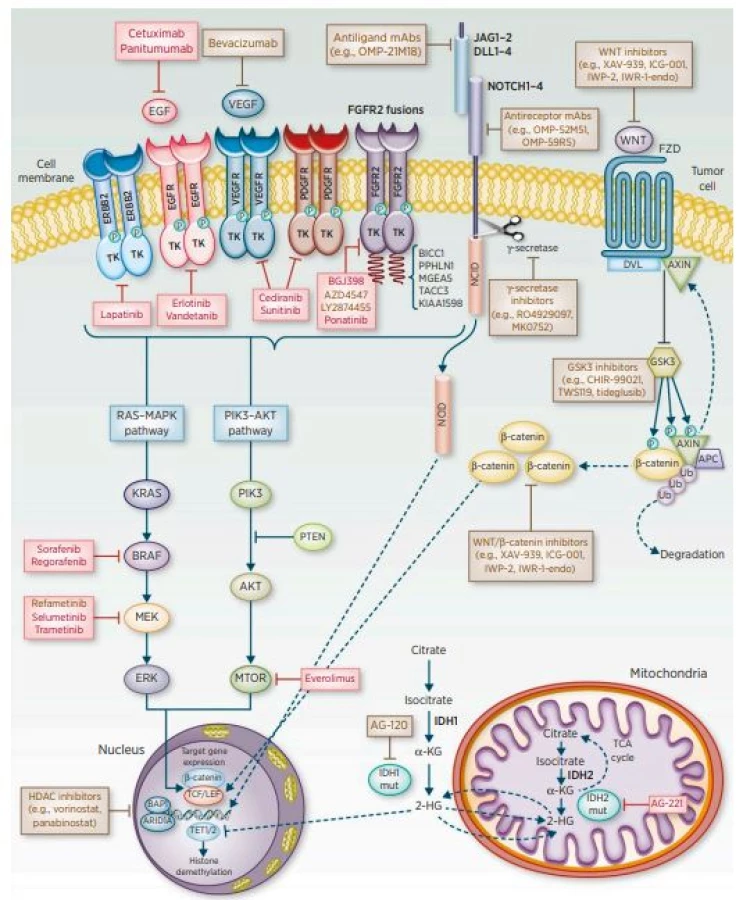
Ve snaze identifikovat nové terapeutické postupy byla čínskou skupinou publikována práce sledující účinky kantaridinu (CAN), možného inhibitoru serin/threoninové proteinové fosfatázy 2A (PP2A) (5). Farmakologické účinky kantaridinu a některých jeho derivátů jsou založeny na inhibici katalytické jednotky serin/threoninových proteinových fosfatáz typu 1 a 2A (6) přes významnou signální dráhu VEGFR2/MEK/ERK. V této studii byl inhibiční účinek kantaridinu sledován na dvou buněčných liniích (QBC939 a Hucc-t1) (5). Po aplikaci kantaridinu bylo pozorováno zvýšení koncentrací reaktivních forem kyslíku (ROS) u buněk QBC939. V obou případech kantaridin významně inhiboval migraci buněk a jejich invazi v závislosti na dávce. Bylo tedy prokázáno, že kantaridin selektivně a účinně inhibuje migraci a invazi buněk CCC, což může poskytnout nový pohled na použití CAN v léčbě CCC.
Snahou zůstává nalezení nových molekulárních cílů, definování genetických a také epigenetických změn pro využití diagnostické, ale hlavně terapeutické.
GENETIKA
Omezené terapeutické možnosti vyplývají jednak z biologické podstaty CCC, jednak z jeho heterogenity zahrnující jak genetické, tak epigenetické změny. V kontextu značného počtu genetických změn začíná být v rámci translačního výzkumu významná aplikace bioinformatiky, která je hybnou silou výzkumu biologie nádoru. Jejím úkolem je zpracování velkého množství dat získaných vysoce sofistikovanými metodami molekulární diagnostiky. Cílem je podpora interdisciplinární komunikace a výzkumu také u CCC (7). Získaná data pomohou k budování databází sloužících k přesné diagnostice a při rozhodování o prognóze i léčbě tohoto onemocnění.
CCC vykazuje značné fenotypové rozdíly v důsledku bodových mutací KRAS a změn genu TP53 a dalších. Následkem těchto genetických změn dochází k premaligní biliární intraepitelové neoplazii (BiIN) s následným vznikem CCC. Dále může dojít k přímému vzniku fenotypu CCC z onkogenně změněných primárních hepatocytů v důsledku ztráty TP53. Tímto mechanismem se hepatocyty mění na cholangiocyty (obr. 2) (8). Genetické mutace tak určují cestu progrese a výsledný fenotyp CCC.
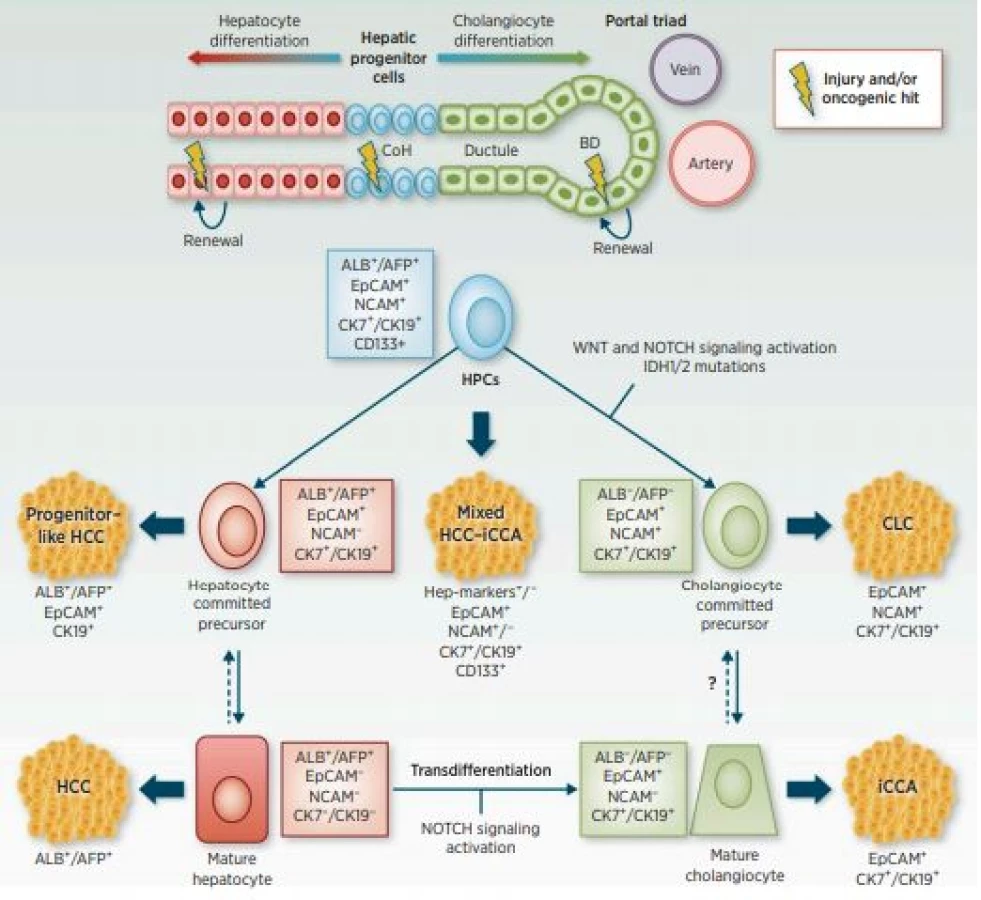
Mimo mutací v genech TP53 a KRAS jsou u CCC často detekovány mutace v genech CTNNB1, ARID1A, PBRM1 a IDH1. Mutace genů RYR3, FBN2 a KCNN3 jsou spojovány s migrací buněk a metastazováním nádoru (9). Mutace IDH1 koreluje s lepší prognózou a snížením růstu nádoru tím, že potlačuje signalizaci Akt. Pacienti s mutací IDH1 by mohli být indikovaní k méně agresivní terapii, v budoucnu i pro terapii přizpůsobenou jejich mutačnímu stavu (10). Časté jsou také ztráty exprese SMAD4 a amplifikace MDM2 (11). Sekvenční technologie odhalily přítomnost rekurentních nových fúzních genů (fúze FGFR2 a ROS1) a somatických mutací v metabolických (IDH1/2) a chromatin remodelačních genech (ARID1A, BAP1). Tyto změny spolu s mutacemi v genech pro KRAS/BRAF/EGFR a amplifikace 11q13 přispěly k lepšímu porozumění molekulárním změnám CCC (12, 13). Na základě genové exprese obsahující jednonukleotidové polymorfismy byly identifikovány další signální dráhy aktivované u CCC.
Dosud byly identifikovány 2 hlavní biologické třídy CCC. První je charakterizována expresí cytokinů a aktivací dráhy STAT3. Druhá třída je charakterizována aktivací onkogenních signalizačních drah (MAPK – mitogenem aktivované proteinkinázy a MET), amplifikací v oblasti 11q13.2, delecí 14q22.1, mutacemi v genech KRAS a BRAF. Tato třída je spojena s horší prognózou a kvůli své molekulární charakteristice bude vyžadovat odlišné přístupy k léčbě (14).
Jako další možný molekulární cíl se jeví epigenetické změny. Je popisována vysoká exprese histonu Z z rodiny H2A (H2A.Z) jakožto vysoce rizikového faktoru špatné prognózy pacientů s CCC. Nicméně mechanismus exprese H2A.Z zatím zůstává neobjasněný. Recentní studie čínských autorů prokázala, že H2A.Z je u CCC nadměrně exprimován a koreluje se špatnou prognózou (15). H2A.Z reguluje buněčnou proliferaci prostřednictvím signalizace proteinu 2/p27/p21 souvisejícího s kinázou H2A.Z/S-fáze. Inhibice H2A.Z snížila proliferaci buněk a vyvolala apoptózu u CCC. Dále downregulace H2AZ snížila tvorbu metastáz potlačením epitelově-mezenchymové tranzice a zvýšila protinádorové účinky cisplatiny při léčbě CCC. Celkově výsledky naznačují, že H2A.Z může být nový perspektivní biomarker a potenciální terapeutický cíl u CCC.
Aberantní methylace DNA (16) jsou u CCC poměrně časté, méně je však známo o jejich vzniku. V recentní studii bylo sledováno 10 kandidátních genů zapojených do oprav DNA (PPP4C – protein phosphatase 4 catalytic subunit), apoptózy (RUNX3 – runt-related transcription factor 3; IRF4 – interferon regulatory factor 4; UCHL1 – ubiquitin C‑terminal hydrolase L1; TP53I3 – tumor protein p53 inducible protein 3; CCND2 – cyclin D2; RASSF1 – Ras association domain family member 1), metabolismu (ALDH1A3 – aldehyde dehydrogenase 1 family member A3; SLC29A1 – solute carrier family 29 member 1) a konečně do angiogeneze (HTATIP2 – human immunodeficiency virus 1 tat interactive protein 2). Vysoká míra methylace byla pozorována u UCHL1, IRF4, CCND2, HTATIP2 a TP53I3. Naopak v normální tkáni byla identifikována nízká methylace u HTATIP2 a UCHL1. Pacienti s CCC mající zvýšenou methylaci HTATIP2 a nízkou methylaci UCHL1 vykazovali delší celkové přežití. Současně byla prokázána methylace UCHL1 jako nezávislého faktoru pro CCC s poměrem rizik (HR) 1,81 (95% CI 1,01–3,25). Kombinace methylačního stavu HTATIP2 a UCHL1 se tak jeví jako potenciální prediktivní biomarker.
Pomocí molekulárně cytogenetických metod byla u CCC zaznamenána delece v oblasti 3p21 genu BAP1 důležitého při tvorbě chromatinu, což naznačuje, že BAP1 může hrát roli v patogenezi těchto nádorů (17). Ztráta BAP1 je popisována u více než poloviny CCC. Ztráta heterozygozity v oblasti 3p21 je popisována u 75 % CCC. Exprese markerů hepatocytů (Hep Par-1) a žlučovodů (cytokeratin 7) je častá u CCC za současné ztráty oblasti 3p21. Tyto chromosomové změny jsou detekovány u CCC na rozdíl od hepatocelulárního karcinomu (HCC), což by naznačovalo možnost jejich využití jako potenciálního diagnostického markeru.
V rámci výzkumu nádorů, CCC nevyjímaje, se do popředí pozornosti dostávají krátké kruhové nekódující ribonukleové kyseliny (ncRNAs), které je možné identifikovat v genomových sekvenčních studiích. I když je lze snadno odhalit, jejich regulace a funkce u nádorových onemocnění doposud není přesně objasněna.
ZÁVĚR
Zlepšení výsledků léčby a prognózy nemocných s CCC lze v budoucnu očekávat od zlepšení resekability, ale hlavně časné diagnostiky a využití nových terapeutických možností na základě molekulární diagnostiky a subtypizace onemocnění. Stejně jako u ostatních nádorových onemocnění bude i u CCC velice důležitý přístup personalizované medicíny.
Poděkování
Podpořeno Výzkumným projektem Univerzity Karlovy PROGRES Q28 (Onkologie).
Práce vznikla za podpory grantu ze strukturálních fondů EU OPP Konkurenceschopnost „Centrum integrované intenzivní péče“ CZ2.16/3.1.00/21533.
Seznam zkratek
BiIN biliární intraepitelová neoplazie
CCC cholangiocelulární karcinom
CI interval spolehlivosti
DNA deoxyribonukleová kyselina
HBV virus hepatitidy B
HCC hepatocelulární karcinom
HCV virus hepatitidy C
HR poměr rizik
MAPK mitogenem aktivovaná proteinkináza
ncRNA nekódující ribonukleová kyselina
Adresa pro korespondenci:
RNDr. Ing. Libor Staněk
Chirurgická klinika 3. LF UK a FNKV
Šrobárova 50, 100 34 Praha 10
Tel.: 732 760 265
e-mail: stanek.libor@seznam.cz
Sources
- Aherne EA, Pak LM, Goldman DA et al. Intrahepatic cholangiocarcinoma: can imaging phenotypes predict survival and tumor genetics? Abdom Radiol 2018; 43(10): 2665–2672.
- Honsová E. Histopathological diagnosis of hepatocellular carcinoma. Gastroent Hepatol 2012; 66(2): 93–98.
- Bělina F. Hilový cholangiokarcinom (Klatskinův tumor) – současné možnosti léčby. Rozhl Chir 2013; 92(1): 4–15.
- Rydlo M, Dvořáčková J, Kupka T et al. The rational diagnostic of cholangiocarcinoma. Vnitř Lék 2016; 62(2): 125–133.
- Zhou H, Xu J, Wang S, Peng J. Role of cantharidin in the activation of IKKα/IκBα/NF-κB pathway by inhibiting PP2A activity in cholangiocarcinoma cell lines. Mol Med Rep 2018; 17(6): 7672–7682.
- Patočka J, Kuča K. Kantaridin: přírodní bioaktivní molekula s dlouhou historií. Kontakt 2013; 15 : 463–469.
- Qian F, Guo J, Jiang Z, Shen B. Translational Bioinformatics for Cholangiocarcinoma: opportunities and Challenges. Int J Biol Sci 2018; 22; 14(8): 920–929.
- Hill MA, Alexander WB, Guo B et al. KRAS and Tp53 mutations cause cholangiocyte - and hepatocyte-derived cholangiocarcinoma. Cancer Res 2018; 15; 78(16): 4445–4451.
- Zhong W, Dai L, Liu J, Zhou S. Cholangiocarcinoma‑associated genes identified by integrative analysis of gene expression data. Mol Med Rep 2018; 17(4): 5744–5753.
- Wang J, Zhang ZG, Ding ZY et al. IDH1 mutation correlates with a beneficial prognosis and suppresses tumor growth in IHCC. J Surg Res 2018; 231 : 116–125.
- Moeini A, Sia D, Bardeesy N et al. Molecular pathogenesis and targeted therapies for intrahepatic cholangiocarcinoma. Clin Cancer Res 2016; 15; 22(2): 291–300.
- Liu ZH, Lian BF, Dong QZ et al. Whole-exome mutational and transcriptional landscapes of combined hepatocellular cholangiocarcinoma and intrahepatic cholangiocarcinoma reveal molecular diversity. Biochim Biophys Acta Mol Basis Dis 2018; 1864 : 2360–2368.
- Akita M, Sofue K, Fujikura K et al. Histological and molecular characterization of intrahepatic bile duct cancers suggests an expanded definition of perihilar cholangiocarcinoma. HPB (Oxford) 2019; 21(2): 226–234.
- Sia D, Hoshida Y, Villanueva A et al. Integrative molecular analysis of intrahepatic cholangiocarcinoma reveals 2 classes that have different outcomes. Gastroenterology 2013; 144(4): 829–840.
- Yang B, Tong R, Liu H, et al. H2A.Z regulates tumorigenesis, metastasis and sensitivity to cisplatin in intrahepatic cholangiocarcinoma. Int J Oncol 2018; 52(4): 1235–1245.
- Nanok C, Jearanaikoon P, Proungvitaya S, Limpaiboon T. Aberrant methylation of HTATIP2 and UCHL1 as a predictive biomarker for cholangiocarcinoma. Mol Med Rep 2018; 17(3): 4145–4153.
- Mosbeh A, Halfawy K, Abdel-Mageed WS et al. Nuclear BAP1 loss is common in intrahepatic cholangiocarcinoma and a subtype of hepatocellular carcinoma but rare in pancreatic ductal adenocarcinoma. Cancer Genet 2018; 224–225 : 21–28.
- Moirangthem A, Wang X, Yan IK, Patel T. Network analyses-based identification of circular ribonucleic acid-related pathways in intrahepatic cholangiocarcinoma. Tumour Biol 2018; 40(9): 1010428318795761.
Labels
Addictology Allergology and clinical immunology Angiology Audiology Clinical biochemistry Dermatology & STDs Paediatric gastroenterology Paediatric surgery Paediatric cardiology Paediatric neurology Paediatric ENT Paediatric psychiatry Paediatric rheumatology Diabetology Pharmacy Vascular surgery Pain management Dental HygienistArticle was published in
Journal of Czech Physicians

2019 Issue 2
- Advances in the Treatment of Myasthenia Gravis on the Horizon
- Possibilities of Using Metamizole in the Treatment of Acute Primary Headaches
- Metamizole at a Glance and in Practice – Effective Non-Opioid Analgesic for All Ages
- Metamizole vs. Tramadol in Postoperative Analgesia
- Spasmolytic Effect of Metamizole
Most read in this issue
- Chirurgická léčba cholangiocelulárního karcinomu
- Patologie cholangiocelulárního karcinomu
- Epidemiologie zhoubných nádorů žlučníku a žlučových cest v České republice
- Umělá inteligence a moderní informační a komunikační technologie vstupují do medicíny