
Molekulárně genetický pohled na podstatu buněčné senescence v kontextu stárnutí organismu
The molecular genetics of cellular senescence in the context of organismal aging
Cellular senescence is a physiological state generally defined as a stable arrest of proliferation by preventing the cells from cycling. Unlike terminally differentiated cells, that also do not show proliferative activity, cellular senescence is stress induced and blocks the proliferation of cells with theoretical ability to divide (such as progenitor, stem or cancer cells) due to the activity of specific signaling pathways. The number of senescent cells increases during the ontogenesis of an organism. Senescent cells are not only associated with aging, but also significantly influence this process – a fact that is becoming increasingly well documented.
Keywords:
aging – cellular senescence – progeria – Werner syndrome – Cockayne syndrome – ataxia telangiectasia
Authors:
Matthew Lacey; Martin Mistrík
Authors‘ workplace:
Ústav molekulární a translační medicíny LF UP a FN Olomouc
Published in:
Čas. Lék. čes. 2020; 159: 88-92
Category:
Review Article
Overview
Buněčná senescence je fyziologický stav obecně definovatelný jako stabilní zástava proliferace, tedy přerušení buněčného cyklu. Na rozdíl od buněk terminálně diferenciovaných, které taktéž nevykazují proliferační aktivitu, je zpravidla podmíněna nějakým typem stresu. Buňky s teoretickou schopností dělení (například progenitorové, kmenové či rakovinné) jsou drženy v senescentním stavu aktivitou specifických signálních drah. Množství senescentních buněk v průběhu ontogeneze organismu narůstá. Buněčná senescence tak nejen provází stárnutí, ale jak se ukazuje, významně tento proces ovlivňuje.
Klíčová slova:
buněčná senescence – stárnutí – progerie – Wernerův syndrom – Cockayneův syndrom – ataxia telangiectasia
ÚVOD
Text se zabývá rolí buněčné senescence (BS) v kontextu stárnutí organismu, se zvláštním zaměřením na události, podmínky a fenomény asociované se stářím, v nichž hrají senescentní buňky kauzální roli.
Stárnutí je proces zhoršujících se funkcí tělesných orgánů a tkání (1) provázený zvýšeným rizikem vzniku život ohrožujících nemocí (2). Proces stárnutí je poháněn především epigenetickými změnami, akumulací DNA lézí včetně poškození telomer a mitochondriální dysfunkcí (3). Jedním za základních jevů provázející stárnutí je pak akumulace senescentních buněk (3).
Senescence je forma přerušení buněčné proliferace. Zpravidla se jedná o setrvalý stav, kdy se buňka již dále nedělí, nicméně zůstává metabolicky aktivní a zároveň nabývá specifický fenotyp (podrobně popsáno dále). Akumulace senescentních buněk je považována za jeden z klíčových faktorů podmiňujících proces stárnutí (3, 4).
EVOLUČNÍ ROLE BUNĚČNÉ SENESCENCE
BS je komplexní a regulovaný proces, který zahrnuje velké množství specifických buněčných drah. Nejedná se tedy o náhodně získanou neschopnost buňky se dále dělit. Z toho důvodu lze u BS předpokládat určitá evoluční vysvětlení, která zahrnují následující procesy:
- a) Zabránění proliferaci buněk s poškozeným genomem. Poškozená DNA, ať už vnějšími či vnitřními faktory, je jedním z nejvýznamnějších induktorů BS. Vyřazení takto postižených buněk z proliferace představuje zásadní mechanismus prevence onkogenní transformace buňky, a tedy vzniku maligního bujení (5–8). Zůstává poněkud nedořešenou otázkou, proč se organismus raději poškozených buněk úplně nezbaví, například procesem buněčné apoptózy. Z hlediska ochrany před malignitou by se totiž jednalo o mnohem lepší řešení, hlavně ve světle celé řady negativních dopadů, jež způsobuje hromadění senescentních buněk v těle (jak je diskutováno dále).
- b) Role BS během embryogeneze a hojení ran (9). Hlavně v procesu hojení, kde BS hraje významnou podpůrnou roli v podobě atrakce buněk imunitního systému, může mít z evolučního hlediska velkou váhu. V minulosti infekce vázané na krvácivá zranění ohrožovaly schopnost jedince předat geny významně více než degenerace organismu v pozdním věku.
- c) Role v napomáhání stárnutí organismu. Přijmeme-li tezi, že samotný proces stárnutí je evolučně výhodný pro přežití druhu, lze roli BS chápat pozitivně. To však nic nemění na faktu, že akumulace senescentních buněk u konkrétního jedince významně snižuje jeho zdatnost.
- d) Zvláštním případem možné pozitivní role BS je akumulace senescentních buněk u tzv. steatózy jater (10). Třebaže se jedná o patologický stav, který časem může přejít k cirhóze a jaterním nádorům, senescentní buňky nahromaděné ve steatotické jaterní tkáni sekretují metaloproteinázy. Tyto enzymy pak sehrávají pozitivní roli při degradaci jaterních fibrotických plaků (3).
- e) Role v aktivaci zánětlivé odpovědi, která je již zmíněná v souvislosti s hojením ran. Nahromadění senescentních buněk obecně vede k aktivaci zánětlivé odpovědi, často pak ke vzniku chronického zánětu s možným negativním i pozitivním dopadem na organismus (1, 11).
SENESCENTNÍ BUŇKY V ONTOGENEZI
Počet senescentních buněk se s narůstajícím věkem zvyšuje (1, 2, 6). Jedná se o fenomén, který je dobře popsán také u primátů a hlodavců (6). Právě hlodavci pak poskytují model pro studium procesů, do nichž jsou senescentní buňky zapojeny. Ukazuje se, že senescentní buňky patří mezi hlavní hybné síly stárnutí organismu (1) a že tento efekt souvisí s fenoménem chronického zánětu (12) (podrobněji diskutováno dále). Přítomnost senescentních buněk je asociována s celou řadou nemocí typických pro pozdní věk, jako jsou ateroskleróza, artritida, nádorové malignity, Alzheimerova choroba, chronická plicní obstrukce, idiopatická plicní fibróza ad. (13), a je jimi podmíněna i obecná degenerace tkání (neschopnost se plnohodnotně obnovovat) (14).
VÝZNAM BUNĚČNÉ SENESCENCE PRO SOUČASNOU MEDICÍNU
Vzhledem k provázanosti BS s řadou degenerativních onemocnění typických pro pozdní věk představují senescentní buňky, podobně jako například maligní buňky, možný terapeutický cíl (1). Jedná se o relevantní téma současné medicíny, protože množství starých lidí se rychle zvyšuje. Předpokládá se, že počet lidí starších 80 let se v období 2015–2050 ztrojnásobí (15). Problém stárnoucí populace se projevuje například růstem počtu pacientů s Alzheimerovou nemocí, kde se taktéž předpokládá trojnásobný nárůst do roku 2050. Navíc terapie Alzheimerovy nemoci je extrémně nákladná (například v USA jsou současné náklady na její léčbu větší než součet nákladů na léčbu malignit a kardiovaskulárních nemocí) (14). Ukazuje se, že cílená likvidace senescentních buněk u hlodavců oddaluje nástup stáří, a hlavně snižuje riziko vzniku degenerativních nemocí typických pro pozdní věk.
Specifický problém pak představuje buněčná senescence indukovaná terapií. BS vzniká hlavně v důsledku onkologické léčby zahrnující DNA poškozující cytostatika a radiační terapii. Také v tomto případě se ukazuje, že eliminace léčbou indukovaných senescentních buněk zlepšuje u hlodavců regeneraci postižených tkání (16).
FAKTORY VEDOUCÍ K BUNĚČNÉ SENESCENCI
BS může být vyvolána různými mechanismy zahrnujícími kritické zkrácení telomer, expresi některých onkogenů, reaktivní kyslíkové radikály a jiné chemické či fyzikální faktory poškozující DNA, mitochondriální dysfunkci a zánět (3). Téměř všechny tyto mechanismy se vyskytují přirozeně během života jedince, což z BS dělá jeho nevyhnutelnou součást (6).
BS se dá klasifikovat do dvou specifických kategorií – replikativní senescence (RS) a stresem indukovaná předčasná senescence (SISP) (9, 17). Fenotyp jednotlivých senescentních buněk se může lišit například ve specifickém sekretomu, který je označován jako se senescencí asociovaný sekretomový fenotyp (SASP) (a je diskutován dále) (6). Senescentní stav je u buněk zpravidla udržován dvěma buněčnými signálními dráhami p53-p21 a pRB-p16INKa (3, 13, 14, 17, 18) (obr. 1). Faktory účastnící se funkce těchto drah jsou vesměs všechny prokázané tumorsupresory, nepřímo se tak potvrzuje role senescence v ochraně organismu proti nádorům.
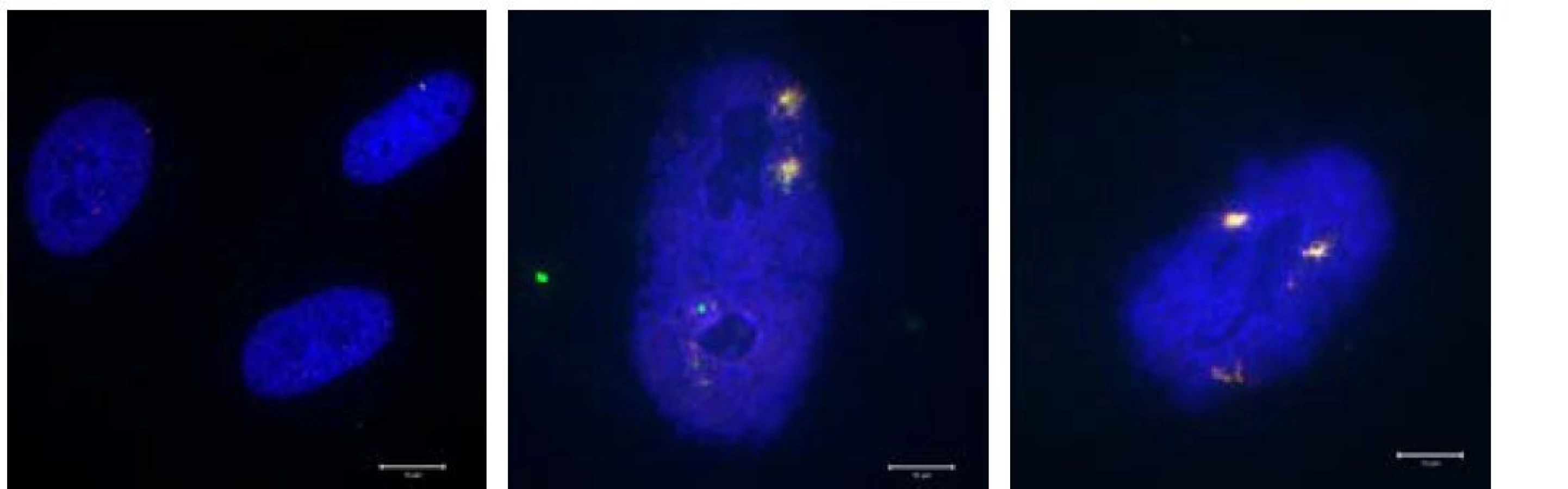
Obrázek zachycuje rozdíly mezi jádry normálně proliferujících buněk (lidské fibroblasty) a jádry buněk senescentních (senescence byla indukována dlouhodobým replikačním stresem). Povšimněte zásadně rozdílně velikosti buněčných jader (modrý kanál – barveno DAPI). V senescentních jádrech je navíc přítomno perzistující DNA poškození, které se je vizualizováno pomocí proteinových markerů 53BP1 (červený kanál) a yH2AX (zelený kanál), které tvoří typické fokusy v místech dvouřetězcových DNA zlomů.
Za normálních podmínek je CDK4/6 kináza (regulátor buněčného cyklu) aktivní, a umožňuje tak buňkám proliferovat. Některé buněčné stresové dráhy však dokážou její aktivitu blokovat a buněčný cyklus tak zastavit. Například DNA poškození aktivuje checkpointové kinázy ATR/ATM, které následně fosforylují protein p53. Tím jej stabilizují a aktivují tak jeho transkripční funkci vedoucí k expresi proteinu p21, který funguje jako účinný CDK4/6 inhibitor. Alternativně může být p53 stabilizován zablokováním MDM2 ubikvitin ligázy, která jej za normálních podmínek označuje (ubikvityluje), což vede k jeho rychlé degradaci. MDM2 je řízena například proteinem ARF, jehož transkripce je aktivována dlouhodobým poškozením DNA a také mitochondriální dysfunkcí. Dlouhodobé poškození DNA aktivuje i transkripci proteinu p16, který působí stejně jako p21 – jako přímý inhibitor CDK4/6.
Replikativní senescence
Jedná se o první popsanou BS (19). Je způsobena kritickým zkrácením chromozomálních telomer. Všechny buňky, které se dělí a zároveň neexprimují specifický enzym, telomerázu, zkracují s každým buněčným cyklem telomerní oblasti. To je způsobeno tím, že DNA polymeráza není schopná během replikace syntetizovat zpožďující se vlákno DNA bez RNA templátu. V místě navázaného RNA templátu (na úplném 3’ DNA konci) tak replikace neproběhne úplně a tím se řetězec DNA nevyhnutelně zkrátí. Tento fenomén je znám také jako „problém koncové replikace lineární DNA“ (20). Dříve nebo později (v závislosti na délce telomerní oblasti) tak replikující se buňky dosáhnou tzv. Hayflickova limitu, kdy je telomerní oblast natolik zkrácena, že přestane vázat specifický proteinový komplex označovaný jako shelterin. Ten slouží k maskování konce telomer před buněčnými systémy rozpoznávající DNA poškození, konkrétně dvouřetězcové zlomy v DNA (3). Jakmile je jednou telomerní oblast rozpoznána jako dvouřetězcový zlom, buňka aktivuje dráhu zahrnující kinázy ATM a ATR, které fosforylují p53 transkripční faktor aktivující produkci inhibitoru buněčného cyklu proteinu p21 (14) (obr. 1). Jelikož zpravidla není v blízkosti kriticky zkrácené telomery jiný volný konec DNA, který by DNA opravný systém mohl připojit (zlom je tedy z principu neopravitelný), je signalizace z telomer perzistující a vede dále k aktivaci p16INK4A dráhy a k stabilnímu zastavení proliferace – tedy buněčné senescenci (8, 9, 14). Alternativně mohou být obnažené konce telomer spojeny, čímž vznikají typické chromozomální fúze. Buňky pak pokračují v proliferaci i s takto poškozeným genomem a stávají se adepty na onkogenní transformaci (21). Replikativní senescence bývala u lidí považována za dominantní formu BS (7), ale některé práce tuto tezi zpochybňují (3). Jakým zásadním způsobem přispívá replikativní senescence oproti SISP k fenoménu stárnutí organismu, je zatím nezodpovězeno (3, 9).
Stresem indukovaná předčasná senescence
Nejčastější formou stresu, která indukuje BS, je akumulované poškození DNA, především pak dvouřetězcové DNA zlomy, které není buňka schopna opravit a tyto léze pak dlouhodobě perzistují (9, 21). Perzistující DNA poškození vyvolá následnou aktivaci drah zastavující buněčný cyklus. Jde tedy v principu o stejný proces, jako byl popsán výše pro kriticky zkrácené telomery. Zdrojem takového poškození mohou být různé endogenní faktory, zahrnující především reaktivní kyslíkové radikály a problémy během replikace DNA vedoucí ke kolapsům replikačních vidlic (tzv. replikačnímu stresu). Poškození DNA a vznik neopravitelných DNA lézí bývá velmi silně indukováno například aberantní aktivací některých onkogenů, např. RAS, MYC, cyklinu E, E2F (22). Ty mohou jednak zvýšit produkci kyslíkových radikálů, především však interferují se správným průběhem replikace DNA. Například onkogenem indukované opětovné spouštění replikačních počátků vede k tzv. hyperreplikaci způsobující vzájemné kolize replikačních vidlic a jejich kolaps, který provází vznik velmi obtížně opravitelných dvouřetězcových zlomů v DNA. Obzvláště v oblastech tzv. běžných fragilních míst na DNA, která jsou typická problematickou replikací, dochází při onkogenním stresu ke vzniku těchto perzistujících DNA lézí (23, 24). Běžná fragilní místa, která jsou evolučně konzervována, třebaže většinově nemají kódující funkci, tak pravděpodobně hrají roli jakýchsi pojistek integrovaných přímo do struktury DNA, schopných vyvolat BS u buněk s významným replikačním stresem (25).
Exogenní faktory vyvolávající BS zahrnují především gama iradiaci, UV záření, vybraná chemoterapeutika a různé karcinogenní látky a jedy (16, 26). Obecným pojítkem těchto faktorů je, že přímo či nepřímo poškozují DNA, a působí tedy genotoxicky. Chemoterapie nádorových onemocnění je významný induktor BS a nejspíše je velkou měrou zodpovědná i za astenii, která je typickým vedlejším efektem a přetrvává i dlouhý čas po ukončení léčby (4, 26). U myších modelů je možné celkovou tělesnou slabost způsobenou chemoterapií potlačit cílenou eliminací senescentních buněk (26). Stejně tak lze napravit i další negativní efekty léčby, jako jsou například srdeční toxicita a útlum krvetvorby (16). Právě oblast zmírnění negativních dopadů chemoterapie nádorových nemocí je horkým kandidátem na využití budoucích léčiv cílících na senescentní buňky – tzv. senolytik. Jejich pozitivní dopad lze přitom očekávat i na další terapií indukované stavy, jako jsou chronický zánět, pokles kognitivních funkcí, diabetes mellitus, hypertenze, dysfunkce ledvin, osteoporóza a sekundární malignity (26–28).
BS A SYNDROMY PŘEDČASNÉHO STÁRNUTÍ
Genetické syndromy předčasného stárnutí – tzv. progerie – se vyznačují akcelerovaným nástupem stařeckého fenotypu (29). Například Hutchinsonova-Gilfordova progerie a Wernerův syndrom jsou typické souborem postižení, jež se vyskytují u starých lidí a zahrnují ztrátu vlasů, vrásky a atrofii kůže, aterosklerózu, ztrátu podkožního tuku, kataraktu a osteoporózu. To vše již v poměrně mladém věku (30, 31). Všechny výše popsané fenotypy jsou do značné míry asociovány s akumulací senescentních buněk.
Přes zjevné podobnosti zůstává částečně nedořešenou otázkou, nakolik jsou fenotypové projevy provázející tyto progerie shodné s procesem přirozeného stárnutí organismu. I přes tyto pochybnosti některé progerie slouží jako modely studia BS, využívány jsou především vybrané myší progeriodní syndromy, které významně zjednodušují logistiku experimentů zkoumající stárnutí organismu (diskutováno dále). Také se nabízejí možnosti léčby těchto syndromů pomocí senolytik, jak naznačují data ze zvířecích modelů.
Ne všechny lidské progerie však byly asociovány s BS. V současnosti je tento fenomén dobře popsán u Hutchinsonovy-Gilfordovy progerie (31, 32), Wernerova syndromu (14, 30), Cockayneova syndromu (14, 33) a ataxia telangiectasia (14). U některých dalších syndromů předčasného stárnutí lze asociaci s BS s velkou pravděpodobností předpokládat. Například u Bloomova syndromu, který je způsoben mutací v BLM helikáze a je charakterizován významnou genetickou nestabilitou (34), lze zvýšenou akumulaci senescentních buněk předpokládat jako důsledek hromadění neopravitelných DNA lézí.
Každý z výše uvedených syndromů má svou specifickou příčinu. Například Hutchinsonova-Gilfordova progerie je způsobena aberantní produkcí proteinu progerinu v důsledku mutace v genu LMNA (31). Progerin se ukazuje jako důležitý faktor regulující vazbu replikačního proteinu PCNA a defektní regulace tohoto procesu způsobuje významnou míru replikačního stresu provázeného kolapsem replikačních vidlic a hromadění DNA poškození (32). Cockayneův syndrom je způsoben mutacemi v genech pro CSA nebo CSB, jejichž produkty jsou zapojeny do DNA opravy na aktivně transkribovaných genech přes nukleotidový excizní opravný systém (33, 35). Wernerův syndrom má příčinu v mutaci WRN helikázy, která hraje roli ve stabilitě běžných fragilních míst a opravě dvouřetězcových DNA zlomů (30, 34). ataxia telangiectasia je způsobena mutací v kináze ATM, která je zapojena do opravy dvouřetězcových DNA zlomů a podílí se na aktivaci tumorsupresorové buněčné dráhy řízené transkripčním faktorem p53 (36). Z výše uvedeného je zřejmé, že akumulace senescentních buněk má i v případě progerií souvislost s nesprávně fungujícími DNA opravnými či signálními dráhami, a jde tak v podstatě o důsledek prostého hromadění DNA lézí.
Progeroidní syndromy jsou známé také u myší, kde se využívají pro studium procesu stárnutí. Užití těchto modelů představuje významné experimentální zjednodušení, protože přirozeně stárnoucí myši jsou z časových důvodů obtížně studovatelné. Například tzv. BubR1 hypomorfická myš je model, u něhož byla přímo studována role senescentních buněk na výsledný fenotyp. Tato myš vykazuje v již raném věku četné fenotypy starých jedinců zahrnující kataraktu, ztrátu pružnosti arteriálních stěn, sarkopenii a později snížené přežití. Myši také vykazují zvýšené hromadění senescentních buněk na specifických místech těla označovaných jako „místa s věkem souvisejících nemocí“, která jsou popsána i u lidí (14). Tyto senescentní buňky jsou typické významně zvýšenou expresí proteinu p16Ink4a. Cílená likvidace těchto buněk pomocí aktivace suicidního transgenu INK-ATTAC umístěného za promotor p16 dokázala prodloužit život těchto myší, a navíc i významně potlačila výše uvedené fenotypy (3, 37). Relativně nedávno byl podobný systém geneticky podmíněné likvidace p16Ink4a pozitivních buněk použit i u normálně stárnoucích myší. I zde se ukázalo, že odstranění senescentních buněk prodlužuje celkové přežití jedinců a i v pozdním věku zvyšuje jejich celkovou zdatnost (3, 9, 38). Podobných výsledků pak bylo u normálních myší dosaženo použitím experimentálních senolytických látek (9).
ZVLÁŠTNOSTI SENESCENTNÍCH BUNĚK A SASP
Jak již bylo uvedeno, senescentní buňky se nedělí, ale zůstávají metabolicky aktivní (7). Jakmile se z replikující buňky stane senescentní, dojde v ní k řadě změn, které ji odlišují od ostatních zdravých buněk. Tyto změny zahrnují nárůst exprese se senescencí asociované beta-galaktosidázy (SA-β-Gal), expresi proteinu p16INKa, změny v morfologii jádra, nárůst celkové buněčné velikosti a vznik tzv. se senescencí asociovaných heterochromatinových fokusů, jež pravděpodobně reflektují specifické změny v expresi některých genů (7, 8, 17). Zajímavé je, že celkový expresní profil senescentních buněk vykazuje i některé podobnosti s buňkami nádorovými, a je tedy možné, že senescentní stav, alespoň v některých případech, mohl předcházet onkogenní transformaci nádorových buněk a vzniku malignity (6, 7, 39).
Senescentní buňky mají také specifický sekretom, který je označován jako se senescencí asociovaný sekretomový fenotyp (SASP) (6, 7). SASP lze považovat z hlediska biologických efektů senescentních buněk za nejdůležitější (6) a má poměrně významný prozánětlivý efekt (7, 8, 13). Faktory sekretované senescentními buňkami zahrnují různé rozpustné signální molekuly, jako jsou interleukiny, růstové faktory a chemokiny, dále různé peptidy a proteiny a také komponenty extracelulární matrix (6). V některých případech jsou sekretovány i aktivní enzymy, jako jsou metaloproteinázy (3, 6), jež mohou mít paradoxně i pozitivní efekt na okolní tkáň (diskutováno výše v sekci evolučního zdůvodnění BS). Obecně je však SASP považován za významný zdroj poškození okolní zdravé tkáně a celého organismu (viz dále). Molekulární mechanismy kontrolující SASP dosud nejsou uspokojivě vysvětleny, i když řada významných buněčných signálních drah, zahrnující například mTOR, NF-κB a p38MAPK, je do sekrece prokazatelně zapojena (7). Předpokládá se také, že SASP je do značné míry kontrolován i typickými změnami v heterochromatinu senescentních buněk (17).
Samotná role SASP pro senescentní buňky pravděpodobně spočívá v udržování proliferačního bloku, čímž plní důležitou tumorsupresorovou funkci. SASP zde funguje jako zpětnovazebná smyčka, kdy sekretované faktory (např. IL-6, IL-8, GRO-α a IGFBP-7) interagují s dráhami podmiňujícími aktivitu p53 a p16 a posilují tak celkový senescentní stav (3, 7, 11). Zajímavé je, že SASP je sám induktorem senescence u okolních zdravých buněk a jeho působení spouští v těchto buňkách i signalizaci typickou pro poškození DNA (2, 3). SASP také významně ovlivňuje imunitní systém a podmiňuje odstraňování existujících senescentních buněk atrakcí adaptivní i vrozené imunity (hlavně makrofágy) (3). Nicméně tento způsob eliminace senescentních buněk zjevně není dostatečně účinný, jak dokazuje jejich hromadění v průběhu ontogeneze (11). Navíc se zde může uplatňovat negativní zpětná vazba, kdy BS utlumuje imunitní systém, který pak o to více selhává v odstraňování senescentních buněk a akceleruje tak některé fenotypy stáří (7).
SASP tedy má významný efekt nejen na okolní tkáň, ale působí i systémově a od určité míry lze hovořit až o jakési formě celkové otravy organismu. Předpokládá se, že se tak významnou měrou podílí na akceleraci nástupu nemocí stáří včetně malignit a je zodpovědný za chronický zánět (6, 7).
ZÁVĚR
Role BS je v organismu velmi komplexní a dosud ne zcela pochopená. I přes jistou evoluční roli tohoto procesu je však stále více zřejmé, že cílené odstranění senescentních buněk, alespoň v určitých fázích ontogeneze, může mít významný pozitivní vliv na zdatnost organismu. Otevírají se tak možnosti pro převratný způsob léčby s věkem asociovaných nemocí, pro pacienty s progeriemi, onkologické pacienty nevratně poškozené chemoterapií a také obecně pro zvyšování kvality života v pozdním věku a jeho celkové prodloužení. V této souvislosti je zásadní zmínit nedávno ukončenou klinickou studii, kdy byla poprvé použita senolytická léčba kombinující látky dasatinib a kvercetin, jejichž podávání pacientům trpícím idiopatickou plicní fibrózou skutečně vedlo ke zlepšení fyzických funkcí (40).
Poděkování
Podpořeno z Evropského fondu pro regionální rozvoj – projekt ENOCH (reg. č.: CZ.02.1.01/0.0/0.0/16_019/0000868).
Čestné prohlášení
Autoři práce prohlašují, že v souvislosti s tématem, vznikem a publikací tohoto článku nejsou ve střetu zájmů a vznik ani publikace článku nebyly podpořeny žádnou farmaceutickou firmou.
Seznam zkratek
- BS buněčná senescence
- GRO růstový onkogen
- IGFBP protein vážící se na inzulinu podobný faktor
- IL interleukin
- mTOR mammalian target of rapamycin
- RS replikativní senescence
- SISP stresem indukovaná předčasná senescence
- SASP se senescencí asociovaný sekretomový fenotyp
- SA-β-Gal se senescencí asociované beta-galaktosidázy
Adresa pro korespondenci:
Mgr. Martin Mistrík, Ph.D.
Ústav molekulární a translační medicíny LF UP a FN Olomouc
Hněvotínská 1333/5, 779 00 Olomouc
Tel.: 585 634 873
e-mail: martin.mistrik@upol.cz
Sources
- Gurău F, Baldoni S, Prattichizzo F et al. Anti-senescence compounds: A potential nutraceutical approach to healthy aging. Ageing Res Rev 2018; 46 : 14–31.
- Franceschi C, Garagnani P, Morsiani C et al. The continuum of aging and age-related diseases: common mechanisms but different rates. Front Med 2018; 5 : 61.
- McHugh D, Gil J. Senescence and aging: causes, consequences, and therapeutic avenues. J Cell Biol 2018; 217(1): 65–77.
- He S, Sharpless NE. Senescence in health and disease. Cell 2017; 169(6): 1000–1011.
- Bártková J, Hořejší Z, Koed K et al. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature 2005; 434(7035): 864–870.
- Coppé JP, Desprez PY, Krtolica A et al. The senescence-associated secretory phenotype: the dark side of tumor suppression. Ann Rev Pathol 2010; 5 : 99–118.
- Watanabe S, Kawamoto S, Ohtani N et al. Impact of senescence-associated secretory phenotype and its potential as a therapeutic target for senescence-associated diseases. Cancer Sci 2017; 108 (4): 563–569.
- Rodier F, Campisi J. Four faces of cellular senescence. J Cell Biol 2011; 192(4): 547–556.
- de Magalhães JP, Passos JF. Stress, cell senescence and organismal ageing. Mech Ageing Dev 2018; 170 : 2–9.
- Ogrodnik M, Miwa S, Tchkonia T et al. Cellular senescence drives age-dependent hepatic steatosis. Nat Commun 2017; 8 : 15691.
- Freund A, Orjalo AV, Desprez PY et al. Inflammatory networks during cellular senescence: causes and consequences. Trends Mol Med 2010; 16(5): 238–246.
- Xia S, Zhang X, Zheng S et al. An update on inflamm-aging: Mechanisms, prevention, and treatment. J Immunol Res 2016; 2016 : 8426874.
- Childs BG, Gluscevic M, Baker DJ et al. Senescent cells: an emerging target for diseases of ageing. Nat Rev Drug Discov 2017; 16(10): 718–735.
- Baker DJ, Petersen RC. Cellular senescence in brain aging and neurodegenerative diseases: evidence and perspectives. J Clin Investig 2018; 128(4): 1208–1216.
- Jaul E, Barron J. Age-related diseases and clinical and public health implications for the 85 years old and over population. Front Public Health 2017; 5 : 335.
- Chang J, Wang Y, Shao L et al. Clearance of senescent cells by ABT263 rejuvenates aged hematopoietic stem cells in mice. Nat Med 2016; 22(1): 78–83.
- Košař M, Bártková J, Hubáčková S et al. Senescence-associated heterochromatin foci are dispensable for cellular senescence, occur in a cell type - and insult-dependent manner and follow expression of p16(ink4a). Cell Cycle 2011; 10(3): 457–468.
- Brooks CL, Gu W. The impact of acetylation and deacetylation on the p53 pathway. Protein Cell 2011; 2(6): 456–462.
- Hayflick L. The limited in vitro lifetime of human diploid cell strains. Exp Cell Res 1965; 37(3): 614–636.
- Blackburn EH. Structure and function of telomeres. Nature 1991; 350(6319): 579–573.
- Fumagalli M, Rossiello F, Clerici M et al. Telomeric DNA damage is irreparable and causes persistent DNA-damage-response activation. Nature Cell Biology 2012; 14(4): 355–365.
- Zeman MK, Cimprich KA. Causes and consequences of replication stress. Nat Cell Biol 2014; 16(1): 2–9.
- Di Micco R, Fumagalli M, Cicalese A et al. Oncogene-induced senescence is a DNA damage response triggered by DNA hyper-replication. Nature 2006; 444(7119): 638–642.
- Sarni D, Kerem B. Oncogene-induced replication stress drives genome instability and tumorigenesis. Int J Mol Sci 2017; 18(7): 1339.
- Béresová L, Veselá E, Chamrád I et al. Role of DNA repair factor xeroderma pigmentosum protein group C in response to replication stress as revealed by DNA fragile site affinity chromatography and quantitative proteomics. J Proteome Res 2016; 15(12): 4505–4517.
- Demaria M, O'Leary MN, Chang J et al. Cellular senescence promotes adverse effects of chemotherapy and cancer relapse. Cancer Discov 2017; 7(2): 165–176.
- Hudson MM, Ness KK, Gurney JG et al. Clinical ascertainment of health outcomes among adults treated for childhood cancer. JAMA 2013; 309(22): 2371–2381.
- Cupit-Link MC, Kirkland JL, Ness KK et al. Biology of premature ageing in survivors of cancer. ESMO Open 2017; 2(5): e000250.
- Navarro CL, Cau P, Lévy N. Molecular bases of progeroid syndromes. Hum Mol Genet 2006; 15(2): R151–R161.
- Shamanna RA, Croteau DL, Lee JH et al. Recent advances in understanding Werner syndrome. F1000Res 2017; 6 : 1779.
- Vidak S, Foisner R. Molecular insights into the premature aging disease progeria. Histochem Cell Biol 2016; 145(4): 401–417.
- Wheaton K, Campuzano D, Ma W et al. Progerin-induced replication stress facilitates premature senescence in Hutchinson-Gilford progeria syndrome. Mol Cell Biol 2017; 37(14): e00659-16.
- Cordisco S, Tinaburri L, Teson M et al. Cockayne syndrome type A protein protects primary human keratinocytes from senescence. J Investig Dermatol 2019; 139(1): 38–50.
- de Renty C, Ellis NA. Bloom’s syndrome: why not premature aging? A comparison of the BLM and WRN helicases. Aging Res Rev 2016; 33 : 36–51.
- Frontini M, Proietti-De-Santis L. Interaction between the Cockayne syndrome B and p53 proteins: implications for aging. Aging 2012; 4(2): 89–97.
- Zhan H, Suzuki T, Aizawa K et al. Ataxia telangiectasia mutated (ATM)-mediated DNA damage response in oxidative stress-induced vascular endothelial cell senescence. J Biol Chem 2010; 285(38): 29662–29670.
- Baker DJ, Wijshake T, Tchkonia T et al. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature 2011; 479(7372): 232–236.
- Baker DJ, Childs BG, Durik M et al. Naturally occurring p16(Ink4a)-positive cells shorten healthy lifespan. Nature 2016; 530(7589): 184–189.
- Cruickshanks HA, McBryan T, Nelson DM et al. Senescent cells harbour features of the cancer epigenome. Nat Cell Biol 2013; 15(12): 1495–1506.
- Hickson LJ, Langhi Prata LGP, Bobart SA et al. Senolytics decrease senescent cells in humans: preliminary report from a clinical trial of dasatinib plus quercetin in individuals with diabetic kidney disease. EBioMedicine 2019; 47 : 446–456.
Labels
Addictology Allergology and clinical immunology Angiology Audiology Clinical biochemistry Dermatology & STDs Paediatric gastroenterology Paediatric surgery Paediatric cardiology Paediatric neurology Paediatric ENT Paediatric psychiatry Paediatric rheumatology Diabetology Pharmacy Vascular surgery Pain management Dental HygienistArticle was published in
Journal of Czech Physicians

- Advances in the Treatment of Myasthenia Gravis on the Horizon
- Possibilities of Using Metamizole in the Treatment of Acute Primary Headaches
- Metamizole at a Glance and in Practice – Effective Non-Opioid Analgesic for All Ages
- Metamizole vs. Tramadol in Postoperative Analgesia
- Spasmolytic Effect of Metamizole
Most read in this issue
- Testing for COVID-19: a few points to remember
- A novel coronavirus (SARS-CoV-2) and COVID-19
- Genetic mechanisms of aging
- The molecular genetics of cellular senescence in the context of organismal aging