
Sekundární imunodeficience jako následek iatrogenního působení
Secondary immunodeficiencies as a result of the iatrogenic impairments
Secondary immunodeficiencies (SID) represent heterogeneous group of acquired impairment of immune systém function with diverse aetiology. It is mostly a combined disorder of both humoral and cellular components of the innate and adaptive immune responses. Except for various pathological states as diabetes mellitus, impairment of liver and kidney functions, protein-energy malnutrition or immunosenescence, also iatrogenic-mediated immune system impairments belong among the most important causes of SID. SID are associated with immunosuppressive or anti-inflammatory treatment used in malignant and autoimmune diseases [(immunosuppressant or disease-modifying drugs, nonsteroidal anti-inflammatory drugs, tyrosine kinase inhibitors or monoclonal antibodies against the CD20 molecule, tumour necrosis factor – (TNF-α) or IL-6 receptor], some other therapeutic procedures (splenectomy or radiotherapy) or present as an adverse events of treatment which at first glance do not interfere with the mechanisms of the immune system (neuroleptic and antiepileptic drugs, angiotensin-converting enzyme inhibitors or anticonvulsants).
Mostly a mild impairment of immunoglobulin production or cellular function is induced, but sometimes clinically significant immunodeficiency develops (especially after treatment with anti-CD20 or anti-TNF-alpha monoclonal antibodies) that already require therapeutic intervention. Therefore, these adverse effects of the treatment should be considered. If patients suffer from clinical symptoms typical for significant immunodeficiency, immunological monitoring is appropriate.
Keywords:
secondary immunodeficiency – radiotherapy – splenectomy – anti-CD20 monoclonal antibodies – anti-TNF-alfa monoclonal antibodies – immunosuppressive drugs – clozapine
Authors:
Z. Chovancová
Authors‘ workplace:
Ústav klinické imunologie a alergologie, Brno
; Masarykova univerzita, Lékařská fakulta a Fakultní nemocnice u sv. Anny v Brně
Published in:
Reviz. posud. Lék., 23, 2020, č. 1-2, s. 22-31
Category:
Original Articles • Review Articles • Case Reports
Overview
Sekundární imunodeficience (SID), tedy získané poruchy funkce imunitního systému, představují heterogenní skupinu onemocnění s rozmanitou etiologií. Většinou se jedná o kombinovanou poruchu humorálních i buněčných složek vrozeného i adaptivního imunitního systému. Mezi nejdůležitější příčiny rozvoje SID patří kromě různých patologických stavů (diabetes mellitus, porucha funkce jater a ledvin, protein-energetická malnutrice, imunosenescence) také iatrogenně navozené poruchy funkce imunitního systému. Ty doprovází imunosupresivní nebo protizánětlivou léčbu používanou při terapii maligních nebo autoimunitních onemocnění (klasická imunosupresiva, chorobu modifikující léčiva, nesteroidní antiflogistika, tyrozin kinázové inhibitory nebo monoklonální protilátky působící proti molekule CD20, tumor nekrotizujícímu faktoru – [TNF α] či receptoru pro IL-6), některé nemedikamentózní léčebné postupy (splenektomii nebo radioterapii), ale také užívání léčiv, u kterých je porušení funkce imunitního systému vedlejším nežádoucím efektem léčby (některá neuroleptika, antiepileptika, inhibitory angiotenzin-konvertujícího enzymu nebo antikonvulziva).
Většinou je navozena mírnější porucha tvorby imunoglobulinů nebo buněčných funkcí, nicméně někdy dochází k rozvoji klinicky významné imunodeficience (zejména při léčbě anti-CD20 nebo anti-TNF α monoklonálními protilátkami), která již vyžaduje terapeutický zásah. Proto je nutné na tyto nežádoucí účinky myslet, a pokud se u pacientů vyskytují klinické příznaky typické pro poruchu funkce imunitního systému, je vhodné je imunologicky sledovat.
Klíčová slova:
sekundární imunodeficience – anti-CD20 monoklonální protilátky – anti-TNF α monoklonální protilátky – imunosupresiva – klozapin – radioterapie – splenektomie
ÚVOD
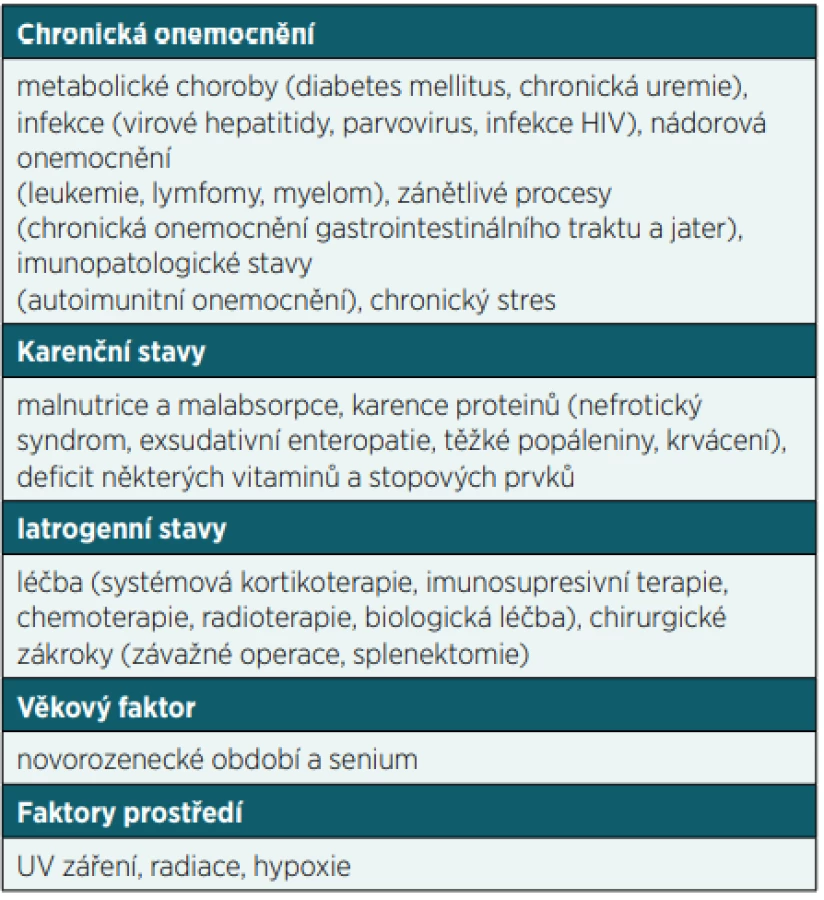
Poruchy funkce imunitního systému můžeme dělit na vrozené (primární) a získané (sekundární). Zatímco primarní imunodeficience (PID) vznikají na základě geneticke poruchy v některém z imunologických mechanismů a manifestují se zejména po narození a v dětském věku, sekundární imunodeficience (SID) představuje stav získané poruchy funkce imunitního systému vyskytující se v dětském i dospělém věku. S výjimkou SID v důsledku HIV infekce a léčby některych maligních onemocnění většinou nejsou příliš závažné. SID sice můžeme dělit podle porušené složky imunitního systému podobně jako PID na protilátkové, buněčné, kombinované, fagocytární nebo komplementové, nicméně většinou jsou porušeny humoralní i buněčné složky imunitního systému současně. Získané poruchy funkce imunitního systému se rozvíjí obecně v důsledku působení vnějších a vnitřních vlivů na organismus nebo v důsledku základního onemocnění včetně jeho léčby. Stavů, které mohou vést k sekundárnímu porušení funkce imunitního systému, je celá řada (tab. 1). Nicméně v tomto sdělení se zaměříme na nejdůležitější formy iatrogenního působení (zejména užívání některých léčiv nebo vlivu jiných terapeutických postupů), které sice řeší základní onemocnění, ale na druhou stranu mohou u pacientů vyvolat získanou poruchu funkce imunitního systému, na což je třeba v rámci péče o tyto pacienty myslet.
SEKUNDÁRNÍ IMUNODEFICIENCE NAVOZENÉ ÚČINKEM LÉČIV
Snížení buněčné nebo protilátkové imunitní odpovědi může způsobit celá řada léků, což může mít za následek mírnější nebo závažnější klinické příznaky sekundární poruchy funkce imunitního systému (tab. 2). U první skupiny léčiv je tlumení aktivity imunitního systému primárním cílem jejich léčebného učinku. Většina těchto molekul se používá v léčbě autoimunitních nebo nádorových onemocnění nebo v rámci prevence rejekce transplantátů.
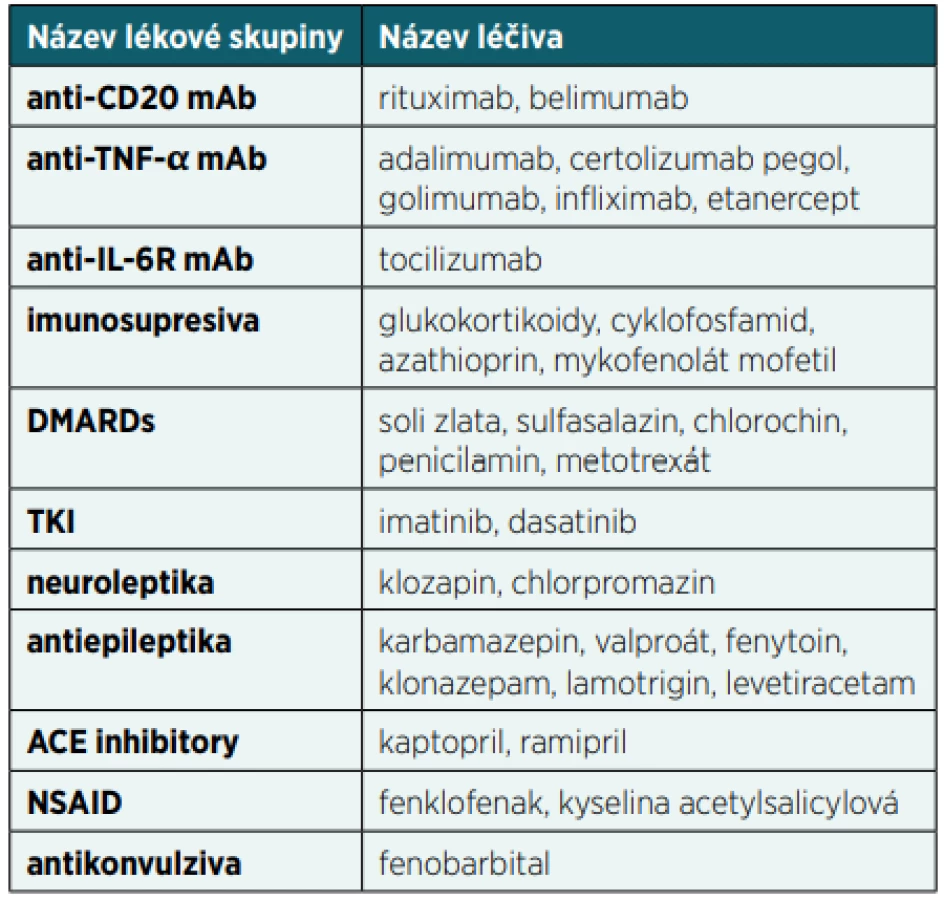
U druhé skupiny léčiv je však tlumení aktivity imunitního systému jen vedlejším nežádoucím efektem léčby. To může být někdy překvapivé, protože se jedná o molekuly, které zdánlivě do mechanismů imunitního systému nezasahují. Po aplikaci některých léčiv dochází převážně k rozvoji sekundární hypogamaglobulinémie, u jiných léčiv převažuje porucha buněčné imunity nebo kombinace obojího [1].
SEKUNDÁRNÍ IMUNODEFICIENCE PO LÉČBĚ ANTIPROLIFERATIVNÍMI LÁTKAMI
Cytotoxická léčiva se používají k léčbě neo-plastického buněčného růstu, ablaci kostní dřeně před transplantací hematopoetických kmenových buněk, v léčbě autoimunitních a zánětlivých onemocnění včetně reakce štěpu proti hostiteli a prevence rejekce transplantatů. Do této skupiny léčiv patři cyklofosfamid, azathioprin, metotrexát a mykofenolat mofetil. Tato léčiva zasahují do syntézy DNA, čímž zastavují buněčný cyklus a indukují apoptózu. Obecně inhibují T-lymfocytární i B-lymfocytární proliferaci, a proto dochází ke snižení imunitní odpovědi na nové antigeny, se kterými se imunitní systém v minulosti nesetkal. Kromě toho také v závislosti na dávce tlumí protilátkovou a buněčnou imunitu, která vznikla předchozi senzibilizací. Tyto léky obecně snižují počty hematopoetických i ostatních buněk, přičemž takto vzniklá cytopenie se dále podílí na rozvoji sekundární imunodeficience a náchylnosti k infekčním komplikacim [2].
Cyklofosfamid patří mezi oxazofosforiny, jejichž mechanismem učinku je alkylace DNA v proliferujících, ale i v neproliferujících buňkách. Tlumí obecně buněčnou i protilátkovou imunitu. Navozuje lymfopenii, přičemž toxičtěji působí na B lymfocyty, což může být následováno rozvojem sekundární hypogamaglobulinémie [3]. Na druhou stranu se ukázalo, že užívání cyklofosfamidu v nízkých dávkách může mít pozitivní imunomodulační efekt. Jeho podávání ovlivňuje homeostázu dendritických buněk a podporuje tvorbu interferonů I. typu, a tím vývoj cytotoxických T lymfocytů (Tc), které hrají důležitou roli v protinádorové imunitě [4]. Dochází též k preferenční eliminaci regulačních T lymfocytů (Treg), které paradoxně tlumí boj vlastního imunitního systému s nádorovým onemocněním [5], i když v poslední době se ukázalo, že snížení aktivity Treg lymfocytů navozené cyklofosfamidem je doprovázeno zvýšením počtu myeloidních supresorových buněk v mikroprostředí nádoru [6, 7].
Azatioprin je tiopurinový derivát, který se používá v léčbě některých autoimunitních onemocnění buď samostatně, nebo v kombinaci s jinými imunosupresivními léčivy. Bylo prokazano, že in vitro stimulace T lymfocytů v přitomnosti azatioprinu nebo jeho metabolitů vede k navození apoptózy T lymfocytů, která je zprostředkovaná přes kostimulační molekulu CD28 na povrchu T lymfocytů. Metabolit azatioprinu (6-thio quanin trifosfat; 6-thio-GTP) se totiž místo GTP váže na malý GTP-vázající protein Rac1, což způsobí přeměnu kostimulačního signálu mediovaného přes molekulu CD28 na signál apoptotický následkem modulace aktivity proteinu Rac1 [8].
Azatioprin snižuje citlivost B lymfocytů k signálům poskytovaným T lymfocyty s následným snížením produkce cytokinů a imunoglobulinů, snižuje počet NK buněk a jejich aktivitu a ovlivňuje cytotoxicitu závislou na protilátkách. U pacientů léčených azatioprinem dochází však jen vzácně k rozvoji hypogamaglobulinémie [9]. Také riziko rozvoje infekčních komplikací je u pacientů léčených azatioprinem ve srovnáný s léčbou cyklofosfamidem nebo kortikoidy menší [10]. Léčba azatioprinem může však vyvolat významný útlum kostní dřeně.
Mykofenolát mofetil prostřednictvím svého aktivního metabolitu kyseliny mykofenolove reverzibilně inhibuje inozinmonofosfát dehydrogenázu, což je klíčový enzym uplatňující se v syntéze purinů. Proto dochází primárně k ovlivnění lymfoidní řady, která je na de novo syntéze purinů závislá. Tím pádem významně ovlivňuje funkci imunitního systému, potlačuje proliferaci lymfocytů a už při nízkých dávkách snižuje zejména B-buněčnou aktivaci, proliferaci a tvorbu plazmatických buněk [11]. Proto může být léčba mykofenolátem mofetilem doprovázená rozvojem signifikantní hypogamaglobulinémie [12, 13]. Kromě toho take inhibuje glykosylaci proteinů adhezivních molekul, a tím omezuje kumulaci lymfocytů a monocytů v místě zánětu. Má také antiproliferativní efekt na fibroblasty, endotelialní buňky a buňky hladkého svalstva [14].
SEKUNDÁRNÍ IMUNODEFICIENCE PO LÉČBĚ KALCINEURINOVÝMI INHIBITORY
Kalcineurinové inhibitory se vážou na cyto-plazmatický protein imunofilin a inhibují jeho vazbu s kalcineurinem, která je zásadní pro aktivaci transkripce IL-2 a tím pro T-buněčnou funkci. IL-2 představuje velmi důležitý cytokin, který hraje nezastupitelnou úlohu v buněčném růstu a proliferaci celé řady buněk imunitního systému, zejména NK buněk a T lymfocytů [15]. Výhodou této skupiny léčiv je to, že významně neovlivňují funkce makrofagů a neutrofilů, čímž snižují náchylnost k infekcím při léčbě těmito imunosupresivy [2].
CYKLOSPORIN A
Cyklosporin A je cyklický peptid izolovaný z vláken houby Tolypocladium inflatum Gams, který má téměř selektivní účinek na T lymfocyty. Znemožňuje tvorbu cytokinů pomocných Th lymfocytů (IL-2 a IFN-γ), které jsou nezbytné pro aktivaci cytotoxických Tc lymfocytů a jejich následnou diferenciaci. Inhibice produkce IL-4 a IL-5 vede k ovlivnění funkce dalších buněčných subpopulací. Zdá se, že chemotaktická ani fagocytární aktivita neutrofilů ovlivněna není, nicméně bylo prokázáno snížení exprese IL-1, IL-6 a TNF α in vitro u lipopolysacharidem aktivovaných makrofágů [16].
SEKUNDÁRNÍ IMUNODEFICIENCE PO LÉČBĚ TYROZIN KINÁZOVÝMI INHIBITORY (TKI)
Molekulárním markerem chronické lymfatické leukémie je přítomnost fuzního genu BCR-ABL1, který je zodpovědný za vznik onkoproteinu Bcr-Abl1 tyrozinkinázy, která hraje hlavní roli v patogenezi tohoto onemocnění. TKI učinně inhibují Bcr-Abl tyrozinkinázu, čímž zastavují proliferaci a indukují apoptózu u Bcr-Abl pozitivních buněčných linií. Tyto molekuly se použivají v léčbě chronické myeloidní leukémie nebo akutní lymfoblastické leukémie [17]. Patří mezi ně imatinib, nilotinib, dasatinib a bosutinib. Ukázalo se, že léčba těmito molekulami se pojí bez přítomnosti lymfopenie s rozvojem sekundární hypogamaglobulinémie [18]. Imatinib snižuje koncentraci IgM paměťových B lymfocytů [19], inhibuje izotypový přesmyk v důsledku downregulace aktivací indukované cytidin deaminázy (AID) [20] a snižuje sérove koncentrace IgA a IgG. Dasatinib snižuje koncentrace celkových IgM imunoglobulinů. Nižší koncentrace imunoglobulinů před léčbou pacienty predisponují k rozvoji sekundární hypogamaglobulinémie po léčbě TKI. Ukázalo se, že pacienti se sníženou koncentrací celkových imunoglobulinů trpěli na signifikantně vyšší počet respiračních infekcí v porovnání s pacienty bez hypogamaglobulinémie, nicméně tyto infekce nebyly závažné [21].
SEKUNDÁRNÍ IMUNODEFICIENCE PO LÉČBĚ GLUKOKORTIKOIDY
Glukokortikoidy jsou steroidní hormony tvořené kůrou nadledvin po stimulaci adrenokortikotropním hormonem (ACTH) uvolňovaným z předního laloku hypofýzy na základě stimulačních podnětů, které aktivují hypotalamo-pitui-tárně-adrenální osu. Léčebně se tyto hormony využívají pro jejich protizánětlivý a imunosupresivní účinek. Odhaduje se, že glukokortikoidy užívá asi 0,5–0,9 % dospělé populace [22]. Kromě celé řady dobře znamých nežadoucích účinku se u pacientů na kortikoterapii může projevit také sekundární porucha funkce imunitního systému.
Léčba glukokortikoidy ovlivňuje zejména buněčnou imunitu. Jejich užívání vede k lymfocytopenii v periferní krvi s maximem za 4−6 hodin s návratem k normalním počtům za 24 hodin po aplikaci v důsledku redistribuce buněk z krevního oběhu do kostní dřeně, hrudního mízovodu, sleziny a lymfatických uzlin [23]. Léčba kortikoidy ovlivňuje negativně zejména T lymfocyty, méně pak B lymfocyty. U léčených pacientů bylo popsáno signifikantníí snížení absolutního a relativního počtu pomocných Th lymfocytů, regulačních Treg lymfocytů a centrálních efektorových i paměťových cytotoxických Tc lymfocytů [24, 25]. Kromě toho dochází také ke snížení počtu naivních a tranzientních B lymfocytů bez ovlivnění počtu paměťových B lymfocytů nebo plazmablastů [24].
U pacientů léčených kortikoidy se můžeme velmi často setkat se sníženou koncentrací, zejména IgG imunoglobulinů, i když někdy dochází také ke snížení IgA nebo IgM třídy [24, 26]. Často se jedná jen o středně těžke snížení koncentrace IgG na hodnoty kolem 4−5 g/l [1]. Zdá se, že riziko snížení koncentrace celkových imunoglobulinů se týká jak nízké dávky dlouhodobě podávané kortikoterapie (> 5 mg denně po dobu více než 2 let) [27], tak vysokých dávek krátkodobě podávané kortikoterapie [28]. Přesný mechanismus, kterým glukokortikoidy snižují tvorbu imunoglobulinů, zatím objasněn nebyl. Mezi pacienty po léčbě glukokortikoidy nebyly pozorovány změny v počtech T nebo B lymfocytů, které by korelovaly se změnami koncentrace imunoglobulinů včetně jejich snížení. A také u pacientů se sníženým počtem nějaké T - nebo B-lymfocytární subpopulace nebyla vždy přítomná hypogamaglobulinémie [24]. Předpokládá se, že by se na poklesu koncentrace imunoglobulinů mohla podílet zvýšená apoptóza plazmatických buněk po vysokých dávkách kortikoidní léčby [29], zvýšený katabolismus imunoglobulinů [30–32] nebo glukokortikoidy potencovaná prostaglandinem mediovaná inhibice funkce B lymfocytů [33]. Nicméně i přes pokles koncentrace celkových IgG imunoglobulinů se zdá být zachovaná tvorba specifických protilátek a pacienti vzhledem k zachovalé funkční rezervě většinou netrpí častějšími závažnými infekcemi [34].
SEKUNDÁRNÍ IMUNODEFICIT PO LÉČBĚ ANTI-CD20 MONOKLONÁLNÍMI PROTILÁTKAMI
B lymfocyty se podílejí na rozvoji celé řady autoimunitních onemocnění, a to tvorbou autoprotilátek, uvolňováním prozánětlivých cytokinů a prezentací antigenů autoreaktivním T lymfocytům. První monoklonální protilátkou namířenou proti účinku těchto buněk byl rituximab (rekombinantní chimérická IgG1 protilátka namířená proti molekule CD20; anti-CD20 mAb). Byla schválena pro léčebné použití v roce 1997 a byla to zároveň první monoklonální protilátka schválená pro léčbu nádorových onemocnění. Molekula CD20 je glykosylovaný fosfolipid, který se za fyziologických okolností vyskytuje na cirkulujících B lymfocytech, ale ne na jejich hematopoetických prekurzorech, plazmablastech nebo plazmatických buňkách [35]. Léčba anti-CD20 mAb je proto cílená jen na cirkulující B lymfocyty a neovlivňuje přímo funkci jiných buněk organismu. Po vazbě anti-CD20 mAb na antigen dochází k depleci cílových CD20-pozitivních cirkulujících B lymfocytů několika mechanismy protilátkami mediovanými (indukce apoptózy, buňkami zprostředkovaná cytotoxicita a komplement dependentní lýza). Anti-CD20 mAb protilátky se proto uplatňují zejména v léčbě chronické lymfocytární leukémie a non-hodgkinských lymfomů vycházejících z B lymfocytů a některých autoimunitních onemocnění, kde má tvorba autoprotilátek patogeneticky potenciál (např. revmatoidní artritida, granulomatóza s polyangiitidou, mikroskopická polyangiitida).
Nicméně deplece počtu B lymfocytů s sebou přináší riziko rozvoje hypogamaglobulinémie. Skutečná frekvence hypogamaglobulinémie po aplikaci anti-CD20 mAb nebo informace o znovuobnovení počtů B lymfocytů v periferní krvi po ukončení léčby není známa, protože v současnosti neexistuje dostatečné množství prospektivních studií. Také klinická signifikance je kontroverzní, protože někteří pacienti trpí v rámci hypogamaglobulinémie na závažné recidivující infekce, ale jiní jsou při podobných hladinách imunoglobulinů asymptomatičtí [36]. Původní studie popisovaly, že u pacientů léčenych anti-CD20 mAb dochází pouze ke snížení koncentrace celkových IgM imunoglobulinů bez ovlivnění koncentrace sérových IgG a IgA imunoglobulinů, přičemž pokles IgM nebyl doprovázen zvýšenou frekvencí infekčních komplikací [37]. Celá řada pozdějších studií však tyto závěry vyvrátila, protože bylo prokázáno, že léčba anti-CD20 mAb může vést také ke klinicky významnému snížení sérové koncentrace IgG imunoglobulinů, což může být doprovázeno signifikantně vyšším výskytem opakovaných závažných bakteriálních i virových infekčních komplikací [38–40]. Bylo prokázáno, že pro rozvoj klinicky významné hypogamaglobulinémie je důležitější pokles imunoglobulinů ve třídě IgG než pokles IgM [41]. Záleží také na koncentraci celkových IgG imunoglobulinů před léčbou, protože jejich snížená koncentrace už před podáním první dávky anti-CD20 mAb predisponuje tyto pacienty k rozvoji klinicky významné hypogamaglobulin émie [36]. Navíc se vyskytuje častěji u pacientů, kteří byli předtím léčeni cyklofosfamidem [42].
U pacientů léčených anti-CD20 mAb pro diagnózu lymfomu se prevalence rozvoje hypogamaglobulinémie pohybovala v rozmezi 39−43 %, nicméně klinicky významná ve smyslu výskytu závažných opakovaných infekcí byla jen u 6,6 % pacientů [43]. Vyšší prevalence infekcí (16,7 %) byla pozorována u skupiny pacientů léčenych anti-CD20 mAb v rámci adjuvantní terapie před autologní transplantací hematopoetických krevních buněk pro diagnózu non-hodgkinského lymfomu [44]. O něco nižší prevalence hypogamaglobulinémie byla pozorována u pacientů léčených pro diagnózu revmatoidní artritidy, kde se po jednom podání vyskytovala u 11,8 % pacientů, přičemž toto procento vzrostlo na 22,2 % u pacientů, kteří dostali více než pět cyklů léčby [45].
Závažnější hypogamaglobulinémie se většinou objevuje při indikacích onkologických, ale byla popsána i při indikacích autoimunitních, i když většinou nedochází k tak závažnému klinicky manifestnímu poklesu IgG [46, 47]. Snížení počtu B lymfocytů v periferní krvi se vyskytuje již za několik dní po podání první dávky terapie, nicméně obnovení jejich počtu je velmi variabilní a záleží na úvodním stavu imunitního systému konkretního pacienta, konkomitantní terapii a dávce anti-CD20 mAb [36]. Navíc se ukázalo, že úprava počtu cirkulujících B lymfocytů nemusí vždy korelovat s úpravou hypogamaglobulinémie [48]. To může částečně souviset se zvýšenou proporcí naivních B lymfocytů oproti izotypově přesmyknutým paměťovým B lymfocytům.
Pacienti léčení anti-CD20 mAb by měli být pravidelně monitorováni, co se týče koncentrace celkových IgG, IgA a IgM imunoglobulinů a B-buněčných počtů po ukončení podávání této léčby [36]. U těch, kteří vykazují anomalie v koncentraci sérovych imunoglobulinů a/nebo počtů B lymfocytů, je vhodné doplnit další analýzu B-buněčných subpopulací ke zjištění, zda došlo k úpravě počtu izotypově přesmyknutých paměťových B lymfocytů. Hlavním kritériem pro zahájení substituční imunoglobulinové léčby by u těchto pacientů měl být výskyt závažných opakovaných infekcí [36].
SEKUNDÁRNÍ IMUNODEFICIT PO LÉČBĚ MOLEKUL NAMÍŘENÝCH PROTI ÚČINKU TNF α
Tumor nekrotizující faktor (z anglickeho „tumor necrosis factor“; TNF) představuje významný cytokin, který je zapojen do velkého množství fyziologických procesů probíhajících v organismu. Kromě zahájení a regulace imunitních reakcí zasahuje také do metabolismu lipidovych kyselin a sacharidů, procesů koagulace a fibrinolýzy, ovlivňuje také endoteliální funkce [49–51]. TNF je produkován celou řadou buněk lidského organismu, a to jak buňkami imunitního systému (makrofágy, granulocyty, mastocyty, NK buňkami nebo T lymfocyty), tak dalšími buňkami našeho organismu (neurony, keratinocyty nebo buňkami hladké svaloviny) [52]. Neporušené fungování procesů mediovaných prostřednictvím TNF α je důležité pro udržení homeostázy organismu. Proto není překvapivé, že zvyšená tvorba nebo aktivita TNF α se uplatňuje v patogenezi celé řady autoimunitních onemocnění (např. systémového lupus erythematodes, revmatoidní artritidy, psoriázy nebo nespecifických střevních zánětů), naopak snížená sekrece nebo funkce tohoto cytokinu vede ke snížení obranyschopnosti organismu vůči některým patogenům [53]. Monoklonální protilátky namířené proti účinkům TNF α inhibují jeho biologickou aktivitu, čímž ovlivňují biologickou odpověď, která je indukována nebo regulována TNF, a to včetně změn hladin adhezivních molekul zodpovědných za migraci leukocytů. Snižují přesun granulocytů do kloubů, snižují výskyt T lymfocytů, B lymfocytů a makrofágů v synoviální výstelce kloubů u pacientů s revmatoidní artritidou nebo některé z nich indukují apoptózu aktivovaných buněk imunitního systému v lamina propria střevní sliznice u pacientů s Crohnovou chorobou [54–56].
Při studiu celkového ovlivnění funkce imunitního systému těmito preparáty bylo zjištěno, že zřejmě nedochází k významnému ovlivnění počtu buněk imunitního systému (granulocytů, monocytů, makrofágů, NK buněk, T lymfocytů nebo B lymfocytů) nebo funkce buněk imunitního systému (tvorba protilátek, proliferace lymfocytů, fagocytární funkce, mechanismy opožděné buněčné přecitlivělosti) [57, 58]. Naopak se ukázalo, že léčba molekulami namířenými proti účinku TNF α podporuje funkci T lymfocytů, protože působí proti inhibičnímu efektu chronického zánětu na tyto buňky [59].
U pacientů léčených molekulami namířenými proti účinku TNF α se mohou vyskytnout závažné a oportunní infekce. Byly popsány infekce způsobené Pneumocystis carinii, Listeria monocytogenes, Legionella pneumophila, Coccidioidomycosis immitis, Histoplasma capsulatum, Aspergillus fumigatus nebo atypickými mykobakterii. Zejména se však jedná o možnou reaktivaci infekce způsobenou virem hepatitidy B nebo Mycobacterium tuberculosis. Celá řada studií už v minulosti jasně prokázala, že TNF α hraje významnou roli v obraně pacientů proti infekci Mycobacterium tuberculosis. Při jeho nedostatečné tvorbě dochází k poruše tvorby granulomů s následnou diseminací infekce [53, 60]. Proto by měli být pacienti před léčbou, během léčby i nějakou dobu po léčbě sledováni stran rizika reaktivace tuberkulózy nebo virové hepatitidy B.
SEKUNDÁRNÍ IMUNODEFICIT PO LÉČBĚ KLOZAPINEM
Klozapin (selektivní antagonista dopaminového receptoru D4 patřícího do rodiny receptorů D2) je atypické antipsychotikum indikované k léčbě pacientů s rezistentní formou schizofrenie nebo Parkinsonovou chorobou doprovázenou psychotickými poruchami, u kterých selhala standardní léčba. Na rozdíl od klasických antipsychotik (chlorpromazinu a haloperidolu) nedoprovází léčbu klozapinem nežadoucí motorické parkinsoidní příznaky (tremor, hypokineze nebo rigidita), avšak závažným známým nežadoucím účinkem je riziko rozvoje letální agranulocytózy, kvůli které jsou pacienti pravidelně sledováni [61]. Nicméně i přesto bylo pozorováno, že pacienti léčení klozapinem trpí vyšší frekvencí respiračních infekcí s nutností léčby antibiotiky včetně pneumonií, což bylo dáváno do souvislosti s různými příčinami (sedací, aspirací nebo kouřením). Teprve nedávno však bylo popsáno, že užívání klozapinu může vést k rozvoji signifikantní hypogamaglobulinémie [62, 63]. Proto by při léčbě tímto preparátem měla být kromě počtu granulocytů sledována i koncentrace sérových imunoglobulinů.
SEKUNDÁRNÍ IMUNODEFICIT U PACIENTŮ PO SPLENEKTOMII
Slezina představuje důležitý imunoaktivní orgán, který se podílí pomocí slezinných makrofágů na odstraňování nepotřebných buněk nebo bakteriálních či parazitárních antigenů nacházejících se přímo v krevním řečišti. Zatímco játra se podílí zejména na odstraňování dobře opsonizovaných bakterií, slezina vychytává méně opsonizované bakterie, mezi které patří zejména obalené mikroorganismy. Slezina je také hlavním místem tvorby imunoglobulinů ve třídě IgM, na které se podílí zejména B lymfocyty marginální zóny. Koncentrace IgM imunoglobulinů po splenektomii klesá [64]. Slezina je také hlavním místem tvorby tuftsinu a properdinu, což jsou dva důležité opsoniny. Tuftsin je tetrapeptid vznikající ve slezině enzymatickým odštěpením z Fc domény těžkého řetězce IgG a významně stimuluje fagocytární a baktericidní aktivitu neutrofilních granulocytů a makrofágů [65]. K funkčnímu hyposplenismu může vést celá řada onemocnění a jiných patologických stavů [66, 67]. Pacienti s vrozenou nebo získanou poruchou funkce sleziny jsou náchylní k infekcím způsobeným obalenými a některými dalšími patogeny (Streptococcus pneumoniae, Haemophilus influenzae typu B, meningokok, salmonela nebo další enterobakterie, virus chřipky), přičemž infekce opouzdřenými patogeny mohou u těchto pacientů vést k progresivní septikemii s mortalitou, která se při sepsi pohybuje mezi 50–70 % [2]. Tento stav je označován jako postsplenektomický septický syndrom (tzv. OPSI syndrom), který byl poprvé popsán počátkem 50. let minulého století [68]. Celoživotní riziko rozvoje tohoto syndromu se odhaduje na 5 %, přičemž z toho 30 % infekcí se projeví v prvním roce po splenektomii a 50 % během prvních dvou let po splenektomii [69]. Proto je v rámci preventivních opatření vhodné tyto pacienty naočkovat vakcinou proti pneumokokům, hemofilům, meningokokům a viru chřipky [70]. Pokud se u těchto pacientů objeví horečka, je vhodné nasadit bezodkladně antibiotika, z nichž jsou doporučována amoxicilin/klavulanat (1 g každých 6–8 hodin), cefuroxim/axetil (500 mg každych 6–8 hodin) nebo moxifloxacin (400 mg každých 24 hodin) [70]. Jistou formou ochrany těchto pacientů by mohlo být též vytvoření národního registru, protože se ukázalo, že u takto dlouhodobě systematicky sledovaných pacientů docházý k signifikantnímu snížení rizika rozvoje infekcí opouzdřenými bakteriemi vzhledem k lepší preventivní i jiné péči o ně [71].
SEKUNDÁRNÍ IMUNODEFICIT JAKO NÁSLEDEK RADIOTERAPIE
Radioterapie zůstává základním pilířem léčby maligního bujení, přičemž asi 60 % pacientů se solidními tumory podstupuje kurativní nebo paliativní ozáření jako část svého léčebného režimu [72]. Účinky radioterapie se neomezují jen na ozářenou lokalitu, ale už dlouhou dobu je známo, že mají systémový charakter, protože ovlivňují mnoho molekulárních signalizačních drah. Ve vztahu k imunitnímu systému se radioterapie chová ambivalentně, nicméně jednotlivé mechanismy nejsou ještě zcela objasněny [73].
Na imunosupresivním efektu radiace se podílí snížení celkového počtu cirkulujících lymfocytů a lymfoidních progenitorů v primárních a sekundárních lymfatických orgánech [74]. Mezi nejvíce radiosenzitivní buňky patří lymfocyty (T lymfocyty, B lymfocyty a NK buňky), dále antigen prezentující buňky (monocyty, makrofágy a dendritické buňky), z nichž posledně jmenované vykazují nejvyšší radiorezistenci [75]. Nicméně ozáření může způsobit funkční poškození dendritických buněk, což může vést k poruchám dendritickými buňkami navozené aktivace nebo tolerance T lymfocytů [76]. Bylo prokázáno, že u pacientů vystavených radiaci dochází k signifikantnímu snížení intersticiálních dendritickych buněk, ktere se významně podílejí na aktivaci Th1 lymfocytů. To by mohla být přičina porušené rovnováhy mezi Th1/Th2 imunitní odpovědí rozvíjející se po ozáření [77]. Kromě toho ozáření způsobuje zvýšení počtu myeloidních supresorových buněk a Treg lymfocytů [74], modifikaci fenotypu makrofágů z prozánětlivého typu M1 na imunosupresivní typ M2 [78] nebo zvýšenou expresi ligandu programované buněčné smrti (PDL-1) a cytotoxického s T lymfocyty asociovaného antigenu (CTLA-4) [74].
Na druhou stranu ionizační záření zvyšuje aktivitu imunitního systému. Dochází při něm k uvolnění proinflamatorních cytokinů, chemokinů, proteinů teplotního šoku a dalších signálů nebezpečí, čímž se zvyšuje nádorová imunogenicita. Ozáření může uvolnit velké množství nádorových neoantigenů, které pak mohou být předkládány T lymfocytům. Dochází též k náboru efektorových buněk do mikroprostředí nádoru. Tímto způsobem radiace aktivuje mechanismy vrozené i získané imunitní odpovědi, což vede ke zvýšené aktivaci imunitního systému proti nádoru jak v místě ozáření, tak systémově [73].
DIAGNOSTIKA SEKUNDÁRNÍCH IMUNODEFICIENCÍ
Na možný rozvoj SID nás klinicky upozorní zvýšená frekvence infekcí. Při porušení protilátkové imunitní odpovědi se můžeme setkat zejména s recidivujícími infekcemi horních a dolních dýchacích cest (sinusitidy, otitidy, pneumonie), které někdy nereagují adekvátně na běžně užívanou léčbu a jsou většinou způsobeny opouzdřenými patogeny. Při porušení buněčné imunity většinou dochází k rozvoji kombinované imunodeficience, která se může projevit infekcemi způsobenými atypickými patogeny nebo viry.
V rámci základního vyšetření pacientů s podezřením na sekundární postižení funkce imunitního systému před odesláním k vyšetření specialistovi by mělo být provedeno vyšetření krevního obrazu a diferenciálního rozpočtu leukocytů, základní biochemické vyšetření včetně stanovení koncentrace celkových bílkovin a albuminu, ale také biochemické vyšetření moče k vyloučení ztrát bílkovin močí. V případě nálezu neutropenie v diferenciálním rozpočtu leukocytů je vhodné pacienta odeslat na vyšetření hematologické. Pokud je podezření na rozvoj klinicky významné sekundární imunodeficience, pak je indikováno vyšetření imunologické. Pravidelné sledování koncentrace celkových imunoglobulinů by mělo být pravidlem v případě léčby preparáty, u kterých je riziko navození klinicky výnamné hypogamaglobulinémie dobře známo. Diagnostika SID je u specialisty kromě anamnézy založena na laboratorních vyšetřeních, ve kterých se zjišťuje koncentrace humoralních složek imunitního systému a počet imunokompetentních buněk.
LÉČBA SEKUNDÁRNÍCH IMUNODEFICIENCÍ
U pacientů s klinickými příznaky sekundární imunodeficience je možno nasadit profylaktickou antibiotickou léčbu. Obecně neni doporučováno aplikování živých vakcín a při používání polysacharidových vakcin je vhodné volit vakciny konjugované. U některých pacientů se závažným poklesem koncentrace celkových IgG imunoglobulinů a klinickými příznaky imunodeficience je vhodná substituční imunoglobulinová léčba. Efekt užívání bakteriálních imunomodulátorů je sporný, každopádně by tyto preparaty neměly být podávány v případě, že k rozvoji sekundární imunodeficience vedlo autoimunitní onemocnění nebo pacient nějakým autoimunitním onemocněním trpí.
ZÁVĚR
Sekundární imunodeficience mohou být kromě různých onemocnění nebo patologických stavů navozeny také iatrogenně. Různý stupeň závažnosti porušení funkce imunitního systému doprovází užívání některých léčiv (zejména v rámci imunosupresivní nebo protizanětlivé léčby, ale také jako nežadoucí účinek medikace, která na první pohled do mechanismů imunitního systému přímo nezasahuje) a další terapeutické přístupy (např. splenektomie nebo radioterapie). Většinou se jedná o lehčí poruchu humoralních nebo buněčných imunitních mechanismů, ale někdy může dojít k rozvoji klinicky významné poruchy funkce imunitního systému (zejména při léčbě anti-CD20 nebo anti-TNF α mAb), která vyžaduje další terapeutické řešení. Proto by se na možný rozvoj iatrogenně způsobené sekundární imunodeficience mělo pomýšlet a tito pacienti by měli být imunologicky vyšetřeni a připadně sledováni.
Čestné prohlášení
Autorka nemá v souvislosti s tématem práce žadný střet zájmů.
Po vzájemné dohodě vedoucích redaktorů a se souhlasem autorů byl článek převzatý z časopisu Postgraduální medicína (219, roč. 21, č. 4, s. 312–318)
Adresa pro korespondenci:
MUDr. Zita Chovancová, Ph.D.
Ústav klinické imunologie a alergologie
Pekařská 664/53
656 91 Brno
e-mail: zita.chovancova@fnusa.cz
Sources
1. Samson, M., Audia, S., Lakomy, D., et al. Diagnostic stratégy for patients with hypogammaglobulinemia in rheumatology. Joint Bone Spine, 2011, 78, p. 241–245.
2. Chinen, J., Shearer, W. T. Secondary immunodeficiencies, including HIV infection. J Allergy Clin Immunol, 2010, 125, p. 195–203.
3. Venhoff, N., Effelsberg, N. M., Salzer, U., et al. Impact of rituximab on immunoglobulin concentrations and B cell numbers after cyclophosphamide treatment in patients with ANCAassociated vasculitides. PLoS One, 2012, 7, e37626.
4. Sistigu, A., Viaud, S., Chaput, N., et al. Immunomodulatory effects of cyclophosphamide and implementations for vaccine design. Semin Immunopathol, 2011, 33, p. 369–383.
5. Ghiringhelli, F., Menard, C., Puig, P. E., et al. Metronomic cyclophosphamide regimen selectively depletes CD4+CD25+ regulátory T cells and restores T and NK effector functions in end stage cancer patients. Cancer Immunol Immunother, 2007, 56, p. 641–648.
6. Sevko, A., Sade-Feldman, M., Kanterman, J., et al. Cyclophosphamide promotes chronic inflammation-dependent immunosuppression and prevents antitumor response in melanoma. J Invest Dermatol, 2013, 133, p. 1610–1619.
7. Becker, J. C., Schrama, D. The dark side of cyclophosphamide: cyclophosphamide-mediated ablation of regulatory T cells. J Invest Dermatol, 2013, 133, p. 1462–1465.
8. Tiede, I., Fritz, G., Strand, S., et al. CD28-dependent Rac1 activation is the molecular target of azathioprine in primary human CD4+ T lymphocytes. J Clin Invest, 2003, 111, p. 1133–1145.
9. Keven, K., Sahin, M., Kutlay, S., et al. Immunoglobulin deficiency in kidney allograft recipients: comparative effects of mycophenolate mofetil and azathioprine. Transpl Infect Dis, 2003, 5, p. 181–186.
10. Bernatsky, S., Hudson, M., Suissa, S. Anti-rheumatic drug use and risk of serious infections in rheumatoid arthritis. Rheumatology (Oxford), 2007, 46, p. 1157–1160.
11. Karnell, J. L., Karnell, F. G., Stephens, G. L., et al. Mycophenolic acid differentially impacts B cell function depending on the stage of differentiation. J Immunol, 2011, 187, p. 3603–3612.
12. Boddana, P., Webb, L. H., Unsworth, J., et al. Hypo-gammaglobulinemia and bronchiectasis in mycophenolate mofetil-treated renal transplant recipients: an emerging clinical phenomenon? Clin Transplant, 2011, 25, p. 417–419.
13. Eickenberg, S., Mickholz, E., Jung, E., et al. Mycophenolic acid counteracts B cell proliferation and plasmablast formation in patients with systemic lupus erythematosus. Arthritis Res Ther, 2012, 14, R110.
14. Goldblum, R. Therapy of rheumatoid arthritis with mycophenolate mofetil. Clin Exp Rheumatol, 1993, 11, p. 117–119.
15. Dhupkar, P., Gordon, N. Interleukin-2: Old and new approaches to enhance immune-therapeutic efficacy. Adv Exp Med Biol, 2017, 995, p. 33–51.
16. Fukata, N., Uchida, K., Kusuda, T., et al. The effective therapy of cyclosporine A with drug delivery system in experimental colitis. J Drug Target, 2011, 19, p. 458–467.
17. Žáčková, D. Tyrozinkinazove inhibitory v lečbě staršich pacientů s chronickou myeloidni leukemii – editorial. Vnitř Lek, 2015, 61, p. 760–761.
18. Santachiara, R., Maffei, R., Martinelli, S., et al. Development of hypogammaglobulinemia in patients treated with imatinib for chronic myeloid leukemia or gastrointestinal stromal tumor. Haematologica, 2008, 93, p. 1252–1255.
19. De Lavallade, H., Khoder, A., Hart, M., et al. Tyrosine kinase inhibitors impair B-cell immune responses in CML through off-target inhibition of kinases important for cell signaling. Blood, 2013, 122, p. 227–238.
20. Kawamata, T., Lu, J., Sato, T., et al. Imatinib mesylate directly impairs class switch recombination through down-regulation of AID: its potential efficacy as an AID suppressor. Blood, 2012, 119, p. 3123–3127.
21. Rajala, H. L. M., Missiry, M. E., Ruusila, A., et al. Tyrosine kinase inhibitor therapy-induced changes in humoral immunity in patients with chronic myeloid leukemia. J Cancer Res Clin Oncol, 2017, 143, p. 1543–1554.
22. Kanis, J. A., Johansson, H., Oden, A., et al. A meta-analysis of prior corticosteroid use and fracture risk. J Bone Miner Res, 2004, 19, p. 893–899.
23. Fauci, A. S., Dale, D. C., Balow, J. E. Glucocorticosteroid therapy: mechanisms of action and clinical considerations. Ann Intern Med, 1976, 84, p. 304–315.
24. Wirsum, C., Glaser, C., Gutenberger, S., et al. Secondary antibody deficiency in glucocorticoid therapy clearly differs from primary antibody deficiency. J Clin Immunol, 2016, 36, p. 406–412.
25. Daien, C. I., Gailhac, S., Mura, T., et al. High levels of memory B cells are associated with response to a first tumor necrosis factor inhibitor in patients with rheumatoid arthritis in a longitudinal prospective study. Arthritis Res Ther, 2014, 16, R95.
26. Hamilos, D. L., Young, R. M., Peter, J. B., et al. Hypogamma-globulinemia in asthmatic patients. Ann Allergy, 1992, 68, p. 472–481.
27. Klaustermeyer, W. B., Gianos, M. E., Kurohara, M. L., et al. IgG subclass deficiency associated with corticosteroids in obstructive lung disease. Chest, 1992, 102, p. 1137–1142.
28. Kawano, T., Matsuse, H., Obase, Y., et al. Hypogamma-globulinemia in steroid-dependent asthmatics correlates with the daily dose of oral prednisolone. Int Arch Allergy Immunol, 2002, 128, p. 240–243.
29. Sharma, S., Lichtenstein, A. Dexamethasone-induced apoptotic mechanisms in myeloma cells investigated by analysis of mutant glucocorticoid receptors. Blood, 2008, 112, p. 1338–1345.
30. Levy, A. L., Waldmann, T. A. The effect of hydrocortisone on immunoglobulin metabolism. J Clin Invest, 1970, 49, p. 1679–1684.
31. Riches, P. G., Hobbs, J. R. Mechanisms in secondary hypogammaglobulinaemia. J Clin Pathol Suppl, 1979, 13, p. 15–22.
32. Butler, W. T., Rossen, R. D. Effects of corticosteroids on immunity in man. I. Decreased serum IgG concentration caused by 3 or 5 days of high doses of methylprednisolone. J Clin Invest, 1973, 52, p. 2629–2640.
33. Galanaud, P., Crevon, M. C., Emilie, D., Abella, A. Effect of hydrocortisone on the in vitro human antibody response: interaction with monocytes and prostaglandins. Clin Immunol Immunopathol, 1983, 29, p. 403–414.
34. Lack, G., Ochs, H. D., Gelfand, E. W. Humoral immunity in steroid-dependent children with asthma and hypogammaglobulinemia. J Pediatr, 1996, 129, p. 898–903.
35. Leandro, M. J. B-cell subpopulations in humans and their dif-ferential susceptibility to depletion with anti-CD20 monoclonal antibodies. Arthritis Res Ther, 2013, 15, S3.
36. Sacco, K. A., Abraham, R. S. Consequences of B-celldepleting therapy: hypogammaglobulinemia and impaired B-cell reconstitution. Immunotherapy, 2018, 10, p. 713–728.
37. McLaughlin, P., Grillo-López, A. J., Link, B. K., et al. Rituximab chimeric anti-CD20 monoclonal antibody therapy for relapsed indolent lymphoma: half of patients respond to a four-dose treatment program. J Clin Oncol, 1998, 16, p. 2825–2833.
38. Lim, S. H., Zhang, Y., Wang, Z., et al. Maintenance rituximab after autologous stem cell transplant for high-risk B-cell lymphoma induces prolonged and severe hypogammaglobulinemia. Bone Marrow Transplant, 2005, 35, p. 207–208.
39. Kosmidis, S., Baka, M., Bouhoutsou, D., et al. Longitudinal assessment of immunological status and rate of immune recovery following treatment in children with ALL. Pediatr Blood Cancer, 2008, 50, p. 528–532.
40. Cabanillas, F., Liboy, I., PaviA, O., RiverA, E. High incidence of non-neutropenic infections induced by rituximab plus fludarabine and associated with hypogammaglobulinemia: a frequently unrecognized and easily treatable complication. Ann Oncol, 2006, 17, p. 1424–1427.
41. Van Vollenhoven, R. F., Fleischmann, R. M., Furst, D. E., et al. Longterm safety of rituximab: final report of the rheumatoid arthritis global clinical trial program over 11 years. J Rheumatol, 2015, 42, p. 1761–1766.
42. Kado, R., Sanders, G., McCune, W. J. Diagnostic and therapeutic considerations in patients with hypogammaglobulinemia after rituximab therapy. Curr Opin Rheumatol, 2017, 29, p. 228–233.
43. Casulo, C., Maragulia, J., Zelenetz, A. D. Incidence of hypogammaglobulinemia in patients receiving rituximab and the use of intravenous immunoglobulin for recurrent infections. Clin Lymphoma Myeloma Leuk, 2013, 13, p. 106–111.
44. Nishio, M., Fujimoto, K., Yamamoto, S., et al. Hypo-gammaglobulinemia with a selective delayed recovery in memory B cells and an impaired isotype expression after rituximab administration as an adjuvant to autologous stem cell transplantation for non-Hodgkin lymphoma. Eur J Haematol, 2006, 77, p. 226–232.
45. De La Torre, I., Leandro, M. J., Edwards, J. C., Cambridge, G. Baseline serum immunoglobulin levels in patients with rheumatoid arthritis: relationships with clinical parameters and with B-cell dynamics following rituximab. Clin Exp Rheumatol, 2012, 30, p. 554–560.
46. Keystone, E., Fleischmann, R., Emery, P., et al. Safety and efficacy of additional courses of rituximab in patients with active rheumatoid arthritis: an open-label extension analysis. Arthritis Rheum, 2007, 56, p. 3896–3908.
47. Králíčková, P., Malá, E., Vokurková, D., et al. Sekundarní humoralní imunodeficience u nemocných se systémovym lupus erythematodes. Vnitř. Lék., 2015, 61, p. 778–794.
48. Kano, G., Nakatani, T., Yagi, K., et al. Complicated pathophysiology behind rituximab-induced persistent hypogammaglobulinemia. Immunol Lett, 2014, 159, p. 76–78.
49. Page, M. J., Bester, J., Pretorius, E. The inflammatory effects of TNF-α and complement component 3 on coagulation. Sci Rep, 2018, 8, p. 1812.
50. Borst, S. E. The role of TNF-alpha in insulin resistance. Endocrine, 2004, 23, p. 177–182.
51. Cawthorn, W. P., Sethi, J. K. TNF-alpha and adipocyte biology. FEBS Lett, 2008, 582, p. 117–131.
52. Tracey, D., Klareskog, L., Sasso, E. H., et al. Tumor necrosis factor antagonist mechanisms of action: a comprehensive review. Pharmacol Ther, 2008, 117, p. 244–279.
53. Mootoo, A., Stylianou, E., Arias, M. A., Reljic, R. TNFalpha in tuberculosis: a cytokine with a split personality. Inflamm Allergy Drug Targets, 2009, 8, p. 53–62.
54. Ten Hove, T., Van Montfrans, C., Peppelenbosch, M. P., Van Deventer, S. J. Infliximab treatment induces apoptosis of lamina propria T lymphocytes in Crohn’s disease. GUT, 2002, 50, p. 206–211.
55. Van Den Brande, J. M., Braat, H., Van Den Brink, G. R., et al. Infliximab but not etanercept induces apoptosis in lamina propria T-lymphocytes from patients with Crohn’s disease. Gastroenterology, 2003, 124, p. 1774–1785.
56. Tak, P. P., Taylor, P. C., Breedveld, F. C., et al. Decrease in cellularity and expression of adhesion molecules by anti-tumor necrosis factor alpha monoclonal antibody treatment in patients with rheumatoid arthritis. Arthritis Rheum, 1996, 39, p. 1077–1081.
57. Pala, O., Diaz, A., Blomberg, B. B., Frasca, D. B lymphocytes in rheumatoid arthritis and the effects of anti-TNF-α agents on B lymphocytes: A review of the literature. Clin Ther, 2018, 40, p. 1034–1045.
58. Moreland, L. W., Bucy, R. P., Weinblatt, M. E., et al. Immune function in patients with rheumatoid arthritis treated with etanercept. Clin Immunol, 2002, 103, p. 13–21.
59. Lee, S. J., Yedla, P., Kavanaugh, A. Secondary immune deficiencies associated with biological therapeutics. Curr Allergy Asthma Rep, 2003, 3, p. 389–395.
60. Mirzaei, A., Mahmoudi, H. Evaluation of TNF-α cytokine production in patients with tuberculosis compared to healthy people. GMS Hyg Infect Control, 2018, 13, Doc09.
61. Seeman, P. Clozapine, a fast-off-D2 antipsychotic. ACS Chem Neurosci, 2014, 5, p. 24–29.
62. Ponsford, M., Castle, D., Tahir, T., et al. Clozapine is associated with secondary antibody deficiency. Br J Psychiatry, 2018, p. 1–7.
63. Lozano, R., Marin, R., Santacruz, M. J., Pascual, A. Selective immunoglobulin M deficiency among clozapine-treated patients: a nested case-control study. Prim Care Companion CNS Disord, 2015, 17.
64. William, B. M., Corazza, G. R. Hyposplenism: a comprehensive review. Part I: basic concepts and causes. Hematology, 2007, 12, p. 1–13.
65. Siebert, A., Gensicka-Kowalewska, M., Cholewinski, G., Dzierzbicka, K. Tuftsin – properties and analogs. Curr Med Chem, 2017, 24, p. 3711–3727.
66. Di Sabatino, A., Carsetti, R., Corazza, G. R. Postsplenectomy and hyposplenic states. Lancet, 2011, 378, p. 86–97.
67. Brigden, M. L. Detection, education and management of the asplenic or hyposplenic patient. Am Fam Physician, 2001, 63, p. 499–506, 508.
68. King, H., Shumacker, H. B. Splenic studies. I. Susceptibility to infection after splenectomy performed in infancy. Ann Surg, 1952, 136, p. 239–242.
69. Price, V. E., Dutta, S., Blanchette, V. S., et al. The prevention and treatment of bacterial infections in children with asplenia or hyposplenia: practice considerations at the Hospital for Sick Children, Toronto. Pediatr Blood Cancer, 2006, 46, p. 597–603.
70. Rubin L. G, Levin M. J, Ljungman P., et al. 2013 IDSA clinical practice guideline for vaccination of the immunocompromised host. Clin Infect Dis, 2014, 58, p. 309–318.
71. Arnott, A., Jones, P., Franklin, L. J., et al. A registry for patients with asplenia/hyposplenism reduces the risk of infections with encapsulated organisms. Clin Infect Dis, 2018.
72. Orth, M., Lauber, K., Niyazi, M., et al. Current concepts in clinical radiation oncology. Radiat Environ Biophys, 2014, 53, p. 1–29.
73. Carvalho, H. A., Villar, R. C. Radiotherapy and immune response: the systemic effects of a local treatment. Clinics (Sao Paulo), 2018, 73, e557s.
74. Venkatesulu, B. P., Mallick, S., LIN, S. H., Krishnan, S. A systematic review of the influence of radiation-induced lymphopenia on survival outcomes in solid tumors. Crit Rev Oncol Hematol, 2018, 123, p. 42–51.
75. Manda, K., Glasow, A., Paape, D., Hildebrandt, G. Effects of ionizing radiation on the immune system with special emphasis on the interaction of dendritic and T cells. Front Oncol, 2012, 2, p. 102.
76. Merrick, A., Errington, F., Milward, K., et al. Immuno-suppressive effects of radiation on human dendritic cells: reduced IL-12 production on activation and impairment of naive T-cell priming. Br J Cancer, 2005, 92, p. 1450–1458.
77. Liu, H., Li, B., Jia, X., et al. Radiation-induced decrease of CD8+ dendritic cells contributes to Th1/Th2 shift. Int Immunopharmacol, 2017, 46, p. 178–185.
78. Formenti, S. C., Demaria, S. Systemic effects of local radiotherapy. Lancet Oncol, 2009, 10, p. 718–726.
79. Compagno, N., Malipiero, G., Cinetto, F., Agostini, C. Immunoglobulin replacement therapy in secondary hypogammaglobulinemia. Front Immunol, 2014, 5, 626.
80. Herriot, R., Sewell, W. A. Antibody deficiency. J Clin Pathol, 2008, 61, p. 994–1000.
Labels
Medical assessment Occupational medicineArticle was published in
Medical Revision

2020 Issue 1-2
Most read in this issue
- Modely zdravotního postižení a kompenzace důsledků zdravotního postižení
- Sekundární imunodeficience jako následek iatrogenního působení
- Současný pohled na odbornou kontrolu zdravotní pojišťovny v podmínkách systému veřejného zdravotního pojištění
- Poznatky revizního lékaře z potvrzování návrhů na lázeňskou léčebně rehabilitační péči