
Antley-Bixlerův syndrom nebo POR deficience?
Antley-Bixler syndrome or POR deficiency?
Antley-Bixler syndrome (ABS) is a rare congenital disorder characterized by numerous craniofacial, skeletal and, in some cases, urogenital abnormalities resulting from disordered steroidogenesis. Known genetic causes in sporadic cases of ABS include dominant mutations in the fibroblast growth factor 2 receptor gene (FGFR2). Recent research shows surprisingly that symptoms of Antley-Bixler syndrome, combined with disordered steroidogenesis and urogenital anomalies, are caused by mutations in the POR gene that encodes NADPH-cytochrome P450 oxidoreductase (CYPOR). CYPOR is a four domain-containing monomeric flavoprotein that contains two flavins, flavin adenine dinucleotide (FAD) and flavin mononucleotide (FMN), a binding site for NADPH, and the N-terminal sequence of 25 amino acids which determines the microsomal localization of the protein. CYPOR is the electron donor to microsomally localized cytochromes P450 that participate in xenobiotic metabolism and steroidogenesis. Mutations in the POR gene lead to apparent diminished activity of some P450 enzymes. Association of CYPOR with ABS discloses new facts about this disease and recent research shows that patients with ABS-like skeletal anomalies, but with mutations in the POR gene and disordered steroidogenesis, represent a new disorder called POR deficiency.
Key words:
Antley-Bixler syndrome, POR deficiency, NADPH-cytochrome P450 oxidoreductase, disordered steroidogenesis.
Autori:
M. Tomková 1; C. C. Marohnic 2; A. Baxová 1; P. Martásek 1
Pôsobisko autorov:
Klinika dětského a dorostového lékařství 1. LF UK a VFN, Praha
1; Department of Biochemistry, The University of Texas Health Science Center at San Antonio, San Antonio, Texas, USA
2
Vyšlo v časopise:
Čas. Lék. čes. 2008; 147: 261-265
Kategória:
Přehledový článek
Súhrn
Antley-Bixlerův syndrom (ABS) je vzácné kongenitální onemocnění charakterizované četnými kraniofaciálními, skeletálními a v některých případech i urogenitálními abnormalitami vyplývajícími z porušené steroidogeneze. Donedávna jedinou známou genetickou příčinou u sporadických případů ABS byly dominantní mutace v genu pro receptor pro fibroblastový růstový faktor 2 (FGFR2). Současný výzkum však velmi překvapivě odhalil, že za projevy Antley-Bixlerova syndromu spojeného s poruchami steroidogeneze a urogenitálními anomáliemi stojí mutace v genu POR kódujícím enzym NADPH-cytochrom P450 oxidoreduktázu (CYPOR). CYPOR je flavoprotein, obsahuje dva flaviny, flavin adenin dinukleotid (FAD) a flavin mononukleotid (FMN) a také sekvenci pro vazbu NADPH. Enzym je monomer organizován do čtyř domén. Sekvence 25 aminokyselin v N-terminální části proteinu zodpovídá za mikrozomální lokalizaci proteinu. CYPOR je hlavním elektronovým dárcem pro mikrozomálně lokalizované cytochromy P450, které se účastní metabolismu xenobiotik a biosyntézy steroidních hormonů. Mutace v genu POR vedou k zdánlivé insuficienci některých P450 enzymů. Asociace CYPOR s ABS odhalila nové skutečnosti ohledně tohoto onemocnění. Dle současných výsledků se zdá, že pacienti se skeletálními anomáliemi charakteristickými pro ABS, kteří však nesou mutaci v genu POR a mají porušenou steroidogenezi, představují nové onemocnění, POR deficienci.
Klíčová slova:
Antley-Bixlerův syndrom, POR deficience, NADPH-cytochrom P450 oxidoreduktáza, poruchy steroidogeneze.
Klinické projevy Antley-Bixlerova syndromu
Antley-Bixlerův syndrom (ABS, OMIM 207410) je vzácně se vyskytující, zato však velmi závažné onemocnění, charakterizované početnými kraniofaciálními, skeletálními a urogenitálními abnormalitami. První případ Antley-Bixlerova syndromu byl popsán v roce 1975 (1). K jeho nejběžnějším fenotypovým projevům patří brachycefalie, hypoplazie středního obličeje, kraniosynostózy, radiohumerální synostózy, ohýbání stehenních kostí a spontánní zlomeniny dlouhých kostí. Tyto malformace se objevují u většiny dosud popsaných případů (do dnešního dne je jich kolem 70). Mezi další projevy, kterými se syndrom může vyznačovat, patří zúžení či dokonce neprůchodnost choán, ztráta sluchu, ageneze ledvin, proptóza, arachnodaktylie, srdeční malformace a u části případů také poruchy steroidogeneze a obojaké pohlavní orgány. Pacienti se většinou dožívají nízkého věku, i když v posledních letech bylo popsáno více postižených jedinců, kteří se dožili dospělosti a mají dobré vyhlídky do dalšího života (2–4). Časná smrt byla zaznamenána přibližně u poloviny známých případů, přičemž hlavní příčinou bylo selhání respiračního systému (5). Rozvoj intelektuálních schopností postižených jedinců je pravděpodobně úzce spjat se dvěma faktory – kraniosynostózy a obstrukce horních dýchacích cest (6). Zdá se, že časné a efektivní zvládnutí těchto komplikací představuje nezbytný předpoklad pro zdravý duševní vývoj pacienta.
Etiologie Antley-Bixlerova syndromu
V současné době se původ ABS vysvětluje genetickou heterogenitou. Na počátku se předpokládal autozomálně recesivní typ dědičnosti. Tyto úvahy vycházely ze tří případů, ve kterých byl ABS zjištěn u sourozenců (7–9), a z případů, ve kterých se postižené dítě narodilo z příbuzenského manželského svazku (10, 11). Zbývající popsané případy se vyskytovaly sporadicky, proto se hledaly jiné možné příčiny vzniku onemocnění.
V roce 1998 Chun et al. (12) publikoval práci, ve které dítě s anomáliemi charakteristickými pro ABS neslo de novo mutaci v heterogenním stavu v genu pro receptor pro fibroblastový růstový faktor 2 (FGFR2). Autoři navrhli autozomálně dominantní typ dědičnosti s možným gonadálním mozaicismem. Ačkoli se objevily námitky, že pacient popsaný Chunem možná nereprezentuje pacienta s ABS (13, 14), asociace kraniosynostózy a kloubní ankylózy s dominantní mutací v FGFR2 byla potvrzena (15).
Dalším záchytným bodem při poznávání etiologie ABS bylo zjištění, že se fenotyp podobný ABS vyskytuje u potomků žen, kterým byly v raných stadiích těhotenství podávány vysoké dávky flukonazolu (16–18). Flukonazol je antimykotikum, které inhibuje biosyntézu ergosterolu v buněčných stěnách plísní. Jeho mechanismus účinku spočívá v tom, že prostřednictvím inhibice enzymu lanosterol-14-alfa-demetylázy (CYP51A1) zabraňuje demetylaci lanosterolu, důsledkem čehož je deplece ergosterolu v buněčných stěnách. V savčích buňkách hraje enzym CYP51A1 významnou roli při syntéze cholesterolu. V následujících studiích se sice nepotvrdil teratogenní účinek flukonazolu (19), ale tato diskuze a také abnormální růst pohlavních orgánů některých pacientů s ABS přiměly vědce ke studiu metabolismu steroidů u postižených jedinců.
Průlom v pochopení etologie ABS představovala práce Reardona et al. (20), v níž referoval o abnormálních steroidních profilech u 7 ze 16 pacientů s ABS, přičemž u jednoho z nich byla zjištěna mutace v FGFR2. Poruchy metabolismu steroidů byly mírného až průměrného charakteru a u pěti žen z této skupiny se zjistily anomálie ve vývoji pohlavních orgánů. Pozorované odchylky v steroidogenezi naznačovaly insuficienci enzymu 21-hydroxylázy (CYP21A2), ale DNA analýza u žádného ze zkoumaných pacientů neodhalila mutaci v genu pro CYP21A2. V této práci bylo poprvé upozorněno na fakt, že by jednotlivé případy ABS mohly být výsledkem mutací dvou různých genů, konkrétně genu pro FGFR2 a nějakého genu způsobujícího defekty v syntéze steroidů. Následovaly další práce o odchylkách v biosyntéze steroidních hormonů (2, 21). Po vyloučení mutací v genech kódujících enzymy, které u sledovaných pacientů prokazovaly nedostatečnou aktivitu (lanosterol-14-alfa-demetyláza, 17 alfa-hydroxyláza, 21-hydroxyláza) (2, 20, 21), byly jako příčina ABS rozpoznané mutace v genu POR kódujícím enzym NADPH-cytochrom P450 oxidoreduktázu (CYPOR) (22–24). CYPOR je hlavní elektronový dárce všech zmíněných enzymů.
Millerova skupina (22) zjistila u čtyř nepříbuzných pacientů s narušenou steroidogenezí 6 alelických variant genu POR (R457H, V492E, A287P, C569Y, V608F a varianta ve splicingu 6. intronu vedoucí k předčasnému vzniku stop kodonu). Tři z nich byli popsáni jako ABS pacienti a u žádného nebyla prokázána mutace v genu pro FGFR2. Arlt et al. (25) objevili jinou mutaci v genu POR (Y181D) tím, že analyzovali tento gen u pacientů s kongenitální adrenální hyperplazií. Několik nových mutací bylo popsáno u japonských pacientů. Ty zahrnovaly inzerce (I444fsX449, L565fsX574) (23), dále deleci (L612_W620delinsR), tichou mutaci (G5G) a missense mutaci (Y578C) (3). U všech popsaných pacientů byla do různé míry porušena steroidogeneze. Oba Adachiho pacienti (23) byli složení heterozygoti v genu POR a nenesli mutaci v genu pro FGFR2. Fukami et al. (3) zjistil u devíti z deseti pacientů homozygotní či složený heterozygotní stav pro pět typů mutací genu POR. Jeden pacient nesl mutaci jen na jedné alele. Analýza genu FGFR2 nebyla provedena. Dalších 11 missense mutací (A115V, T142A, Q153R, M263V, Y459H, A503V, G539R, L565P, R616X, V631I a F646del) bylo zjištěno ve studii, při které bylo analyzováno 32 pacientů s ABS. Cílem studie bylo zjistit distribuci mutací POR a FGFR2 (24). Sekvenace exonů POR a FGFR2 ukázala kompletní genetickou segregaci mutací POR a FGFR2. Patnáct pacientů neslo mutace v genu POR na obou alelách, čtyři na jedné alele, deset neslo mutace na jedné alele v genech FGFR2/3 a u tří pacientů nebyla nalezena žádná mutace. Tímto zjištěním Huang et al. jasně potvrdili dvojí původ ABS a značný podíl mutací v genu POR na jeho vzniku. Naposledy popsal tři nové POR mutace (Q201X, A462_S463insIA a E580Q) Homma et al. (26).
V únoru tohoto roku vstoupil do hry ještě jeden protein, který může hrát důležitou roli v etiologii ABS. Hughes et al. (27) referoval o mikrozomálně lokalizovaném hemoproteinu PGRMC1, který se váže na některé členy rodiny cytochromu P450 a pozitivně moduluje jejich funkci. PGRMC1 je v pořadí jen třetím známým proteinem (kromě CYPOR a cytochromu b5), který se váže na cytochromy P450.
NADPH-cytochrom P450 oxidoreduktáza
Enzym NADPH-cytochrom P450 oxidoreduktáza je membránově vázáný flavoprotein obsahující dva flaviny, flavin adenin dinukeotid (FAD) a flavin mononukleotid (FMN). Hlavní funkcí CYPOR je poskytovat redukující ekvivalent z NADPH všem členům mikrozomálních cytochromů P450 (obr. 1) a současně i řadě dalších akceptorů, jakými jsou například mikrozomálně lokalizovaný enzym hemové degradace, hem oxygenáza (28), elongáza mastných kyselin (29), skvalenová epoxidáza (30) nebo cytochrom b5 (31). Interakcí s mikrozomálními cytochromy se CYPOR účastní oxidativního metabolismu xenobiotik, léčiv a steroidů – a jinými biochemickými cestami, také metabolismu prostaglandinů a mastných kyselin.
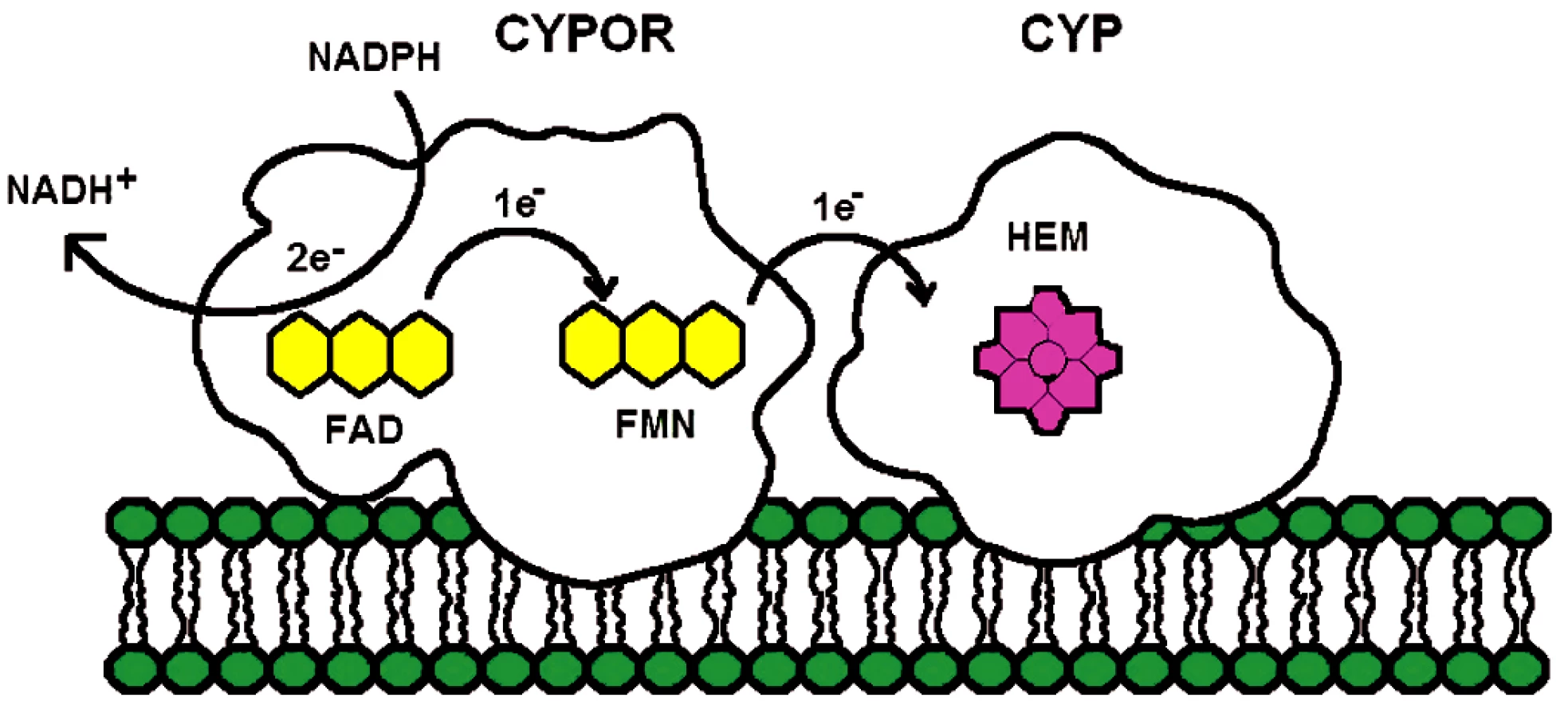
Enzym NADPH-cytochrom P450 oxidoreduktáza byl poprvé izolován a purifikován z jaterních buněk Horeckerem v roce 1950 (32). Tyto počáteční studie nedovolovaly lokalizovat enzym do žádné subcelulární organely. V roce 1956 objevili Strittmatter a Velick (33) při studiu aktivit NADH samostatnou mikrozomální frakci, která katalyzovala redukci cytochromu c dvacetinásobně rychleji s NADPH než s NADH. To dle jejich návrhu mohlo být díky aktivitě enzymu popsaného Horeckerem. Mikrozomální lokalizace CYPOR byla potvrzena ve studii Williamse a Kamina (34) a současně Phillipse a Langdona (35). Další důležitou událost v historii studia CYPOR představovalo zjištění, že enzym je flavoprotein obsahující jak FAD, tak FMN (35) a nejen FAD, identifikovaný již na samém počátku při izolaci enzymu. Lidskou a myší CYPOR poprvé sekvenoval Shephard et al. (37). Použitím Southern blot analýzy zjistili, že CYPOR je kódována jediným genem lokalizovaným na dlouhém raménku 7. chromozómu a pomocí in situ hybridizace upřesnili lokalizaci na 7q11.23. Gen pro CYPOR obsahuje 15 exonů. A nakonec, nedávno popsaná krystalová struktura CYPOR (38) ukázala, že enzym je monomer a je organizován do čtyř domén (obr. 2A). Sekvence 25 aminokyselin v N-terminální části proteinu je zodpovědná za mikrozomální lokalizaci proteinu a jeho vazbu na membránu.
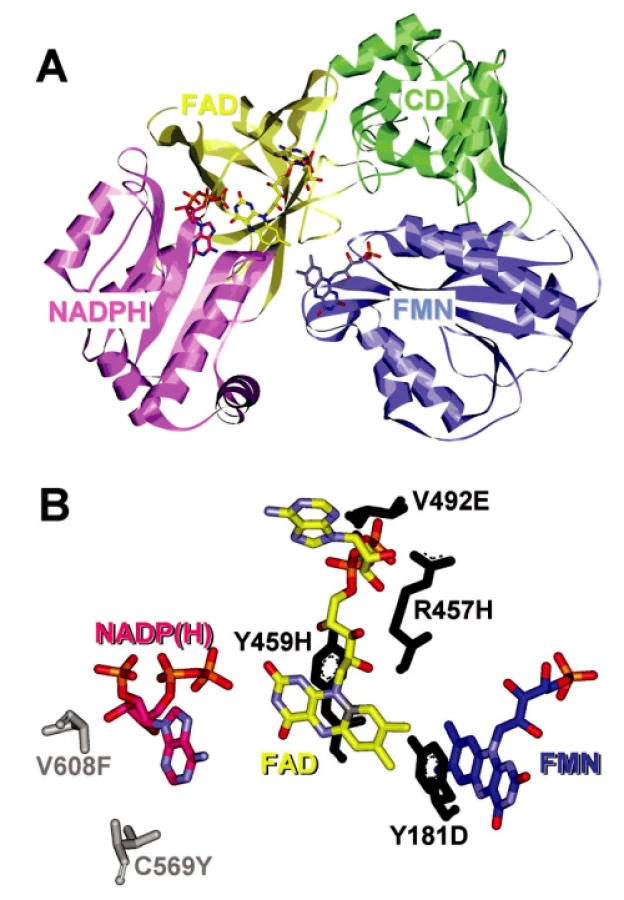
Fenotypové projevy různých mutací genu POR mají rozmanitý charakter. Bylo prokázáno, že pro embryo myši je nefunkční gen POR letální (39). Popsané mutace u pacientů s ABS jsou pravděpodobně mírnější povahy a zajišťují zbytkové aktivity CYPOR.
Porucha ve funkci NADPH-cytochrom P450 oxidoreduktázy vede ke zdánlivé deficienci CYPOR dependentních enzymů, CYP17A1 (17-alfa-hydroxyláza a 17,20-lyáza), CYP21A1 (21-hydroxyláza) a CYP51A1 (lanosterol-14-alfa-demetyláza). V nedávné práci Marohnic et al. prokázali, že některé mutace v POR snižují její afinitu k nezbytnému kofaktoru FAD (40). Je tedy porušen vztah koenzym – kofaktor a CYPOR tak nemůže poskytovat redukující ekvivalent svým akceptorům. Cytochromy P450 nemohou plnit svou funkci a jeví se jako deficientní. Co se týče steroidogeneze, ta je primárně inhibována právě sníženou aktivitou těchto enzymů (obr. 3).
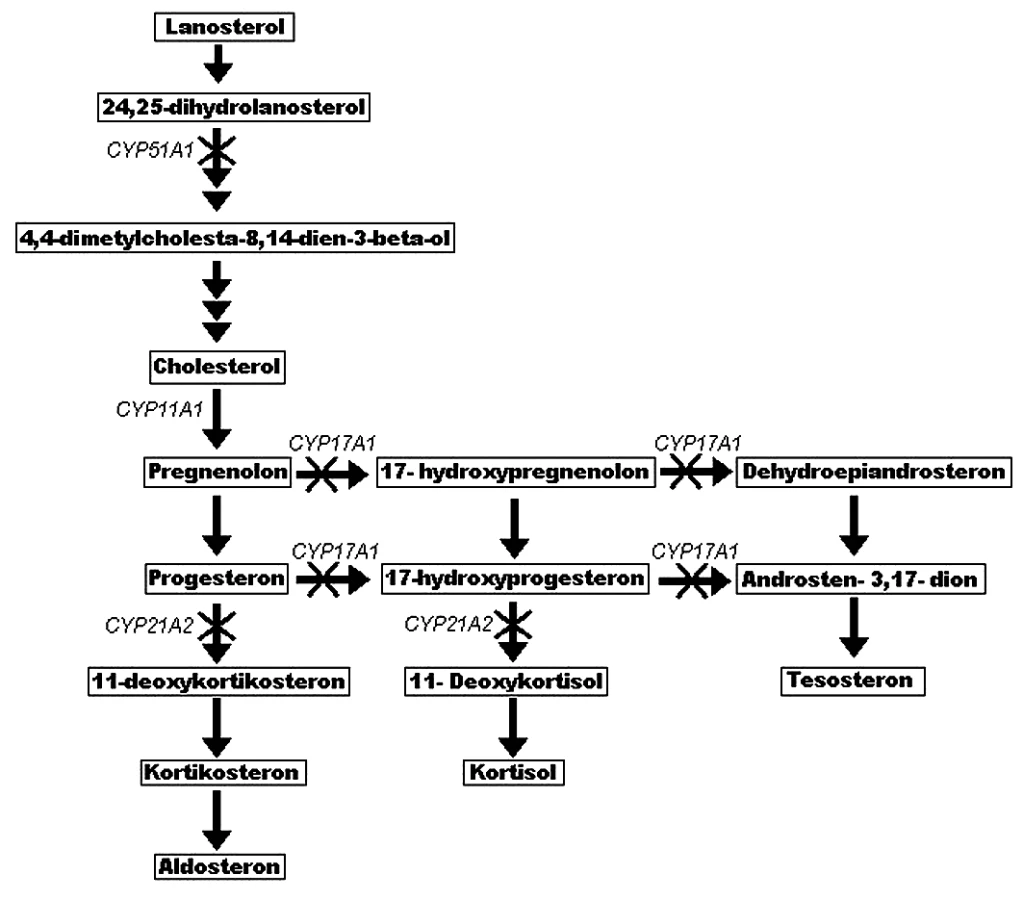
Vliv mutací v genu POR na vznik skeletálních abnormalit není jasný. Dle letálního vlivu POR -/- na embryo myši, u které byly pozorovány různé abnormality neurální trubice, srdce a končetin (39), je možné usuzovat, že vážné poškození CYPOR může být zodpovědné za skeletální projevy u některých jedinců s ABS a být příčinou prenatálního úmrtí plodu. Tato teorie byla poprvé vyslovena v práci Flücka et al. (22). Bylo také dokázáno, že u embrya myši s narušenou aktivitou CYPOR dependentního cytochromu metabolizujícího kyselinu retinovou (CYP26A1) dochází k poruchám vaskulogeneze a vývoje hlavy (41). CYP26A1 plní důležitou funkci při metabolismu kyseliny retinové, čímž chrání embryo před možnými teratogenními účinky této kyseliny. V současnosti se bádání zaměřuje také na studium metabolismu cholesterolu, protože je zřejmé, že některé nemoci s porušenou biosyntézou cholesterolu zahrnují i skeletální deformace (42). Tvorba cholesterolu je závislá na dvou CYPOR dependentních enzymech, CYP51A1 a skvalenové epoxidáze. Předpokládá se, že vznik anomálií bude mít souvislost hlavně se sníženou aktivitou CYP51A1 (43), ale základní mechanismus jejich vzniku musí být ještě objasněn.
Závěr
Je důležité si uvědomit, že klinický obraz pacientů s mutacemi v genu POR je velmi variabilní. Předpokládá se, že mutace v genu POR jsou běžnější, než je relativně nízká incidence ABS. Mírnější mutace můžou vést pouze k poruchám steroidogeneze, zvýšené senzitivitě na toxiny a léčiva (22) nebo k lehčím projevům syndromu. Výskyt mírnějších forem ABS potvrzují i některé studie (4). Většina popsaných POR missens mutací se nachází v oblastech důležitých pro vazbu kofaktorů NADPH, FAD a FMN a ve funkčně důležitých místech (obr. 2B), zatímco zdánlivé polymorfizmy se nacházejí v méně strukturálně významných oblastech (24, 25). Různé mutace genu POR ovlivňují aktivitu mikrozomálních P450 jiným způsobem a vedou k odlišným fenotypovým projevům, proto jsou nesmírně důležité studie, které by objasnily korelaci mezi variacemi v genu POR a fenotypem pacientů. Nové poznatky o etiologii ABS vedly k tomu, že v současnosti je už tendence užívat termín ABS jen pro pacienty se skeletálními deformacemi bez poruchy steroidogeneze a bez genitálních anomálií. Pro případ, kdy se u pacientů kromě skeletálních a kraniofaciálních malformací vyskytují i poruchy steroidogeneze a růstu genitálií a také mutace v genu POR, se již užívá nový název – POR deficience (24, 26, 44). Jak je vidět, lepší pochopení biologické a genetické podstaty onemocnění, vlivu různých polymorfismů v genu POR na fenotyp postižených a jejich frekvence v populaci vyžaduje další studium.
Zkratky
ABS – Antley-Bixlerův syndrom
CYP17A1 – 17-alfa-hydroxyláza
CYP21A2 – 21-hydroxyláza
CYP26A1 – cytochrom P450 metabolizující kyselinu retinovou
CYP51A1 – lanosterol-14-alfa-demetyláza
CYPOR – NADPH-cytochrom P450 oxidoreduktáza
FAD – flavin adenin dinukleotid
FGFR2 – receptor pro fibroblastový růstový faktor 2
FMN – flavin mononukleotid
POR – gen pro NADPH-cytochrom P450 oxidoreduktázu
Práce vznikla díky podpoře grantů GA UK 252000100007 a VZ 64165.
prof. MUDr. Pavel Martásek, DrSc.
Klinika dětského a dorostového lékařství 1. LF UK a VFN
Ke Karlovu 2, 121 08 Praha 2
fax: +420 224 967 099, e-mail: pavel.martasek@gmail.com
Zdroje
1. Antley, R. M., Bixler, D.: Trapezoidocephaly, midface hypoplasia and cartilage abnormalities with multiple synostoses and skeletal fractures. Birth Defects Orig. Art. Ser., 1975, 11, s. 397–401.
2. Adachi, M., Asakura, Y., Tachibana, K., Shackleton, C.: Abnormal steroidogenesis in three patients with Antley-Bixler syndrome: apparent decreased activity of 17alpha-hydroxylase, 17,20-lyase and 21-hydroxylase. Pediatr. Int., 2004, 46, s. 583–589.
3. Fukami, M., Horikawa, R., Nagai, T. et al.: Cytochrome P450 Oxidoreductase Gene Mutations and Antley-Bixler Syndrome with Abnormal Genitalia and/or Impaired Steroidogenesis: Molecular and Clinical Studies in 10 Patients. J. Clin. Endocr. Metab., 2005, 90, s. 414–426.
4. Williamson, L., Arlt, W., Shackleton, C. et al.: Linking Antley-Bixler syndrome and congenital adrenal hyperplasia: a novel case of P450 oxidoreductase deficiency. Am. J. Med. Genet., 2006, 140A, s. 1797–1803.
5. Hassell, S., Butler, M. G.: Antley-Bixler syndrome: report of a patient and review of literature. Clin. Genet., 1994, 46, s. 372–376.
6. Bottero, L., Cinalli, G., Labrune, P. et al.: Antley-Bixler syndrome: Description of two new cases and a review of the literature. Childs. Nerv. Syst., 1997, 13, s. 275–281.
7. Schinzel, A., Savoldelli, G., Briner, J. et al.: Antley-Bixler syndrome in sisters: a term newborn and a prenatally diagnosed fetus. Am. J. Med. Genet., 1983, 14, s. 139–147.
8. LeHeup, B. P., Masutti, J. P., Droulle, P., Tisserand, J.: The Antley-Bixler syndrome: report of two familial cases with severe renal and anal anomalies. Europ. J. Pediat., 1995, 154, s. 130–133.
9. Suzuki, K., Kanda, Y., Sugiyama, K. et al.: Antley-Bixler syndrome in a sister and brother. Jpn. J. Hum. Genet., 1987, 32, s. 247–252.
10. Yasui, Y., Yamaguchi, A., Itoh, Y. et al.: The first case of the Antley-Bixler syndrome with consanguinity in Japan. Jpn. J. Hum. Genet., 1983, 28, s. 215–220.
11. Feigin, E., Udassin, R., Seror, D. et al.: Antley-Bixler syndrome and esophageal atresia in a patient with trisomy 21. Clin. Genet., 1995, 47, s. 53–55.
12. Chun, K., Siegel-Bartelt, J., Chitayat, D. et al.: FGFR2 mutation associated with clinical manifestations consistent with Antley-Bixler syndrome. Am. J. Med. Genet., 1998, 77, s. 219–224.
13. Gorlin, R. J.: Patient described by Chun et al. may not present Antley-Bixler syndrome. (Letter). Am. J. Med. Genet., 1999, 83, s. 64.
14. Gripp, K. W., Zackai, E. H., Cohen, M. M., Jr.: Not Antley--Bixler syndrome. (Letter). Am. J. Med. Genet., 1999, 83, s. 65–66.
15. Tsai, F. J., Wu, J. Y., Yang, C. F., Tsai, C. H.: Further evidence that fibroblast growth factor receptor 2 mutations cause Antley-Bixler syndrome. Acta Paediatr., 2001, 90, s. 595–597.
16. Lee, B. E., Feinberg, M., Abraham J. J., Murthy, A. R.: Congenital malformation in an infant born to a woman treated with fluconazole. Pediatr. Infect. Dis. J., 1992, 11, s. 1062–1064.
17. Pursley, T. J., Blomquist, I. K., Abraham, J. et al.: Fluconazole-induced congenital anomalies in three infants. Clin. Infect. Dis., 1996, 22, s. 336–340.
18. Aleck, K. A., Bartley, D. L.: Multiple malformation syndrome following fluconazole use in pregnancy: report of an additional patient. Am. J. Med. Genet., 1997, 72, s. 253–256.
19. Jick, S. S.: Pregnancy outcomes after maternal exposure to fluconazole. Pharmacotherapy, 1999, 19, s. 221–222.
20. Reardon, W., Smith, A., Honour, J. W. et al.: Evidence for digenic inheritance in some cases of Antley-Bixler syndrome? J. Med. Genet., 2000, 37, s. 26–32.
21. Kelley, R. I., Kratz, L. E., Glaser, R. L. et al.: Abnormal sterol metabolism in a patient with Antley-Bixler syndrome and ambiguous genitalia. Am. J. Med. Genet., 2002, 110, s. 95–102.
22. Flück, C. E., Tajima, T., Pandey, A. V. et al.: Mutant P450 oxidoreductase causes disordered steroidogenesis with and without Antley-Bixler syndrome. Nat. Genet., 2004, 36, s. 228–230.
23. Adachi, M., Tachibana, K., Asakura, Y. et al.: Compound heterozygous mutations of cytochrome P450 oxidoreductase gene (POR) in two patients with Antley-Bixler syndrome. Am. J. Med. Genet. A, 2004b, 128A, s. 333–339.
24. Huang, N., Pandey, A. V., Agrawal, V. et al.: Diversity and function of mutations in p450 oxidoreductase in patients with Antley-Bixler syndrome and disordered steroidogenesis. Am. J. Hum. Genet., 2005, 76, s. 729–749.
25. Arlt, W., Walker, E. A., Draper, N. et al.: Congenital adrenal hyperplasia caused by mutant P450 oxidoreductase and human androgen synthesis: analytical study. Lancet, 2004, 363, s. 2128–2135.
26. Homma, K., Hasegawa, T., Nagai, T. et al.: Urine Steroid Hormone Profile Analysis in Cytochrome P450 Oxidoreductase Deficiency: Implication for the Backdoor Pathway to Dihydrotestosterone. J. Clin. Endocrinol. Metab., 2006, 91, s. 2643–2649.
27. Hughes, A. L., Powell, D. W., Bard, M. et al.: Dap1/PGRMC1 binds and regulates cytochrome P450 enzymes. Cell. Metab., 2007, 5, s. 143–149.
28. Schacter, B. A., Nelson, E. B., Marver, H. S., Masters, B. S.: Immunochemical evidence for an association of heme oxygenase with the microsomal electron transport system. J. Biol. Chem., 1972, 247, s. 3601–3607.
29. Ilan, Z., Ilan, R., Cinti, D. L.: Evidence for a new physiological role of hepatic NADPH:ferricytochrome (P-450) oxidoreductase. Direct electron input to the microsomal fatty acid chain elongation system. J. Biol. Chem., 1981, 256, s. 10066–10072.
30. Ono, T., Bloch, K.: Solubilization and partial characterization of rat liver squalene epoxidase. J. Biol. Chem., 1975, 250, s. 1571–1579.
31. Enoch, H. G., Strittmatter, P.: Cytochrome b5 reduction by NADPH-cytochrome P-450 reductase. J. Biol. Chem., 1979, 254, s. 8976–8981.
32. Horecker, B. L.: Triphosphopyridine nucleotide-cytochrome c reductase in liver. J. Biol. Chem., 1950, 183, s. 593–605.
33. Strittmatter, P., Velick, S. F.: The isolation and properties of microsomal cytochrome. J. Biol. Chem., 1956, 221, s. 253–264.
34. Williams, C. J., Kamin, H.: Microsomal triphosphopyridine nucleotide-cytochrome c reductase of liver. J. Biol. Chem., 1962, 237, s. 587–595.
35. Phillips, A. H., Langdon, R. G.: Hepatic triphosphopyridine nucleotide-cytochrome c reductase: isolation, characterization, and kinetic studies. J. Biol. Chem., 1962, 237, s. 2652–2660.
36. Iyanagi, T., Mason, H. S.: Some properties of hepatic reduced nicotinamide adenine dinucleotide phosphate-cytochrome c reductase. Biochemistry, 1973, 12, s. 2297–2308.
37. Shephard, E.A., Phillips, I. R., Santisteban, I. et al.: Isolation of a human cytochrome P–450 reductase cDNA clone and localization of the corresponding gene to chromosome 7q11.2. Ann. Hum. Genet., 1989, 53, s. 291–301.
38. Wang, M., Roberts, D. L., Paschke, R. et al.: Three-dimensional structure of NADPH-cytochrome P450 reductase: prototype for FMN- and FAD-containing enzymes. Proc. Natl. Acad. Sci. USA., 1997, 94, s. 8411–8416.
39. Shen, A. L., O’Leary, K. A., Kasper, C. B.: Association of multiple developmental defects and embryonic lethality with loss of microsomal NADPH-cytochrome P450 oxidoreductase. J. Biol. Chem., 2002, 277, s. 6536–6541.
40. Marohnic, C. C., Panda, S. P., Martásek, P., Masters, B. S.: Diminished FAD binding in the Y459H & V492E Antley--Bixler syndrome mutants of human cytochrome P450 reductase. J. Biol. Chem., 2006, 281, s. 35975–35982.
41. Ribes, V., Fraulob, V., Petkovich, M., Dolle, P.: The oxidizing enzyme CYP26A1 tightly regulates the availability of retinoic acid in the gastrulating mouse embryo to ensure proper head development and vasculogenesis. Dev. Dyn., 2007, 236, s. 644–653.
42. Herman, G. E.: Disorders of cholesterol biosynthesis: prototypic metabolic malformation syndromes. Hum. Mol. Genet., 2003, 12, s. R75–R88.
43. Debeljak, N., Fink, M., Rozman, D.: Many facets of mammalian lanosterol 14alpha-demethylase from the evolutionarily conserved cytochrome P450 family CYP51. Arch. Biochem. Biophys., 2003, 409, s. 159–171.
44. Miller, W. L., Huang, N., Pandey, A. V. et al.: P450 oxidoreductase deficiency: a new disorder of steroidogenesis. Ann. N. Y. Acad. Sci., 2005, 1061, s. 100–108.
Štítky
Adiktológia Alergológia a imunológia Angiológia Audiológia a foniatria Biochémia Dermatológia Detská gastroenterológia Detská chirurgia Detská kardiológia Detská neurológia Detská otorinolaryngológia Detská psychiatria Detská reumatológia Diabetológia Farmácia Chirurgia cievna Algeziológia Dentální hygienistkaČlánok vyšiel v časopise
Časopis lékařů českých

- Metamizol jako analgetikum první volby: kdy, pro koho, jak a proč?
- Fixní kombinace paracetamol/kodein nabízí synergické analgetické účinky
- Tramadol a paracetamol v tlumení poextrakční bolesti
- Realita liečby bolesti v paliatívnej starostlivosti v Nemecku
Najčítanejšie v tomto čísle
- Antley-Bixlerův syndrom nebo POR deficience?
- Deset let nového anatomického názvosloví
- Vliv psychického stresu na zdravotní stav obviněného v průběhu trestního řízení
- Lékařská etika a etikoterapie