
Kongenitální deficit surfaktantu v důsledku mutace v genu pro ABCA3 jako příčina fatálního respiračního selhání
Congenital surfactant deficiency due to ABCA3 mutations leading to fatal respiratory failure in a newborn
Inherited disorders of surfactant metabolism present as acute severe respiratory failure in the neonatal period or as chronic respiratory insufficiency in later infancy and childhood. We report the first case of genetically proved surfactant deficiency in term newborn in the Czech Republic leading to lethal respiratory failure. This is also the first proof of these mutations at all. The two heterozygous mutations in ABCA3 gene – in exon 24 M 1227R and in exon 29 Ins1510fs/ter1519 have never been published yet.
Key words:
newborn, surfactant deficiency, ABCA3 mutations
Autoři:
M. Navratilova 1; H. Hornychová 2; Z. Kokštein 1; J. Malý 1
Působiště autorů:
Dětská klinika FN a LF UK, Hradec Královépřednosta prof. MUDr. M. Bayer, CSc.
1; Fingerlandův ústav patologie FN a LF UK, Hradec Královépřednosta prof. MUDr. A. Ryška, Ph. D.
2
Vyšlo v časopise:
Čes-slov Pediat 2013; 68 (3): 161-166.
Kategorie:
Kazuistika
Souhrn
Vrozené poruchy metabolismu surfaktantu se manifestují jako akutní závažná respirační selhání v novorozeneckém období, nebo jako chronická respirační insuficience v kojeneckém a dětském věku. Popisujeme první případ donošeného novorozence v České republice s geneticky prokázaným deficitem surfaktantu vedoucím k letálnímu respiračnímu selhání. Zároveň jde celosvětově o první průkaz heterozygotních mutací M 1227R v exonu 24 a Ins1510fs/ter1519 v exonu 29 v ABCA3 genu.
Klíčová slova:
novorozenec, deficit surfaktantu, mutace ABCA3
ÚVOD
Plicní surfaktant je unikátní komplex fosfolipidů a proteinů syntetizovaný, skladovaný a secernovaný pneumocyty II. řádu. Téměř 90 % plicního surfaktantu je tvořeno fosfolipidy, převážně fosfatidylcholinem. Zbývajících cca 10 % tvoří surfaktantové proteiny A, B, C a D (SP-A, SP-B, SP-C, SP-D). SP-B a SP-C spolu s dipalmytoylfosfatidylcholinem snižují povrchové napětí v alveolech a zabraňují jejich kolapsu na konci výdechu. SP-A a SP-D se podílejí na obranyschopnosti plic proti bakteriím, virům i mykotickým agens. Pro funkci surfaktantu jsou významné i další molekuly. Granulocyty-makrofágy kolonie stimulující faktor (GM-CSF) spolu se SP-A přispívají k recyklaci a katabolismu surfaktantu. ATP-binding casette transporter A3 (ABCA3) je transportní protein zodpovědný za přesun fosfolipidů do lamelárních tělísek – organel pneumocytů II. řádu, ve kterých probíhá konečné zpracování surfaktantových komponent před jejich sekrecí. Thyroidní transkripční faktor 1, kódovaný NKX2.1 genem, reguluje expresi mnoha genů esenciálních pro vývoj a funkci plic včetně těch, které kódují SP-B a SP-C [1].
Vrozené poruchy metabolismu surfaktantu jsou způsobeny mutacemi v genech kódujících výše zmíněné molekuly. Genová mutace pak vede ke kvantitativnímu nebo, zřejmě častěji, ke kvalitativnímu deficitu surfaktantu. Klinicky se manifestují jako závažné respirační selhání v novorozeneckém období, nebo jako chronická respirační insuficience v pozdním kojeneckém či dětském věku s variabilním průběhem a tíží onemocnění [1]. Typický pacient s mutacemi genů pro SP-B nebo ABCA3 je donošený novorozenec, u kterého se krátce (hodiny) po narození rozvíjí respirační selhání s nutností ventilační podpory, včetně nekonvenčních modů (HFOV – vysokofrekvenční oscilační ventilace, ECMO – extrakorporální membránová oxygenoterapie) a jen minimálně či přechodně reagující na podání exogenního surfaktantu. Na rtg hrudníku nalezneme difuzní zastínění plic se vzdušným bronchogramem připomínající obraz syndromu dechové tísně (RDS – respiratory distress syndrome) nedonošených novorozenců. U dětí s mutacemi v SP-C, ABCA3 a NKX2.1 genech je klinická prezentace variabilní, včetně doby prvních příznaků, které mohou začít už v novorozeneckém období jako mírné respirační potíže, nebo dojde k rozvoji pozvolna progredující hypoxemie a respiračního selhání v kojeneckém či dětském věku. Na rtg hrudníku bývá obraz plicního intersticiálního procesu [1, 2].
KAZUISTIKA
Na jednotku intenzivní a resuscitační péče pro novorozence Dětské kliniky FN a LF UK Hradec Králové byl přeložen ze spádové porodnice chlapec, který se narodil ve 38. gestačním týdnu s porodní hmotností 3670 g. Gravidita i porod proběhly bez komplikací, beta-hemolytický streptokok skupiny B v pochvě matky nebyl kultivačně zachycen. Chlapec byl prvním potomkem nepříbuzných rodičů, kteří již měli zdravé děti s jinými partnery. Po dobré adaptaci na porodním sále (Apgar skóre 5-10-10) došlo u dítěte ve 30. minutě života k rozvoji dechové tísně (intermitentní grunting, tachypnoe 70/min, potřeba oxygenoterapie) vyžadující transport do perinatologického centra. Pro zmíněné potíže byla již během transportu zahájena distenční dechová podpora aplikací kontinuálního přetlaku v dýchacích cestách (NCPAP) s potřebou FiO2 do 30 %.
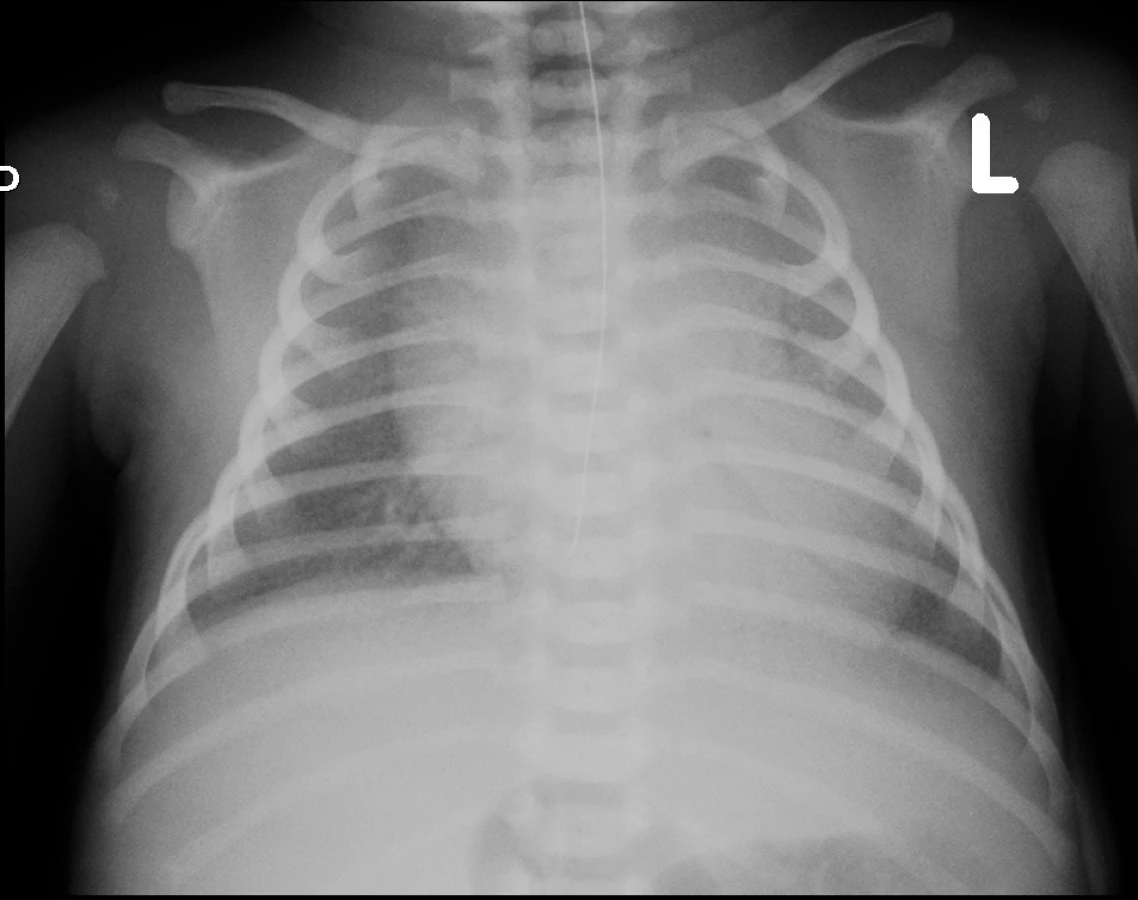
Na vstupním rtg hrudníku 5 hodin po narození byl nález zmnožené intersticiální kresby při uspokojivé vzdušnosti plic. UZ vyšetřením srdce byla vyloučena vrozená srdeční vada a perzistující plicní hypertenze. Pro elevaci interleukinu 6 v séru (184 ng/l) byla zahájena léčba antibiotiky (ampicilin/sulbactam). Ve stáří 14 hodin došlo k progresi dechových potíží s významnou potřebou oxygenoterapie a při současné globální respirační insuficienci (arteriální Astrup: pH 7,229; pCO2 7,45 kPa; pO2 5,02 kPa) byla zahájena UPV s potřebou FiO2 až 100 %. Kontrolní rtg nález (obr. 1) byl hodnocen jako syndrom dechové tísně a ve stáří 15 hodin byl podán surfaktant v dávce 130 mg/kg. Prakticky okamžitě došlo k významnému zlepšení ventilačních parametrů a snížení FiO2 k 21 %, což trvalo téměř 2 hodiny. Poté opět narůstala potřeba kyslíkové léčby, chlapec začal být oběhově nestabilní, hypotenze byla korigována zvýšeným přívodem tekutin a kombinací katecholaminů (dopamin, dobutamin). Při FiO2 téměř 100 % byla ve stáří 23 hodin zahájena HFOV. Laboratorní výsledky v této době připouštěly možnost časné infekce, proto byla ATB léčba upravena na kombinaci (ampicilin, gentamicin). Po zahájení HFOV se pacient po ventilační i oběhové stránce stabilizoval, během 8 hodin postupně klesla potřeba oxygenoterapie ke 30 % a byla ukončena podpora oběhu. Materiály odebírané ke standardním mikrobiologickým vyšetřením byly sterilní, rovněž PCR vyšetření dostupných infekčních agens z tracheálního aspirátu přineslo negativní výsledek (Mycoplasma hominis, Mycoplasma pneumoniae, Chlamydia trachomatis, Chlamydia pneumoniae, Ureaplasma species, průkaz DNA Streptococcus agalactiae). V průběhu 2. týdne života byla, při prakticky neměnném klinickém stavu s nutností HFOV a významné frakce kyslíku (FiO2 40–60 %), s velkými rozpaky zahájena léčba makrolidovými antibiotiky (clarithromycin) a antimykotikem (fluconazol), bez zjevného efektu na klinický průběh. Na konci 2. týdne jsme se pokusili ovlivnit průběh onemocnění podáním kortikoidů, rovněž bez klinického zlepšení.
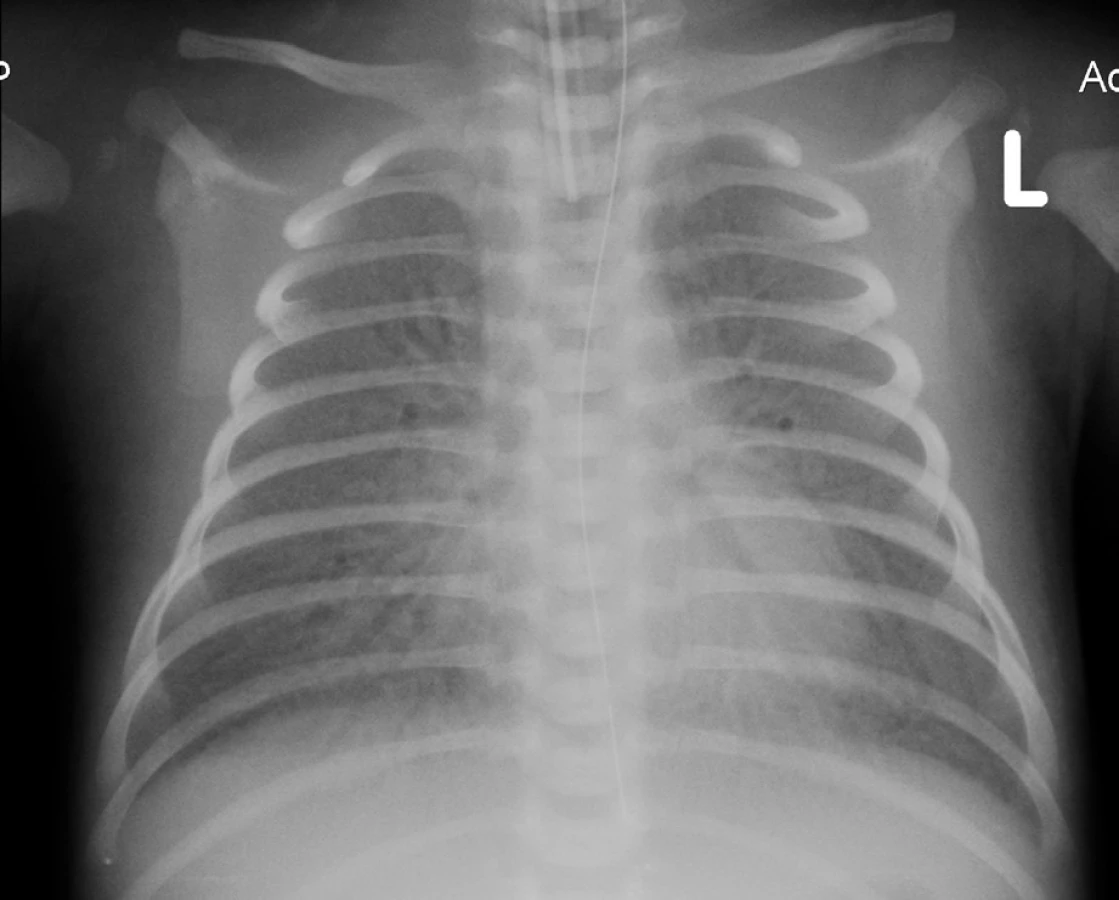
Na základě klinického průběhu onemocnění a rtg plic (obr. 2) jsme 9. den života vyslovili podezření na vrozený deficit metabolismu surfaktantu. Byla provedena bronchoskopie s normálním makroskopickým nálezem na sliznici přehlédnutelných bronchů a odběrem bronchoalveolární laváže. Cytologický nález nesvědčil pro obraz alveolární proteinózy (PAP). Byly však zastiženy četné makrofágy s jemně pěnitou cytoplazmou a ojediněle i pigmentem (obr. 3), tedy nález svědčící pro diagnózu deskvamativní intersticiální pneumonie (DIP) a tím vrozené poruchy metabolismu surfaktantu.
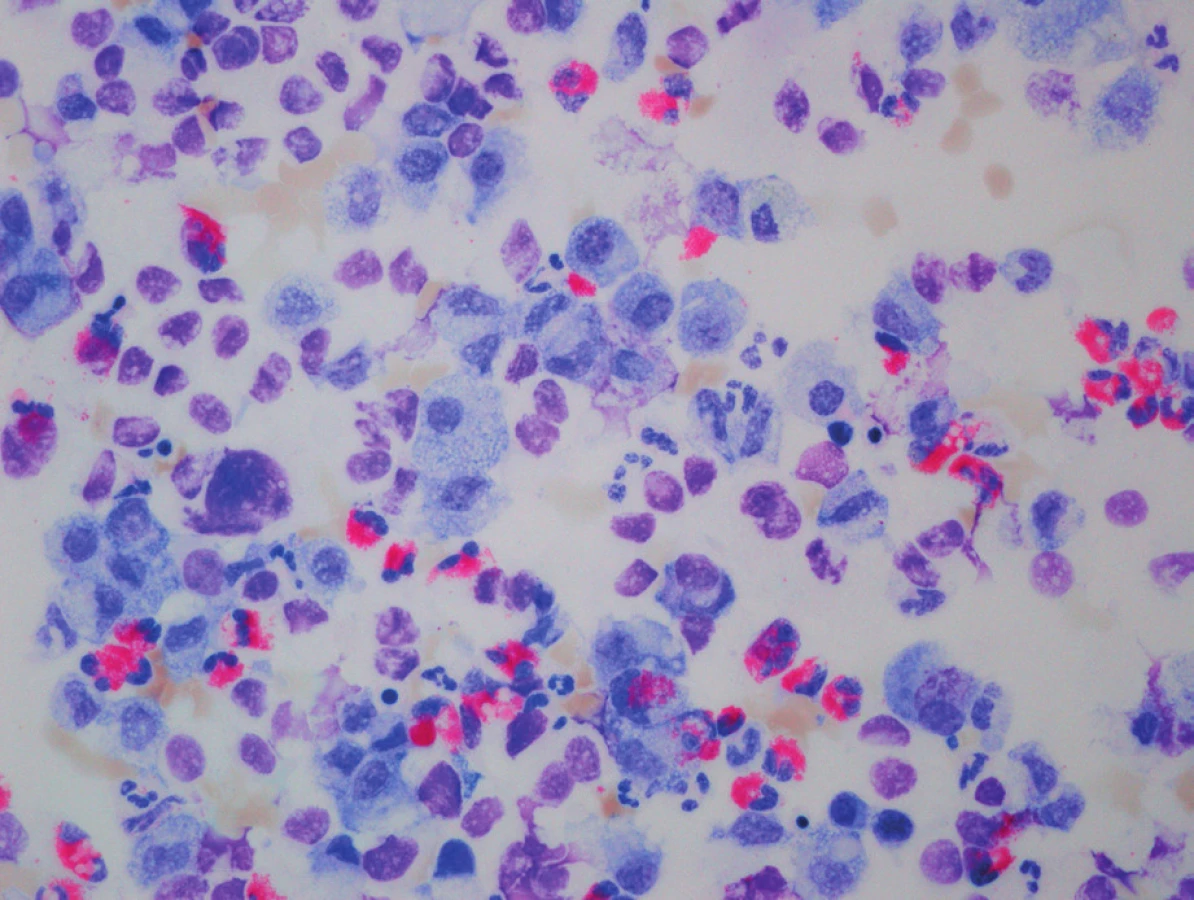
Analýza DNA izolované z krve byla provedena v Gene Analysis Service v Berlíně. Deficit SP-C byl podle klinického průběhu nejméně pravděpodobný, proto nebyl vyšetřován. Vzhledem k akutní manifestaci onemocnění u našeho novorozence byla nejprve provedena analýza SP-B genu a vyšetřením exonů 2, 4, 5, 7 byl deficit SP-B vyloučen. Postupnou sekvenční analýzou genu ABCA3 (exonů 5, 14, 21, 23, 24, 30 a 31 v prvním kroku, exonů 4, 7, 9, 11, 22, 25, 26, 28 ve druhém kroku, exonů 6, 8, 10, 12, 13, 15, 18, 29 ve třetím kroku a exonů 16, 17, 19, 20, 27 a 32 ke kompletaci vyšetření) byly odhaleny 2 heterozygotní mutace. V exonu 24 byla detekována mutace M 1227R (missence mutace se záměnou báze ATG>AGG v kodonu 1227). Již tato první heterozygotní mutace ve významné části genu byla považována za možnou příčinu klinických obtíží. Druhá heterozygotní mutace Ins1510fs/ter1519 (frameshift mutace s předčasnou terminací v kodonu 1519) byla nalezena v exonu 29. Výše uvedené mutace nebyly doposud popsány, proto byly výsledky konzultovány s profesorem Lawrencem Nogeem (Johns Hopkins University School of Medicine, Baltimore, USA). S odkazem na literaturu [4, 5] byly mutace našeho pacienta vzhledem k jejich genové lokaci a klinickému průběhu shledány letálními, s infaustní prognózou pro pacienta.
V průběhu DNA diagnostiky se klinický stav pacienta neměnil, vyjma 1 dne nebylo možné pacienta převést na konvenční formu UPV a hodnoty acidobazické rovnováhy byly při uvedené léčbě ve fyziologických mezích. Po opakovaných diskusích v rámci celoklinických seminářů i s rodiči bylo rozhodnuto o zadržení resuscitační péče s cílem zajištění maximálního možného komfortu pacienta. Chlapec zemřel v přítomnosti rodičů 33. den života.
DISKUSE
ABCA3 patří do skupiny ATP-binding casette proteinů, které aktivně transportují řadu substancí přes biologické membrány. Jedná se o velký integrální membránový protein, který je vysoce exprimován v plicích a je součástí membrány lamelárních tělísek pneumocytů II. řádu [3, 4]. Gen kódující tento protein je lokalizován na chromozomu 16 (16p13.3). ABCA3 importuje surfaktantové fosfolipidy – fosfatidylcholin a fosfatidylglycerol z cytosolu do lamelárních tělísek [3]. Nedostatek těchto látek ve vlastním surfaktantu vede k jeho snížené funkci [1]. Frekvence onemocnění v populaci v důsledku recesivních mutací není známá, i když z předběžných studií se zdá, že ABCA3 deficity jsou mezi poruchami metabolismu surfaktantu nejčastější. Do roku 2010 bylo identifikováno více než 180 mutací v souvislosti s letálním RDS u novorozenců či chronickou respirační insuficiencí u dětí [1].
V současné době neexistuje biochemický marker, který by pomohl v diagnostice ABCA3 deficitu. Z tracheálního aspirátu lze teoreticky vyšetřit imunoreaktivní SP-B metodou ELISA či Western blot [6]. Toto vyšetření je limitováno jeho obtížnou dostupností. V diagnostice může pomoci histopatologické vyšetření plicní tkáně. Genetické poruchy metabolismu surfaktantu mohou nabývat tří základních histologických obrazů. Nejčastější je plicní alveolární proteinóza (PAP), kdy nacházíme alveolární prostory kompletně vyplněné homogenním amorfním materiálem. Tento morfologický obraz je charakteristický pro mutace genu pro SP-B, méně často se vyskytuje i při mutaci ABCA3 genu, někdy však může provázet i virové infekce spojené s rozsáhlou nekrózou alveolárního epitelu. Dalším histologickým nálezem spojeným s poruchou metabolismu surfaktantu je chronická pneu-monitida kojenců (CPI – chronic pneumonitis of infancy). Bývá nalézána u kojenců či menších dětí a nejčastěji je spojena s mutací genu pro SP-C. Poslední třetí typ histologického obrazu, který bývá patrný u poruch metabolismu surfaktantu, je deskvamativní intersticiální pneumonie (DIP). Alveoly jsou vyplněny makrofágy s pigmentem, který je jen velmi slabě pozitivní při průkazu železa. Tento obraz je poměrně málo specifický, mimo defekty metabolismu surfaktantu (zejména ABCA3 deficit), může odpovídat také toxické reakci, aspiraci nebo virové infekci [7]. PAP i DIP lze prokázat i ze vzorku broncholaveolární laváže (BAL). To bylo důvodem k provedení bronchoskopie a vyšetření BAL u našeho pacienta. Cílem bylo získání vzorků k histologickému a případně elektronmikroskopickému vyšetření. PAP nebyla prokázána, ale nález byl kompatibilní s diagnózou DIP. Elektronmikroskopickým vyšetřením morfologie lamelárních tělísek lze odlišit SP-B a ABCA3 deficity. Zatímco při SP-B deficitu bývají v cytosolu pneumocytů II. řádu nebo uvnitř plicních sklípků velká lamelární tělíska ohraničená membránou obsahující malé vezikuly, při ABCA3 deficitu jsou lamelární tělíska malá, s excentricky uloženými denzními inkluzemi [3]. Jednoznačnou metodou pro průkaz vrozených deficitů surfaktantu zůstává sekvenční analýza genomové DNA. Pacientova DNA byla po zajištění patřičných náležitostí odeslána do Gene Analysis Service v Berlíně, kde byl diagnostikován ABCA3 deficit průkazem dvou výše uvedených heterozygotních mutací.
[Adresa k zapamatování: Gene Analysis Service GmbH, Motzener Str. 49, 12277 Berlin, Germany, tel +49-30-84707430, www.gene-analysis-service.de, looman@gene-analysis-service.de].
Kauzální léčba vrozených poruch metabolismu surfaktantu neexistuje, u chronicky probíhajících plicních procesů způsobených vrozeným deficitem surfaktantu je možným řešením transplantace plic. Její indikace je závislá na kombinaci mnoha faktorů. Těmi jsou klinický průběh onemocnění, absence extrapulmonálních orgánových dysfunkcí, možnost transportu do dětského transplantačního centra a přání rodičů. Cílené vyšetření DNA rodičů vzhledem ke znalosti genotypu dítěte není nutné, pro další graviditu je možné nabídnout prenatální poradenství včetně preimplantační diagnostiky. Vyšetření ostatních nevlastních sourozenců na nosičství mutace v ABCA3 genu je třeba zvážit při plánování potomků, riziko opakování této nemoci pro jejich děti je málo pravděpodobné.
ZÁVĚR
Kongenitální deficity surfaktantu jsou raritním onemocněním. Je však třeba na ně myslet v souvislosti s etiologicky nevysvětlitelným respiračním selháním, zvláště u donošených novorozenců, ale také u kojenců a starších dětí. Jednoznačným vyšetřením, které vede k diagnóze, je DNA analýza s přímým průkazem mutací.
Poděkování
Poděkování patří dr. Alfredu C. Loomanovi, molekulárnímu biologovi z Gene Analysis Service GmbH v Berlíně, za rychlou diagnostiku ABCA3 deficitu u našeho pacienta a prof. Lawrenci Nogeemu z Division of Neonatology, Department of Pediatrics, Johns Hopkins University School of Medicine, Baltimore, USA, za spolupráci při interpretaci výsledků DNA analýzy.
Podpořeno programem PRVOUK P37/12.
Došlo: 1. 5. 2012
Přijato: 30. 7. 2012
Korespondující autor
MUDr. Jan Malý, Ph.D.
Dětská klinika FN a LF UK
Sokolská 581
500 05 Hradec Králové
e-mail: malyj@lfhk.cuni.cz
Zdroje
1. Hamvas A. Evaluation and management of inherited disorders of surfactant metabolism. Chin Med J (Peking) 2010; 123 (20): 2943–2947.
2. Bullard JE, Wert SE, Whitsett JA, et al. ABCA3 mutations associated with pediatric interstitial lung disease. Amer J Respir Crit Care Med 2005; 172: 1026–1031.
3. Wert SE, Whitsett JA, Nogee LM. Genetic disorders of surfactant dysfunction. Pediatr Dev Pathol 2009; 12 (4): 253–274.
4. Matsumura Y, Ban N, Ueda K, Inagaki N. Characteriza-tion and classification of ATP-binding cassette transporter ABCA3 mutants in fatal surfactant deficiency. J Biol Chem 2006; 281: 34503–34514.
5. Shulenin S, Nogee LM, Annilo T, et al. ABCA3 gene mutations in newborns with fatal surfactant deficiency. New Engl J Med 2004; 350: 1296–1303.
6. Hamvas A. Inherited surfactant protein–B deficiency and surfactant protein-C associated disease: Clinical features and evaluation. Sem Perinatol 2006; 30: 316–326.
7. Jones KD, Dishop MK, Colby TV. Developmental and Pediatric Lung Disease in Practical Pulmonary Patho-logy. 2nd ed. Leslie KO, Wick MR (eds). Philadelphia: Saunders, 2011: 91–116.
8. Ciantelli M, Ghirri P, Presi S, et al. Fatal respiratory failure in a full-term newborn with two ABCA3 gene mutations: a case report. J Perinatol 2011; 31: 70–72.
Štítky
Neonatológia Pediatria Praktické lekárstvo pre deti a dorastČlánok vyšiel v časopise
Česko-slovenská pediatrie

2013 Číslo 3
- Realita liečby bolesti v paliatívnej starostlivosti v Nemecku
- Využití hodnoticích skóre a objektivních nástrojů při léčbě astmatu
- Nech brouka žít… Ať žije astma!
Najčítanejšie v tomto čísle
- Hirschsprungova choroba a její genetické příčiny
- Novorozenecký ikterus
- Nutriční screening při přijetí k hospitalizaci – NutriAction
- GERD – nemoc z gastroezofageálního refluxu