
Extrémní hypokalémie u familiární periodické paralýzy – kazuistika
Extreme hypokalaemia in familial periodic paralysis – case report
Periodic paralysis (PP) is a rare neuromuscular disorder characterized by episodic muscle weakness with abnormal levels of potassium in the blood.
We present a case report of a 17-year-old boy with a familial form of hypokalaemic periodic paralysis. The disease manifested as caudocranially progressive symmetrical muscle weakness of the limbs with rapid onset in the early morning hours shortly after awakening. Three days prior, the boy had noticed increased tiredness of the lower limbs. His past medical history was unremarkable. Extremely low potassium was observed in the laboratory (1.1 mmol/l; normal 3.3–4.7 mmol/l) with concomitant marked hypophosphatemia (0.43 mmol/l; normal 0.94–1.55 mmol/l), normal magnesaemia (0.80 mmol/l; normal 0.62–0.91 mmol/l), normal natremia and chloridaemia (Na 139 mmol/l; Cl 108 mmol/l). The parameters of the acid-base balance were undisturbed. The markers of muscle metabolism (creatine kinase and transaminases) were normal, myoglobin was slightly increased. With the therapeutically achieved normalization of potassium, the above-mentioned clinical symptoms resolved within three hours.
Upon exclusion of the secondary causes of hypokalaemia (especially thyrotoxicosis and renal tubular disorders) the diagnosis of hypokalaemic periodic paralysis was suspected. Molecular genetic testing detected a heterozygous point mutation in SCN4A gene located on chromosome 17 (17q23), encoding the alpha-subunit of the sodium channel, thus confirming the diagnosis of familial hypokalemic periodic paralysis of type 2. The identical pathogenic variant was found in the asymptomatic father of the patient.
Conclusion: The diagnosis of primary PP is based on the characteristic clinical presentation and confirmed by genetic testing. The absence of previous spontaneously receding episodes of muscle weakness in the family or personal history does not exclude the primary (familial) form of PP.
Keywords:
hypokalaemia – muscle weakness – periodic paralysis
Autori:
I. Pospíšilová 1,3; D. Šišková 2; M. Bloomfield 3,4; R. Vyhnánek 3; L. Fajkusová 5; J. Zídková 5; M. Magner 3,6; K. Bořecká 1
Pôsobisko autorov:
Oddělení klinické biochemie, Thomayerova nemocnice, Praha
1; Oddělení dětské neurologie, Thomayerova nemocnice, Praha
2; Pediatrická klinika 1. LF UK a Thomayerovy nemocnice, Praha
3; Ústav imunologie 2. LF UK a FN Motol, Praha
4; Centrum molekulární biologie a genové terapie IHOK, FN Brno
5; Klinika pediatrie a dědičných poruch metabolismu 1. LF UK a VFN, Praha
6
Vyšlo v časopise:
Čes-slov Pediat 2020; 75 (6): 337-341.
Kategória:
Súhrn
Periodické paralýzy (PP) jsou vzácné neuromuskulární poruchy projevující se epizodickou svalovou slabostí s abnormálními hladinami draslíku v krvi. V práci prezentujeme kazuistiku 17letého chlapce s familiární formou hypokalemické periodické paralýzy.
Onemocnění se manifestovalo rychle se rozvíjející kaudo-kraniálně progredující symetrickou svalovou slabostí končetin pozorovanou od ranního probuzení. Tři dny před rozvojem obtíží pociťoval větší únavu DK. Rodinná i osobní anamnéza chlapce byla bez pozoruhodností. Laboratorně byla zjištěna extrémně nízká kalémie (1,1 mmol/l; norma 3,3–4,7 mmol/l) s doprovodnou zřetelnou hypofosfatémií (0,43 mmol/l; norma 0,94–1,55 mmol/l) při normální magnezémii (0,80 mmol/l; norma 0,62–0,91 mmol/l), natrémii a chloridémii (Na 139 mmol/l; Cl 108 mmol/l). Parametry acidobazické rovnováhy byly v normě. Z ukazatelů svalového metabolismu byl pouze mírně zvýšený myoglobin (148,5 µg/l; norma 28–72 µg/l), aktivita kreatinkinázy a transamináz byla v normě. Po terapeuticky dosažené normalizaci kalémie došlo během tří hodin k úpravě výše zmiňovaných klinických obtíží do normy.
Po vyloučení sekundárních příčin hypokalémie (zejména tyreotoxikózy a renální tubulární poruchy) bylo vysloveno podezření na diagnózu hypokalemické periodické paralýzy. Molekulárně genetickým vyšetřením byla následně potvrzena heterozygotní bodová missense mutace v genu SCN4A lokalizovaném na chromosomu 17 (17q23), kódujícím alfa-podjednotku sodíkového kanálu, tzv. familiární hypokalemická periodická paralýza 2. typu. Identická patogenní varianta byla nalezena taktéž u dosud asymptomatického otce.
Závěr: Diagnóza primární PP je založena na charakteristické klinické prezentaci, která je následně potvrzená genetickým testováním. Absence předchozích, spontánně ustupujících atak svalové slabosti v rodinné i osobní anamnéze nevylučuje primární (familiární) formu PP.
Klíčová slova:
hypokalémie – svalová slabost – periodická paralýza
ÚVOD
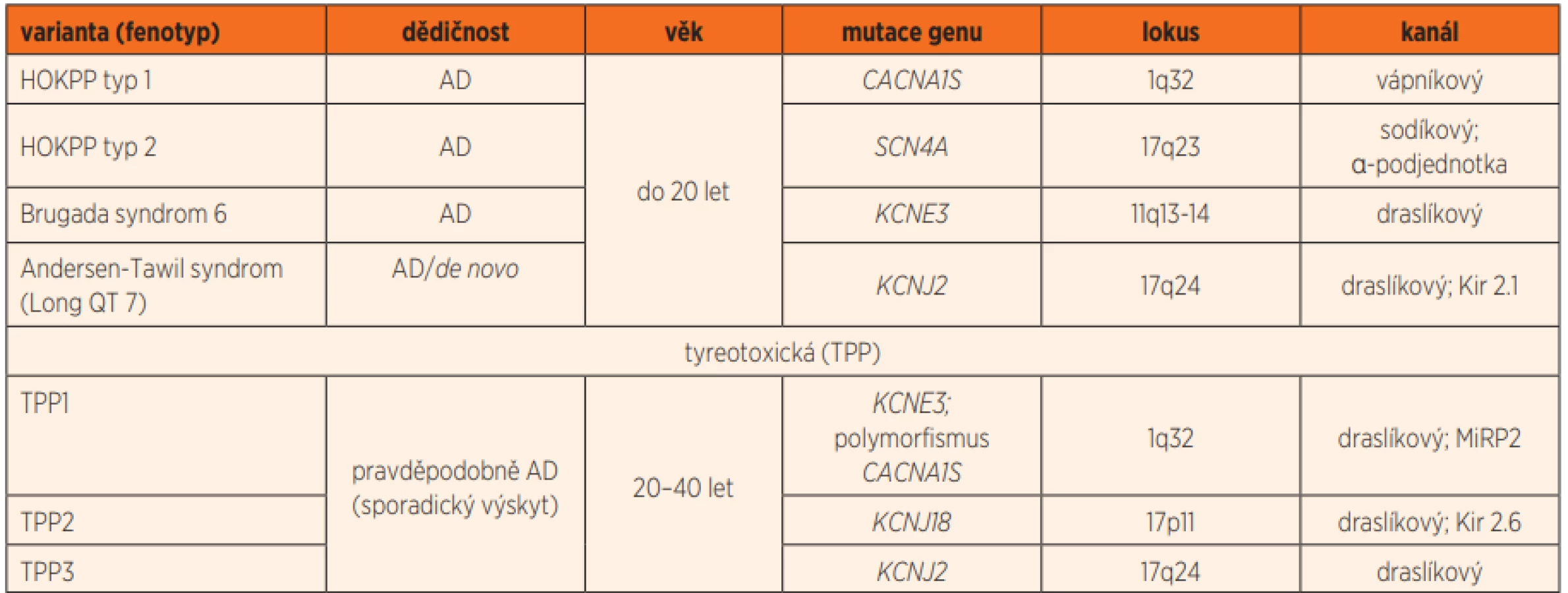
Periodická paralýza (PP) je skupina heterogenních poruch různých etiologií provázených epizodickou, krátkodobou (několik hodin až dní trvající) slabostí kosterního svalstva. Rozlišujeme primární (vrozené) nebo sekundární (získané) formy periodické paralýzy. Primární formy PP představují skupinu vrozených, autosomálně dominantně dědičných kanalopatií spojených s mutacemi genů kódujících membránové iontové kanály kosterního svalstva. K primárním formám PP patří hypokalemická paralýza (hypoPP), hyperkalemická paralýza (hyperPP) a Andersenův-Tawilův syndrom (prodloužený QT interval, kraniofaciální dysmorfie a hypokalemická PP). Existují také úzce související nemoci, jejichž rysy se překrývají s hypoPP a hyperPP, včetně paramyotonia congenita a normokalemické PP. Ve většině případů jsou tato onemocnění způsobena mutacemi sodíkového iontového kanálu [1].
Hypokalemická PP (HOKPP, OMIM 170400, 613345, 613119, 170390) je nejčastější forma PP s prevalencí 0,4–1/100 000 osob [1, 2]. Jedná se o onemocnění s autosomálně dominantním typem dědičnosti s neúplnou penetrancí, postihující 90 % mužů a pouze 50 % žen. Vlastní klinické příznaky se typicky rozvíjí mezi první a druhou dekádou života, na HOKPP je však třeba myslet i ve velmi časném věku. HOKPP je potvrzena identifikací heterogenní patogenní varianty v genu CACNA1S (typ 1, 40–60 %) nebo SCN4A (typ 2, 7–14 %), viz tab. 1. Přibližně 1/3 pacientů splňující diagnostická kritéria HOKPP nemá identifikovanou patogenní variantu v žádném z těchto známých genů. Byly popsány i ojedinělé případy mutace de novo.
Prezentujeme formou kazuistiky případ vzácné formy familiární hypokalemické periodické paralýzy 2. typu, která se u 17letého chlapce poprvé manifestovala nejspíše v důsledku vystupňovaného emočního vypětí.
KAZUISTIKA
Sedmnáctiletý chlapec byl přivezen RZP pro difuzní svalovou slabost končetin spojenou s bolestí při snaze o pohyb, rozvíjející se rychle od časného rána. Zvýšenou únavu dolních končetin pozoroval již tři dny. Rodinná a osobní anamnéza chlapce byla bez pozoruhodností; podobné potíže nikdy neměl, dlouhodobě byl bez větší fyzické zátěže. V době vzniku obtíží se připravoval k přijímacím zkouškám na VŠ, při rozhovoru s matkou byla odkryta dlouhodobě tíživá rodinná situace a její eskalace v posledních dnech.
Při vstupním klinickém vyšetření chlapec polehával, nebyl schopen samostatného stoje ani chůze. Byla zřejmá povšechná chabost končetin při snaze o aktivní či pasivní pohyb, hyperventiloval s projevy mírné tetanie (brnění kolem úst a konců prstů horních končetin). Pacient byl bez známek probíhajícího infektu, eutrofického habitu (BMI 21,2 kg/m2), afebrilní (TT 36,0 °C), eupnoický (DF 20/min, saturace O2 98 %), normotenzní (TK 120/70 mmHg), avšak se sklonem k bradykardii (50–60/min). V objektivním neurologickém nálezu dominovala izolovaná motorická slabost končetin, bez hranice čití, bez slabosti mimického a bulbárního svalstva, dýchacího svalstva a bránice.
V laboratorním vyšetření byla přítomna extrémní hypokalémie (1,1 mmol/l) při normální natrémii a chloridémii (Na 139 mmol/l; Cl 108 mmol/l). Mimo extrémní hypokalémii byla zřejmá zřetelná hypofosfatémie (0,43 mmol/l; norma 0,94–1,55 mmol/l). Magnezémie byla v mezích normy (0,80 mmol/l; norma 0,62–0,91 mmol/l), taktéž glykémie (5,6 mmol/l) byla v normě. Z ukazatelů svalového metabolismu byl pouze mírně zvýšený myoglobin (148,5 µg/l; norma 28–72 µg/l), aktivita kreatinkinázy byla v normě (2,63 µkat/l; norma 0,03–3,97 µkat/l), transaminázy nebyly zvýšeny. Parametry acidobazické rovnováhy v arterializované kapilární krvi byly v normě (pH 7,412; pO2 8,96 kPa; pCO2 4,70 kPa; HCO3- 22,0 mmol/l). Hypokalémii odpovídaly změny na EKG: intermitentně zaznamenán prodloužený PQ interval (max. 240 ms; norma 120–200 ms), mírně oploštělé T vlny v hrudních svodech, alternující QTc interval (370–450 ms), ojediněle izolovaná komorová extrasystola, pozitivní U vlny nebyly zachyceny.
V rámci vyloučení sekundární příčiny hypokalémie byla vyšetřena funkce štítné žlázy (TSH 5,480 mU/l, norma 0,510–4,300 mU/l; fT4 17,88 pmol/l, norma 12,60–21,00 pmol/l). Fyziologické pH krve, absence hypertenze, fyziologické hodnoty natrémie, ranního kortizolu (520 nmol/l) nesvědčily pro endokrinní příčinu nízké kalémie. Screeningové toxikologické vyšetření moče na neznámou noxu bylo negativní. Výpočet frakční exkrece iontů z jednorázové porce moče před zahájením terapie nebyl proveden. Chemické vyšetření moče a sedimentu bylo však s fyziologickým nálezem, včetně pH, a nesvědčilo pro tubulární poruchu. Stolice byla formovaná a pravidelná, chlapec neměl jiné gastrointestinální příznaky, nebyla proto pravděpodobná významná střevní ztráta K+.
Na základě biochemických výsledků byla zahájena parenterální suplementace kalia, na které svalová slabost rychle ustupovala a při hodnotách draslíku 3,0 mmol/l zcela vymizela.
Bylo indikováno genetické vyšetření se zaměřením na okruh periodických obrn. Molekulární analýzou byla následně potvrzena heterozygotní bodová missense mutace v genu SCN4A (NM_000334.4:c.2014C>T p.(Arg672Cys)). Vzhledem k absenci zjevné genetické zátěže v rodinné anamnéze byla zvažována mutace de novo, identická mutace iontového kanálu byla však nalezena u dosud asymptomatického otce.
Po propuštění chlapce se obdobná epizoda nezopakovala, při kontrolních vyšetřeních byly hodnoty kalémie v referenčním rozmezí.
DISKUSE
Hypokalemická PP je charakterizována epizodickými atakami fokální nebo generalizované svalové slabosti s potvrzeným sérovým K+ <3,5 mmol/l. Hypokalémie je obvykle doprovázená hypofosfatémií a hypomagnezémií; základní parametry acidobazické rovnováhy nebývají narušeny. Ukazatele svalového metabolismu mohou, ale i nemusí být zvýšeny.
Postižení svalů je výraznější v proximálních svalových skupinách, je bilaterální, symetrické, rychle progredující s ascendentní propagací (dolní končetiny bývají postižené před horními končetinami). Šlacho-svalové reflexy jsou normální nebo lehce snížené, nejsou však vyhaslé. Bolesti svalů mohou, ale i nemusí být přítomny. Ataky svalové ochablosti bývají variabilní intenzity a délky trvání, nejsou provázeny poruchou vědomí, bulbární symptomatologií, sfinkterovými obtížemi ani senzitivním neurologickým deficitem. V průběhu epizody svalové slabosti/paralýzy zpravidla nebývají postiženy dýchací svaly, mohou však být přítomny závažné poruchy srdečního rytmu charakteru komorových arytmií, vyskytující se převážně u Andersenova-Tawilova syndromu. Záchvatům mohou předcházet prodromální příznaky jako únava, parestézie či změny chování. Mohou vznikat spontánně; spouštěčem vlastní ataky může být taktéž strava (především bohatá na sodík, sacharidy), emoční vypětí či nadměrná fyzická zátěž, tedy situace spojené s uvolňováním katecholaminů a inzulinu.
Diagnóza HOKPP je založena na přítomnosti charakteristických klinických a laboratorních příznaků; viz tab. 2 [3]. Absence předchozích atak svalové slabosti v rodinné i osobní anamnéze nevylučuje familiární (primární) formu periodické paralýzy, jak vyplývá i z naší kazuistiky. HOKPP je autosomálně dominantně dědičné onemocnění s neúplnou penetrancí. Pravděpodobnost manifestace HOKPP závisí nejen na identifikaci typu postiženého genu (ne vždy je však mutace odhalena), ale i na pohlaví postižené osoby.

Vzácnou formou hypokalemické periodické paralýzy je tyreotoxická PP (TPP, OMIM 188580). Patofyziologickým podkladem TPP je genetická nebo získaná porucha Na+/K+-ATPázy podmiňující přesun K+ z extracelulárního prostoru intracelulárně u vnímavých jedinců v přítomnosti nadbytku hormonů štítné žlázy. Většina případů se vyskytuje u Asiatů, je však stále častěji pozorována i v kavkazské populaci [4–6].
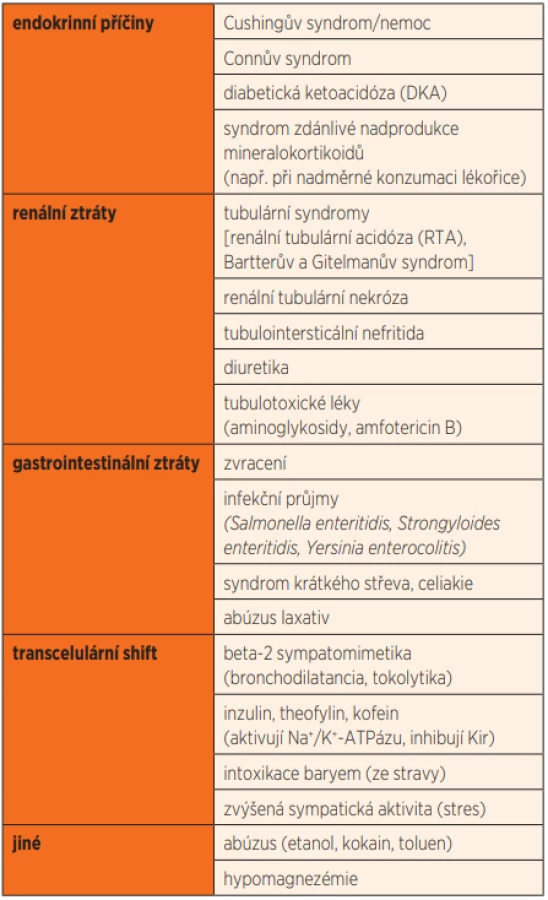
Hypokalémie provázená paralýzou může být taktéž příznakem/projevem jiného onemocnění. Nejznámější příčiny sekundárně podmíněné hypokalémie jsou uvedeny v tabulce 3 [7]. Může být způsobena sníženým příjmem, poruchou distribuce kalia mezi extracelulární a intracelulární tekutinou a/nebo zvýšenými ztrátami kalia stolicí nebo močí, které mohou být hormonálně podmíněny. Zvýšený transcelulární přesun K+ může způsobit řada spouštěčů, a to umocněním činnosti Na+/K+-ATPázy, ev. v součinnosti s inhibicí iontového kanálu Kir (inward rectifiers potassium channels). Patří sem již zmiňované stavy zvýšené aktivity sympatiku s uvolňováním katecholaminů a inzulinu, strava bohatá na sacharidy a kofein, dále také hormony štítné žlázy a nadledvin; ty způsobují změny membránového potenciálu buněk kosterní svaloviny a vedou k dysfunkci sodíkového kanálu vyúsťující v „distribuční“ hypokalémii [8].
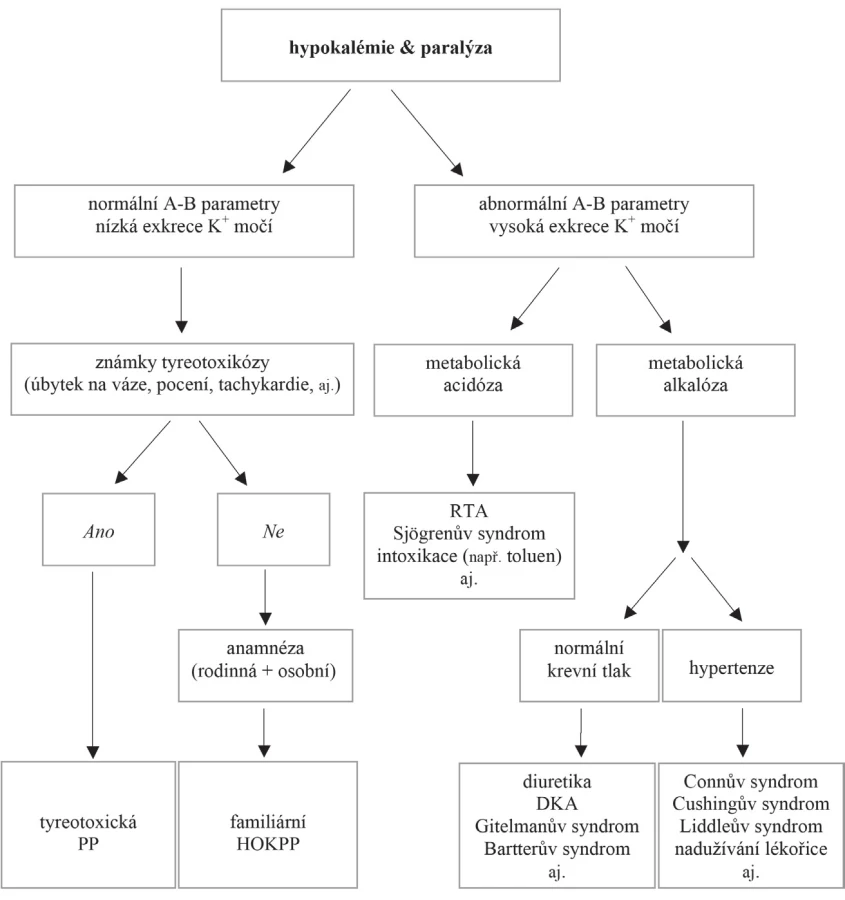
V diferenciální diagnostice hypokalémie provázené paralýzou je nutné kromě stanovení sérové koncentrace kalia vyšetřit i parametry acidobazické rovnováhy, hladinu magnezia a odpady K+ do moči. Rutinní metodou k měření vylučování kalia do moči (kaliurie) je sběr moči po dobu 24 hodin. U pacientů s těžkou hypokalémií však vzhledem k urgentní potřebě substituovat deficitní ionty nelze 24hodinový sběr moči provést.
Využití jednoduchých výpočtů z jednorázového vzorku moče vycházejících ze sérových (S) a močových (U) koncentrací K+ a kreatininu (Kr) může být nápomocné k odlišení extrarenálních a renálních ztrát K+. Jedná se o kalkulaci poměru draslíku a kreatininu (UK/UKr) a frakční exkrece draslíku (FEK [%] = [UK × SKr]/[UKr × SK] × 100). Výpočet transtubulárního gradientu pro draslík (TTKG = [UK/Uosmolalita]/[SK/Sosmolalita]) vyžaduje navíc změření osmolality v séru i v moči, pro validní vyhodnocení TTKG je třeba, aby byla Uosmolalita > Sosmolalita. Tyto výpočty však mají svá omezení zejména u stavů provázených hypovolémií, u nichž se v důsledku aktivace osy renin-angiotenzin-aldosteron (RAA) zvyšuje renální exkrece kalia. V těchto případech může být navíc užitečný výpočet (UK/UK + UNa); hodnota nad 0,6 svědčí pro zapojení osy RAA.
Normální parametry acidobazické rovnováhy a kaliurie <20 mmol/24 h a/nebo UK/UKr <1,5; FEK <6 % a TTKG <4 korespondují s extrarenální příčinou hypokalémie [8–10], tedy i možnou familiární formou PP; při abnormalitách v parametrech acidobazické rovnováhy a vyšších hodnotách výše zmiňovaných výpočtů se pravděpodobně na hypokalémii podílejí jiná získaná onemocnění, jak vyplývá z obrázku 1.
Léčebný přístup HOKPP zahrnuje jak zvládnutí akutních atak v podobě parenterální či perorální substituce deficitních iontů, tak vlastní prevenci dodržováním režimových opatření ve smyslu pravidelného stravování s omezením nadbytku jednoduchých cukrů, dostatku kvalitního spánku a přiměřené fyzické aktivity.
ZÁVĚR
Prezentovaná kazuistika popisuje klinický obraz a laboratorní nálezy sedmnáctiletého chlapce se vzácnou formou primární HOKPP. Poukazuje na možnou přítomnost familiární formy tohoto onemocnění i při vstupně nevýznamné rodinné i osobní anamnéze. Předkládá jednoduché diferenciálně diagnostické schéma, které může být nápomocné v rozlišení primární a sekundární formy HOKPP.
Podpořeno grantem RVO-VFN 64165/2012.
MUDr. Klára Bořecká
Oddělení klinické biochemie
Thomayerova nemocnice
Vídeňská 800
140 59 Praha 4
e-mail: klara.borecka@ftn.cz
Zdroje
1. Statland JM, Fontaine B, Hanna MG, et al. Review of the diagnosis and treatment of periodic paralysis. Muscle Nerve 2018; 57 (4): 522–530.
2. Jabor A, et al. Vnitřní prostředí. 1. vyd. Praha: Grada Publishing, a. s., 2008: 432–435.
3. Weber F, Lehmann-Horn F. Hypokalemic periodic paralysis. 2002 Apr 30 [Updated 2018 Jul 26]. In: Adam MP, Ardinger HH, Pagon RA, et. al. (eds). Gene Reviews® [Internet]. Seattle (WA): Univerzity of Washington, 1993–2020.
4. Lin SH, Huang CL. Mechanism of thyreotoxic periodic paralysis. J Am Soc Nephrol 2012; 23 (6): 985–988.
5. Park S, Kim TY, Sim S, et al. Association of KCNJ2 genetic variants with susceptibility to thyreotoxic periodic paralysis in patients with Grawes‘ disease. Exp Clin Endocrinol Diabetes 2017; 125 (2): 75–78.
6. Lam L, Nair RJ, Tingle L. Thyreotoxic periodic paralysis. Proc (Bayl Univ Med Cent), 2006 Apr; 19 (2): 126–129.
7. Kardalas E, Paschou SA, Anagnostis P, et al. Hypokalemia: a clinical update. Endocr Connect 2018,7: R135–146.
8. Doležal Z, Novotná D, Schneiderová H, et al. Tyreotoxická periodická paralýza. Pediatr praxi 2016; 17 (6): 379–382.
9. Srivastava RN, Bagga A. Pediatric Nefrology. 5th ed. India: Yaypee Brothers Medical Publishers (P) Ltd., 2011: 101–105.
10. Kliegman RM, Stanton BF, Geme JW, et al. Nelson Textbook of Pediatrics. 19th ed. Philadelphia: Elsevier Saunders, 2011: 224.
Štítky
Neonatológia Pediatria Praktické lekárstvo pre deti a dorastČlánok vyšiel v časopise
Česko-slovenská pediatrie

2020 Číslo 6
- Realita liečby bolesti v paliatívnej starostlivosti v Nemecku
- Využití hodnoticích skóre a objektivních nástrojů při léčbě astmatu
- Nech brouka žít… Ať žije astma!
Najčítanejšie v tomto čísle
- Extrémní hypokalémie u familiární periodické paralýzy – kazuistika
- Škrkavka jako vzácná příčina nočních bolestí břicha u dětí – kazuistika
- Mozkový absces u dětí – souhrnný článek a dvě kazuistiky
- Vaskulitida jako vzácný projev infekce Sarcoptes scabiei – kazuistika