
Srovnávací proteomická analýza krevní plazmy pacientů s mnohočetným myelomem léčených režimy s bortezomibem
Comparative Plasma Proteomic Analysis of Patients with Multiple Myeloma Treated with Bortezomib-based Regimens
Backgrounds:
Recently, the term biomarker has become, especially in connection with the term clinical proteomics, one of the most frequent terms in the field of biomedical research. The aim of this work was to select an appropriate pre-fractionation method of blood plasma prior to a subsequent proteomic analysis of low-abundant fraction of proteins by two dimensional gel electrophoresis (2-DE) and mass spectrometry to improve the resolution of 2-DE maps and protein identification.
Materials and Methods:
First, we compared two prefractionation methods (MARS versus ProteoMiner) preceding 2-DE analysis using 10 blood plasma samples. Based on the results of the comparative experiments, low-abundant plasma protein fractions from 18 multiple myeloma patients treated with bortezomib were analyzed. Patients were divided into two groups: a group resistant to chemotherapy (9 patients – disease progression, stable disease) and a group with positive clinical response (9 patients – complete and partial remission).
Results and Conclusion:
Samples prefractioned by ProteoMiner method yielded 2-DE maps with a significantly increased number of detected protein spots, as compared to immunodepletion method MARS (Multiple Affinity Removal System). Between groups of chemoresistant and sensitive patients treated with bortezomib, 15 differently intense spots were revealed by image analysis. These spots were found to correspond to 10 proteins, as confirmed by mass spectrometry. Seven proteins had significantly lower protein level in the group of chemosensitive patients (serum amyloid P, fibrinogen – gamma chain, retinol-binding protein 4, complement factor C4-A, apolipoprotein E, carboxypeptidase N and complement factor H-related protein 1) and 3 proteins showed significantly higher levels of protein (or were only detected) in the group of chemosensitive patients (serum paraoxonase 1, alpha-1-antitrypsin and complement factor B).
Key words:
multiple myeloma – proteomics – two dimensional gel electrophoresis – plasma – protein – bortezomib
This work was supported by grants of Ministry of Education, Youth and Sports of the CR (LC06027 a MSM0021622434) and IGA MZ (NS9683) and NT12130) and grant of The Czech Science Foundation GAP304/10/1395..
The authors declare they have no potential conflicts of interest concerning drugs, products, or services used in the study.
The Editorial Board declares that the manuscript met the ICMJE “uniform requirements” for biomedical papers.
Submitted:
3. 6. 2011
Accepted:
8. 9. 2011
Autori:
J. Čumová 1,2; L. Jedličková 3; D. Potěšil 3,4; O. Šedo 4; K. Stejskal 3; A. Potáčová 1; Z. Zdráhal 3; R. Hájek 1,5,6
Pôsobisko autorov:
Babákova myelomová skupina, Ústav patologické fyziologie, LF MU Brno
1; Oddělení klinických laboratoří, Nemocnice Blansko
2; Oddělení funkční genomiky a proteomiky, Ústav experimentální biologie, PřF MU Brno
3; Výzkumná skupina Proteomika CF, Mendelovo centrum genomiky a proteomiky rostlin, Středoevropský technologický institut, MU Brno
4; Laboratoř experimentální hematologie a buněčné imunoterapie, Oddělení klinické hematologie, FN Brno
5; Interní hematoonkologická klinika, LF MU a FN Brno
6
Vyšlo v časopise:
Klin Onkol 2012; 25(1): 17-25
Kategória:
Původní práce
Súhrn
Východiska:
Termín biomarker se v posledních letech stal, zvláště pak ve spojení s termínem klinická proteomika, jedním z nejfrekventovanějších pojmů v oblasti biomedicínského výzkumu. Předmětem této práce byl výběr vhodné metody frakcionace krevní plazmy umožňující oddělení majoritních proteinů od nízkoabundantní proteinové frakce před následnou proteomickou analýzou pomocí dvourozměrné gelové elektroforézy (2-DE) s cílem zlepšit rozlišení 2-DE mapy a identifikaci proteinů hmotnostní spektrometrií.
Materiál a metody:
Nejprve bylo provedeno paralelní srovnání dvou metod frakcionace krevní plazmy (depleční kolona MARS vs ProteoMiner) na souboru 10 vzorků. Na základě výsledků srovnávacích experimentů byla k analýze vzorků 18 pacientů nemocných mnohočetným myelomem a léčených režimy s bortezomibem použita frakcionační metoda ProteoMiner v kombinaci s 2-DE. Soubor pacientů byl rozdělen na dvě skupiny: skupina pacientů rezistentních na chemoterapii (9 pacientů – progrese onemocnění, stabilizace onemocnění) a skupina pacientů s pozitivní léčebnou odpovědí (9 pacientů – kompletní a parciální remise).
Výsledky a závěr:
Metoda ProteoMiner umožnila na 2-DE mapách signifikantně zvýšit počet proteinových spotů ve srovnání s imunodepleční metodou MARS (Multiple Affinity Removal System). Mezi skupinami pacientů chemorezistentních a pacientů citlivých na léčbu bortezomibem bylo pomocí analýzy obrazu 2-DE gelů zjištěno 15 rozdílových proteinových spotů, které byly analyzovány hmotnostní spektrometrií. V těchto spotech bylo identifikováno 10 proteinů. Sedm proteinů vykazovalo signifikantně nižší hladinu proteinu ve skupině chemosenzitivních pacientů (sérový amyloid P, fibrinogen – gama řetězec, retinol-vazebný protein 4, komplement faktor C4-A, apolipoprotein E, karboxypeptidáza N, komplement faktoru H – příbuzný protein 1) a 3 proteiny vykazovaly signifikantně vyšší hladinu proteinu nebo byly detekovány pouze ve skupině chemosenzitivních pacientů (sérová paraoxonáza 1, alfa-1-antitrypsin a komplement faktor B).
Klíčová slova:
mnohočetný myelom – proteomika – elektroforéza gelová dvourozměrná – plazma – protein – bortezomib
Úvod
Moderní medicína využívá k diagnostice nemocných stanovení hladin látek, které jsou pro dané procesy charakteristické (tzv. „biomarkery“). Mezi typické tělní tekutiny, u kterých je prováděna analýza prognostických a prediktivních biomarkerů nádorových onemocnění, patří krevní sérum, plazma, moč a mozkomíšní mok [1,2]. Méně často se provádí rozbor slin, bronchoalveolární laváže, amniotické nebo folikulární tekutiny. Lidská krevní plazma představuje komplexní tělní tekutinu, kde jsou navíc proteiny přítomny v řádově rozdílných koncentracích (až 1010) [3]. Zdrojem diverzity proteinů je např. alternativní sestřih mRNA a posttranslační modifikace [4,5]. Dvourozměrná gelová elektroforéza (2-DE) je již klasickou metodou pro separaci proteinových směsí před jejich identifikací hmotnostní spektrometrií (MS) [6–8]. Metoda má omezenou kapacitu vzhledem k množství a velikosti analyzovaných proteinů a neumožňuje detekci proteinů s nízkou hladinou exprese [9,10]. Většina potenciálních biomarkerů se v plazmě vyskytuje právě v nízkých koncentracích a nadbytek albuminu a protilátek jejich analýzu komplikuje. Proto je odstranění abundantních proteinů z krevní plazmy (deplece) často prvním a rozhodujícím krokem celého postupu. Odstranění abundantních proteinů by mělo být v ideálním případě úplné, kvantitativní a především reprodukovatelné. Pro tyto účely se často využívají depleční kolonky, které vyvazují většinově zastoupené proteiny na bázi specifických interakcí s imobilizovanými protilátkami. Např. imunodepleční metodou Multiple Affinity Removal System (MARS) lze odstranit 6 nebo 14 nejhojněji zastoupených lidských proteinů krevní plazmy [11,14], séra [15,16] či moče [17]. Některé výsledky prací však poukazují na neúplnou depleci vysoce zastoupených proteinů [16,18,19]. Bellei et al popsali odstranění některých necílených proteinů z frakce nízkoabundantních proteinů [20]. Tu et al. také popsali, že některé proteiny lidské plazmy, jako je alfa-1-kyselý glykoprotein 1, alfa-1-kyselý glykoprotein 2 a alfa-1-antichymotrypsin, se rovněž vážou na MARS kolonu [21].
Novým přístupem prepurifikace (frakcionace) krevní plazmy je afinitní nedepleční metoda ProteoMinerTM (Bio-Rad Laboratories, USA), která využívá rozdílů ve vazebných afinitách přítomných proteinů k hexapeptidům [22–24]. Hexapeptidy jsou, podobně jako protilátky u deplečních kolonek, imobilizovány na vhodném nosiči uloženém v prepurifikační koloně. V koloně jsou pak přítomny ve stejném množství odlišné hexapeptidy z rozsáhlé hexapeptidové knihovny. Každý hexapeptid přitom vykazuje rozdílnou proteinovou specifitu. Výsledkem je omezená celková vazebná kapacita kolony pro konkrétní proteiny. Vysoce abundantní proteiny jsou tak na koloně zachyceny jen částečně, kdežto proteiny o nízké vstupní koncentraci jsou naopak zachyceny poměrově více. Při celém procesu je navíc zachována informace o relativní kvantitě proteinu, což umožňuje relativní srovnání více vzorků. Nutno však dodat, že existuje i práce poukazující na možný odlišný princip funkce metody ProteoMiner [25].
Cílem této práce bylo srovnání obou metod frakcionace krevní plazmy nezbytné před 2-DE proteomickou analýzou nízkoabundantní frakce proteinů s cílem zvýšit počet rozlišitelných proteinových spotů obrazu 2-DE mapy, které by umožnilo identifikovat proteinové změny charakteristické pro pacienty senzitivní a chemorezistentní na podané antimyelomové léčivo.
Materiál a metody
Zpracování odebrané krve
Vzorky krevní plazmy byly získány od 28 pacientů nemocných mnohočetným myelomem (MM) a léčených na Interní hematoonkologické klinice LF MU a FN MU Brno po podepsání informovaného souhlasu s odběrem biologického materiálu. Lidská krev byla odebrána do uzavřeného odběrového systému S-Monovette (2,7ml zkumavka s K3EDTA, Sarstedt, Německo) a centrifugována 15 min při 2 200 g 20 °C. Plazma byla rozdělena do alikvotů po 0,5 ml a zamražena do –80 °C pro následnou analýzu. Vzorky 10 pacientů byly použity pro srovnávací studii frakcionačních postupů, vzorky druhé skupiny 18 pacientů vybrané pro vlastní proteomickou analýzu byly pak zpracovány zvoleným postupem. Základní charakteristika těchto 18 pacientů, u kterých byla provedena srovnávací 2-DE analýza, je uvedena v tab. 1.
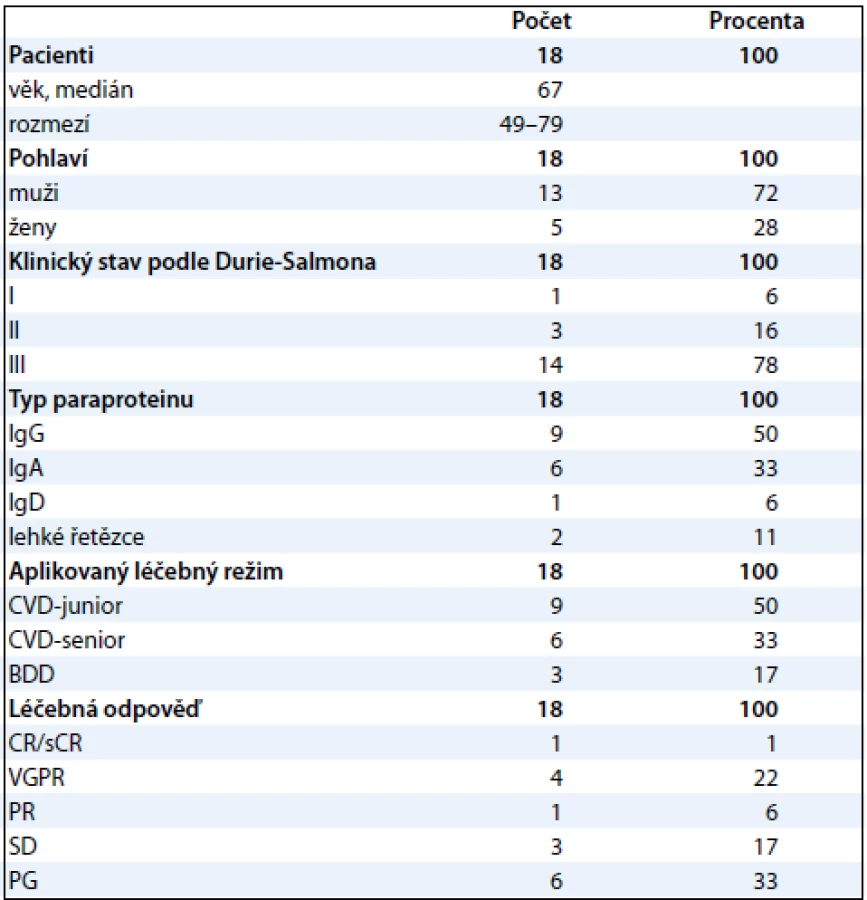
Frakcionace proteomu krevní plazmy
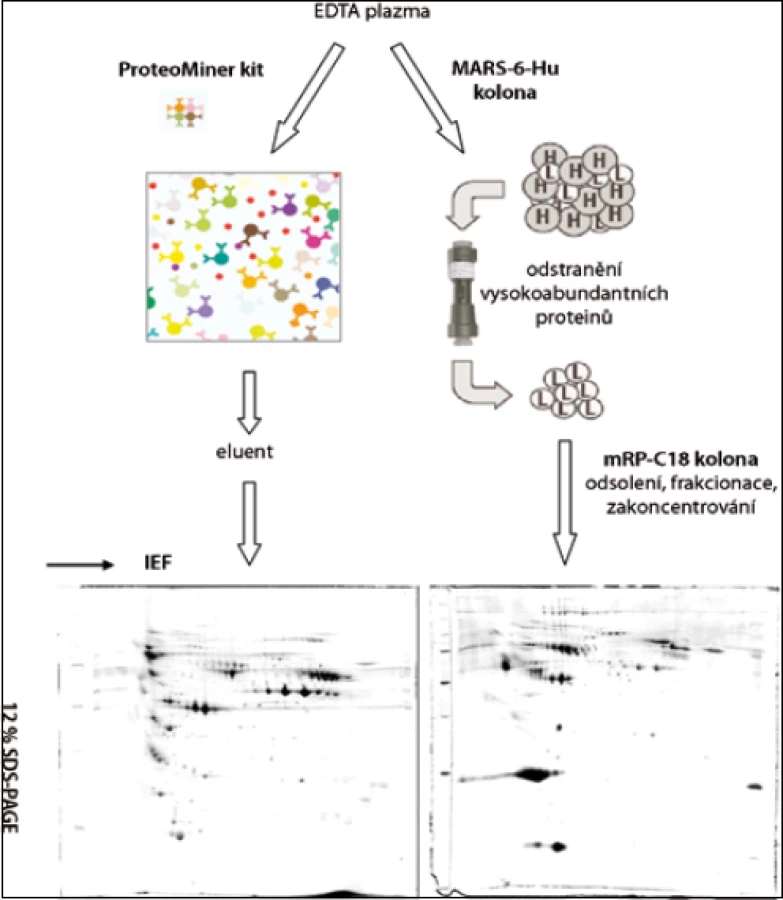
Po rozmražení byla plazma centrifugována (5 min při 16 000 g, 20 °C) přes 0,22 µm filtr (Agilent Technologies, USA). Frakce nízkoabundantních proteinů 10 vzorků krevní plazmy byly získány paralelně metodou ProteoMiner (Bio-Rad, USA) a metodou afinitní chromatografie na koloně MARS-6-Hu (4,6 × 100 mm; Agilent Technologies, USA). U obou metod bylo postupováno podle pokynů výrobce (obr. 1).
ProteoMiner
Alikvóty plazmy (1 ml od každého pacienta) byly pipetovány do ProteoMiner centrifugačních kolon a inkubovány 2 hod na rotátoru SB3 Stuart (BioCote, UK) při 20 °C. Po promytí kolony (3 × 5 min) pufrem obsahujícím 150 mmol/l NaCl, 10 mmol/l NaH2PO4 pH 7,4 (3 × 5 min, 1 ml pufru) byly frakce nízkoabundantních proteinů z kolony vymyty elučním pufrem (Bio-Rad, USA) obsahujícím 8 mol/l močovinu a 2% 3-3[(3-cholamidopropyl)dimethylamonio]propan-1-sulfonát (CHAPS), 3 × 15 min při 750 rpm, 20 °C se 100 µl pufru. Všechny 3 eluované frakce od každého pacienta byly spojeny do jedné frakce.
Metoda MARS (Multiple Affinity Removal System)
Metodou MARS bylo odstraněno celkem 6 vysokoabundantních proteinů lidské krevní plazmy: albumin, IgG, IgA, transferin, alfa-1-antitrypsin a haptoglobin. Vzorek plazmy od každého pacienta byl zpracován paralelně dvakrát. Plazma byla 4× naředěna v pufru A (Agilent Technologies, USA): 80 µl plazmy + 240 µl pufru A. Nízkoabundantní frakce proteinů byla získána promytím kolony pufrem A při průtoku 0,5 ml/min a frakce vysokoabundantních proteinů byla získána promytím kolony pufrem B při průtoku 1 ml/min (obr. 2). Jednotlivé frakce nízkoabundantních proteinů (500–600 µl) byly odsoleny kapalinovou chromatografií na koloně mRP-C18 s makroporézní silikou (4,6 × 50 mm; Agilent Technologies, USA) při 80 °C a průtoku 0,75 ml/min. Mobilní fáze A obsahovala 0,1% (v/v) kyselinu trifluoroctovou (TFA, Sigma-Aldrich, USA) a 3% (v/v) acetonitril (ACN, Merck, Německo) ve vodě, mobilní fáze B pak 97% (v/v) ACN a 0,08% (v/v) TFA ve vodě. Gradientová eluce začínala na 10 % mobilní fáze B a lineárně rostla na 70 % mobilní fáze B v prvních 5 minutách. V průběhu následující jedné minuty obsah mobilní fáze B vzrostl na 100 % a na této hodnotě zůstal další 2 minuty. Poté následovala reekvilibrace kolony 10 % mobilní fáze B. Frakcionace byly provedeny na kapalinovém chromatografu od firmy Waters. Na vakuové odparce SpeedVac (Thermo Scientific, USA) byly získané frakce obsahující nízkoabundantní proteiny odpařeny a proteinové pelety byly rozpuštěny v 60 µl IPG8 pufru obsahujícím 7 mol/l močovinu, 2 mol/l thiomočovinu, 4% CHAPS, 60 mmol/l dithiotreitol (DTT), 0,8% (v/v) amfolyt Pharmalyte pH 3–10, 0,003% (w/v) bromfenolovou modř (vše Sigma-Aldrich, USA). Celková koncentrace proteinů byla stanovena pomocí soupravy BioRad Protein Assay podle metody Bradfordové [26] na spektrofotometru SmartSpec Plus, s použitím standardu γ-globulinu (vše Bio-Rad, USA).
Dvourozměrná gelová elektroforéza (2-DE)
2-DE separace vzorku od každého pacienta byly prováděny v duplikátu. Proteiny krevní plazmy (100 µg/315 µl IPG8 pufru) byly naneseny na komerčně dostupné proužky s imobilizovaným nelineárním pH gradientem (ReadyStrip IPG Strips o délce 18 cm, pH 3–10, Bio-Rad, USA). Přes noc probíhala pasivní rehydratace vzorku na zvolený proužek (16 hod, 20 °C). První rozměr 2-DE byl proveden na izoelektrickém fokuzátoru Protean IEF cell (Bio-Rad, USA) při teplotě 20 °C v následujících krocích: 1) 150 V, 30 min, 2) 1 500 V, 3 hod, 3) 3 500 V, 3 hod, 4) 5 000 V, 80 kVh, 5) 500 V, 24 hod. Po izolektrické fokuzaci (IEF) byla při 20 °C provedena v IPG proužku 1) redukce proteinů 2% (w/v) DTT (10 min) a 2) alkylace proteinů 2,5% (w/v) jodacetamidem (IAA, Sigma-Aldrich, USA), 10 min, které byly rozpuštěny v ekvilibračním pufru obsahujícím 6 mol/l močovinu, 0,375 mol/l Tris(hydroxymethyl)-aminomethan pH 8,8 (Tris-HCl, Serva, USA), 2% (w/v) dodecylsulfát sodný (SDS, Serva, USA); 30% (v/v) glycerol. IPG proužek byl přenesen na 12% polyakrylamidový gel o rozměrech 18 × 20 cm s molekulovým standardem (Precision Plus Protein Standards, Bio-Rad, USA, 1 µl standardu naneseného na wicks) a zalit 1% agarózou (Fluka, USA). Elektroforéza probíhala v systému Protean Plus Dodeca Cell (Bio-Rad, USA) v elektroforetickém pufru obsahujícím 19,2 mmol/l glycin (Bio-Rad, USA), 19 mmol/l SDS, 2,5 mmol/l Tris-HCl pH 8,3 za konstantního napětí 50 V, 1 hod a následně 100 V, 17 hod (zdroj PowerPac Universal, Bio-Rad, USA). 2-DE nádoba byla chlazena na 10 °C (Julabo, Labortechnik, Německo).
Vizualizace 2-DE gelů a analýza obrazu
Gely byly fixovány 2 × 30 min ve fixačním pufru (200 ml/gel) obsahujícím 10% (v/v) ethanol a 7% (v/v) kyselinu octovou (vše Penta, Česká republika). Separované proteiny byly v gelu vizualizovány fluorescenčním barvením Sypro Ruby (Bio-Rad, USA) a snímány na laserovém skeneru TyphoonTM FLA-7000 (Fuji Photo Film Co., Ltd., USA). Analýza obrazu 2-DE gelů byla provedena pomocí programu PDQuest 8. 0. 1 (Bio-Rad, USA). Při vyhodnocení byla provedena normalizace dat, která sloužila k vyrovnání intenzit jednotlivých gelů. Obraz tzv. Master gelu byl vytvořen jako syntetický obraz normalizovaných dat všech 36 2-DE gelů vytvořených z tzv. matchsetu. Pomocí analytických setů byly gely kvalitativně, kvantitativně a statisticky vyhodnoceny (Studentův t-test na hladině významnosti 0,05). Pomocí analýzy byly nalezeny rozdílové proteinové spoty, jejichž intenzita byla 2× nebo 4× snížená nebo zvýšená mezi skupinami pacientů citlivých a chemorezistentních na bortezomib. Zahrnuty jsou kvalitativní změny přesahující desetinásobek hodnoty pozadí.
Identifikace proteinů pomocí hmotnostní spektrometrie
Oblasti gelu obsahující rozdílně zastoupené proteiny mezi 2 skupinami pacientů byly vyřezány pomocí přístroje EXQuestTM Spot Cutter (Bio-Rad, USA). Po odbarvení pak byly proteiny v gelu proteolyticky naštěpeny trypsinem (Promega, USA). Vzniklá směs peptidů byla analyzována pomocí MALDI-MS, resp. MALDI-MS/MS, a v případě dosažení nesignifikantního výsledku identifikace proteinu následně i kapalinovou chromatografií s tandemovou hmotnostní spektrometrií (LC-MS/MS).
Analýzy technikou MALDI-MS a MS//MS analýzy byly provedeny na přístroji Ultraflex III (Bruker Daltonik, Německo). Vzorek (1 µl) byl na vzorkovací desce AnchorChip smíchán s 0,6 µl roztoku matrice (2 mg/ml, kyselina α-kyano-4-hydro-xyskořicová, Bruker Daltonik, Německo) ve směsi 2,5% TFA a 100% ACN, 1 : 2 v/v) [27].
Analýzy tryptických digestů technikou LC-MS/MS byly provedeny na systému EASY-nLC (Proxeon, Thermo Scientific, USA) spojeném on-line s HCTultra PTM Discovery System (Bruker Daltonik, Německo) hmotnostním spektrometrem s iontovou pastí vybaveným nanosprejem. Po odsolení a prekoncentraci na předkoloně (Jupiter Proteo, 100 µm × 30 mm, Pheno-menex, USA) byly vzorky separovány na kapilární koloně (X-Bridge BEH 130 C18, 100 µm × 100 mm, Waters, USA) pomocí 0,1% FA/ACN gradientu. Prekoncentrační a separační kolony použité k LC separaci byly plněny podle dříve popsaného postupu [28]. Hmotnostní spektrometr pracoval v pozitivním módu v m/z rozsahu 300–1 500 pro MS a 100–2 500 pro MS/MS spektra.
Ke zpracování MS a MS/MS dat byl použit prohledávací algoritmus MASCOT 2.2 (MatrixScience, UK). Databázové hledání bylo provedeno proti NCBI databázi (neredundantní, taxonomie Homo sapiens). U peptidového mapování na základě MALDI-TOF MS dat byla nastavena tolerovaná chyba určení molekulové hmotnosti peptidu na 30 ppm, u MALDI-MS/MS dat pak na 0,5 Da. Při prohledávání ESI-MS/MS dat byla nastavena tolerance 0,5 Da pro MS i MS/MS data s korekcí na hmotnost prekurzorového iontu na jeden 13C atom. Pro všechna hledání byla nastavena oxidace methioninu a karbamidometylace jako variabilní modifikace a jedno povolené vynechané štěpné místo pro trypsin.
Pro určení funkce identifikovaných proteinů byla využita databáze UniProtKB/Swiss-Prot (http://www.uniprot.org/, [29]) a PANTHER klasifikační systém (http://www.pantherdb.org/, [30]).
Statistická analýza dat
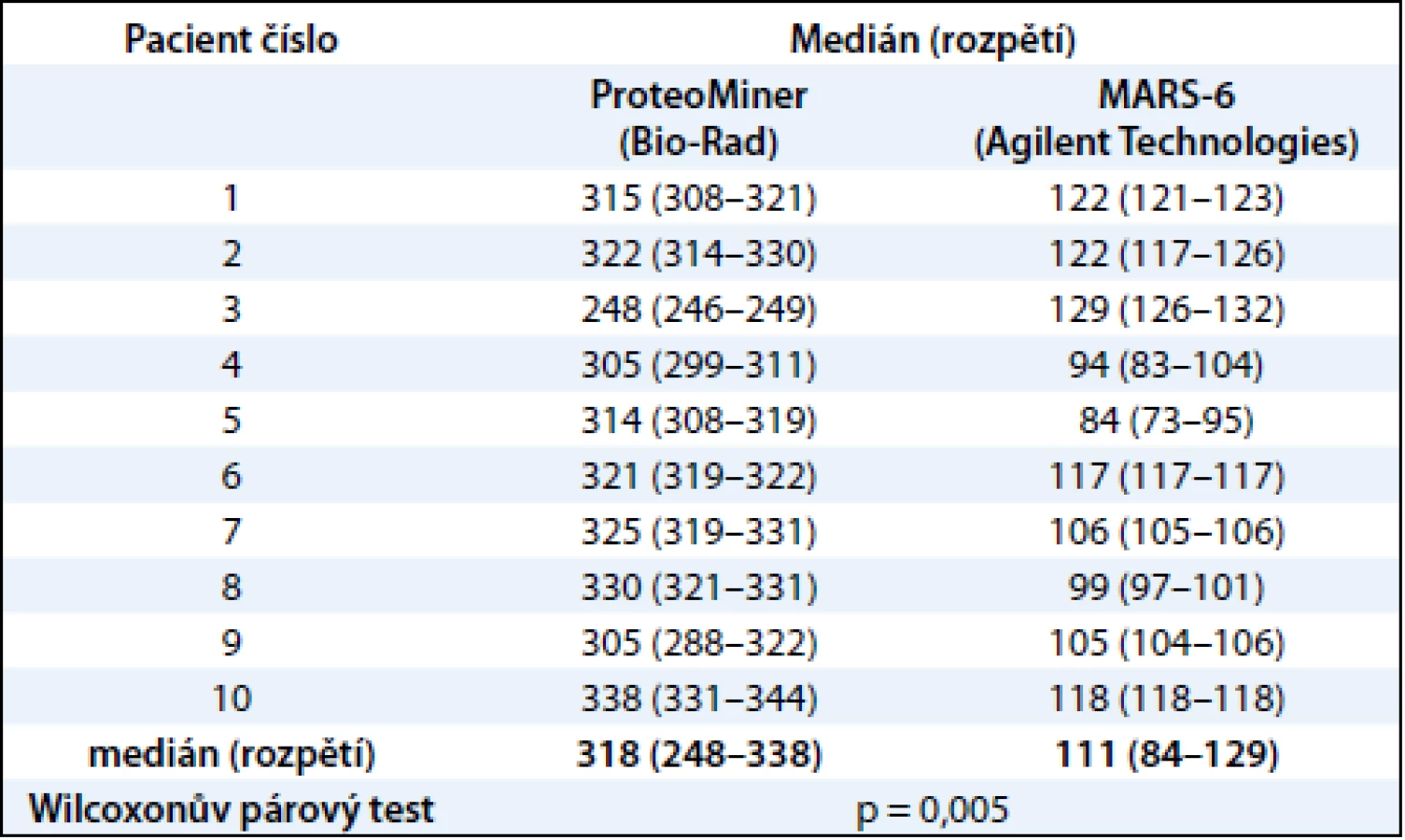
Statisticky významný rozdíl mezi počtem proteinových spotů získaných metodami ProteoMiner a MARS-6-Hu byl určen Wilcoxonovým párovým testem na hladině významnosti 0,05 (program Statsoft Statistica 10.0).
Výsledky
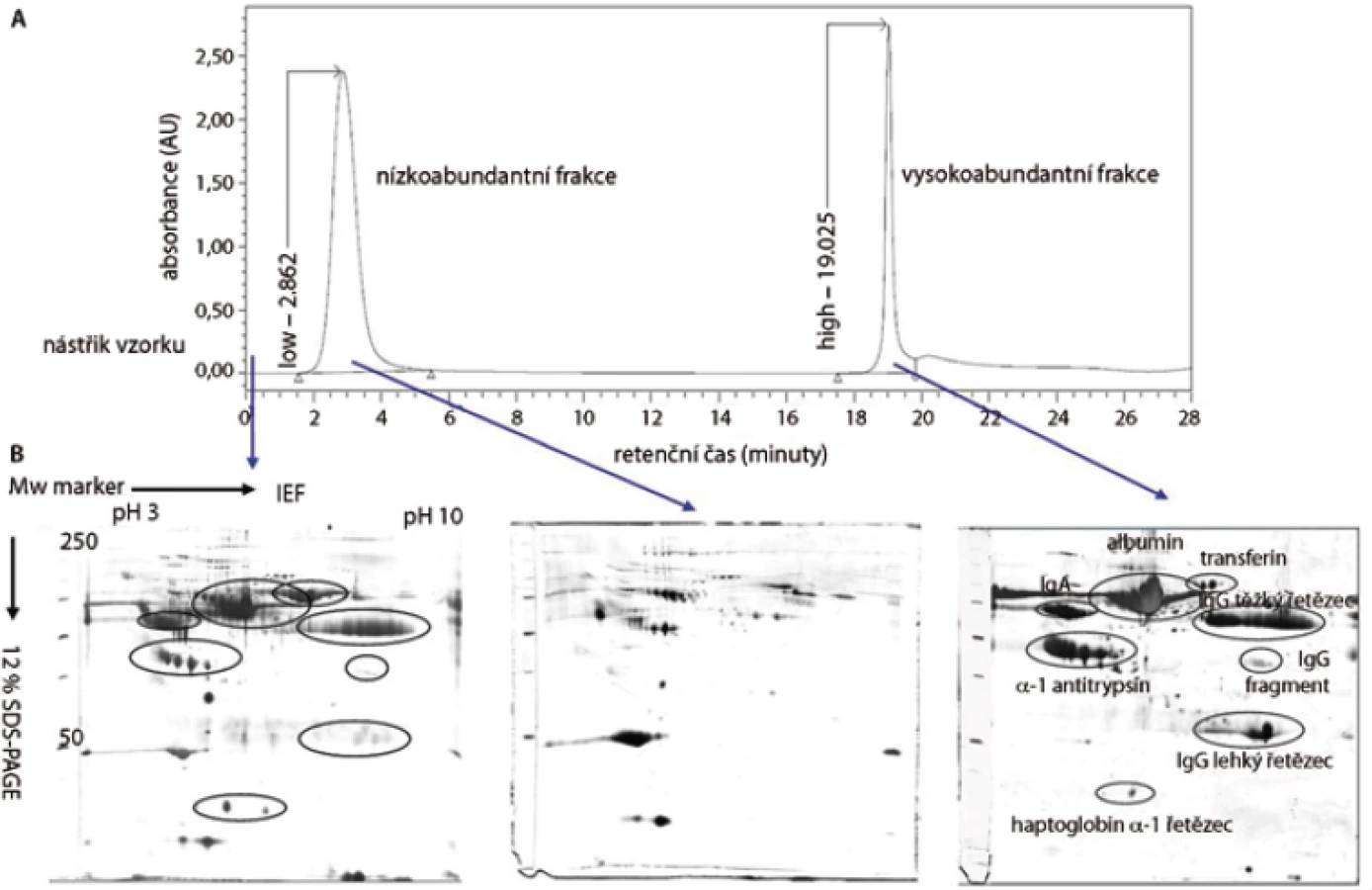
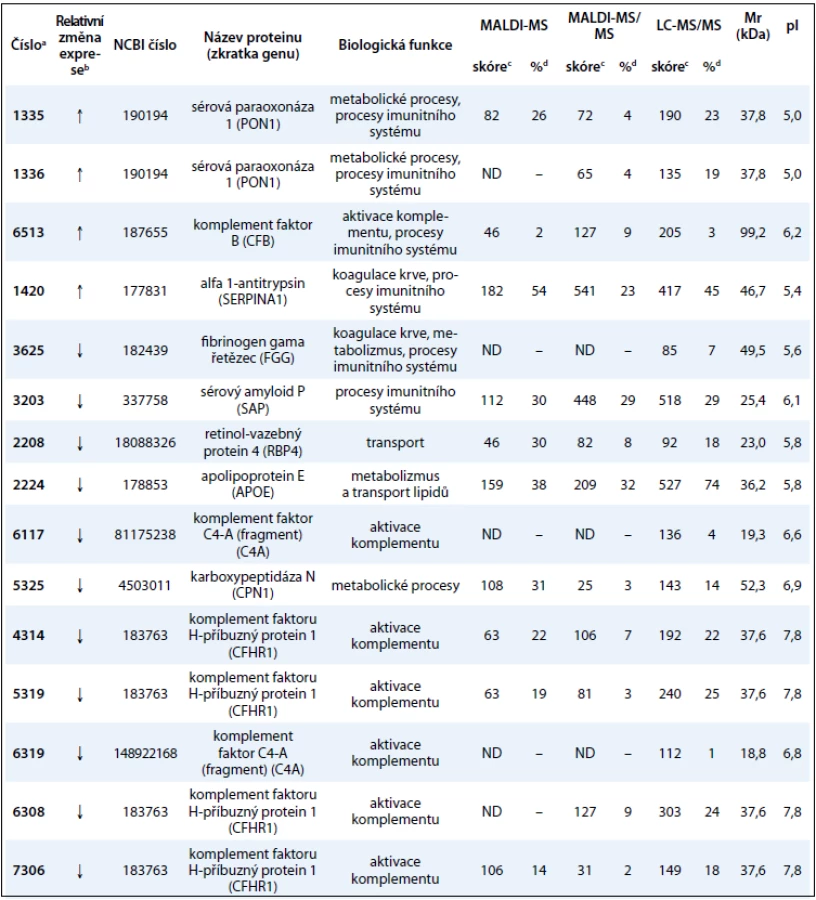
V této práci jsme porovnali u 10 vzorků krevní plazmy dvě metody (MARS, ProteoMiner) umožňující odstranění vysokoabundantních proteinů krevní plazmy a zisk obohacené frakce nízkoabundantních proteinů (obr. 1). Metodou MARS bylo odstraněno celkem 6 majoritních proteinů krevní plazmy (obr. 2). Frakce nízkoabundantních proteinů získané oběma metodami byly poté separovány metodou 2-DE. U frakcí získaných metodou ProteoMiner bylo na 2-DE mapách detekováno o přibližně 200 proteinových spotů víc než u frakcí získaných metodou MARS, p < 0,005. Celkový přehled o počtu identifikovaných proteinových spotů na 2-DE gelech pro obě metody uvádí tab. 2. Na základě těchto výsledků byla metoda ProteoMiner vybrána pro další 2-DE analýzy vzorků krevní plazmy 18 pacientů nemocných MM. Rozdíly v proteinových mapách mezi skupinami pacientů citlivých a rezistentních na podaný bortezomib byly určeny pomocí programu PDQuest. Celkem bylo nalezeno 15 rozdílových proteinových spotů (obr. 3), ve kterých bylo identifikováno MS technikami 10 proteinů (tab. 3). Tři proteiny vykazovaly signifikantně nižší hladinu proteinu v celé skupině 9 chemosenzitivních pacientů – sérový amyloid P (spot 3203), fibrinogen – gama řetězec (spot 3625), retinol-vazebný protein 4 (spot 2208). Dvě formy komplementu faktoru C4-A (spoty 6117, 6319) a apolipoprotein E (spot 2224) vykazovaly signifikantně nižší hladinu proteinu u skupiny 8, resp. 6 chemosenzitivních pacientů. U šesti z devíti chemosenzitivních pacientů byla pozorována signifikantně zvýšená exprese tří proteinů – dvě formy sérové paraoxonázy (spoty 1335, 1336), alfa-1-antitrypsin (spot 1420) a komplement faktor B (spot 6513). U celé skupiny 9 chemorezistentních pacientů byly detekovány signifikantně zvýšené hladiny 2 proteinů: izoformy komplementu faktoru H – příbuzného proteinu 1 (spoty 4314, 5319, 6308, 7306) a karboxypeptidáza N (spot 5325).
Diskuze a závěr
Proteomické přístupy využívané při hledání nových biomarkerů krevní plazmy kombinují postupy založené na frakcionaci, separaci, detekci a identifikaci přítomných proteinů. Aby bylo možné detekovat nízkoabundantní proteiny krevní plazmy, je třeba nejdříve odstranit většinově zastoupené proteiny. K tomuto účelu je na trhu dostupných několik analytických metod [13,31,32]. V naší práci jsme se zaměřili na srovnání dvou prepurifikačních metod umožňujících detekci nízkoabundantních proteinů pomocí 2-DE. Použili jsme dvě afinitní metody: Multiple Affinity Removal System (MARS) a ProteoMiner. Metoda ProteoMiner představuje odlišný, nedepleční systém pro zkoncentrování méně zastoupených proteinů a oproti metodě MARS umožnila detekci přibližně trojnásobného počtu proteinových spotů na 2-DE gelech (318 vs 111 identifikovaných spotů, p < 0,005). Výhodami metody ProteoMiner jsou dobrá reprodukovatelnost, jednoduchost postupu a přímá kompatibilita s dalšími proteomickými metodami, jako jsou např. diferenční gelová elektroforéza (2D-DIGE) a desorpce/ionizace laserem z upravených povrchů (SELDI-TOF-MS) [33]. Bylo prokázáno, že saturace kuliček vysokoabundantními proteiny neovlivňuje kvantifikaci obohacené frakce nízkoabundantních proteinů [34], proto jsme tuto metodu zvolili jako vhodnou prepurifikační metodu před 2-DE analýzou proteinů krevní plazmy.
Pomocí metod ProteoMiner/2-DE/MS byly identifikovány signifikantní změny v expresi proteinů mezi skupinami pacientů senzitivních a rezistentních na bortezomib. Identifikované proteiny patří především do skupiny běžně se vyskytujících proteinů, které se v plazmě nacházejí v poměrně vysoké koncentraci. Podle jejich specifické funkce se jedná o transportní proteiny (APOE, FGG, RBP4), metabolické proteiny (CPN1, PON1, CFHR1, C4A, CFB, SERPINA1), signální proteiny (FGG, C4A, CFHR1, CFB), proteiny aktivující komplementový systém (CFB, CFHR1, C4A), proteiny imunitní odpovědi (SAP, FGG, PON1) a proteiny aktivní při koagulaci krve (FGG, CFHR1).
U některých identifikovaných proteinů již byl popsán vztah k onemocněním nesouvisejícím s mnohočetným myelomem. Aktivovaná komplementová kaskáda je mechanizmem, který představuje prospěšnou obrannou reakci organizmu, ale také může vyvolávat širokou škálu patologických reakcí od zánětu až po degenerativní onemocnění nebo k nim přispívat. Aktivace komplementu byla popsána např. u alergické reakce typu III, kdy je komplement aktivován imunokomplexy, u systémového lupus erythematodes, u diabetes mellitus I. typu, u hemolytických anémií, revmatoidní artritidy a dny [35].
Alfa-1-antitrypsin je 52 kDa protein akutní fáze, typický serin proteázový inhibitor vyskytující se v plazmě o koncentraci 0,9–2,0 g/l. Protein je tvořený v játrech, odkud je uvolňován do krevního řečiště. Zabezpečuje ochranu organizmu před účinky enzymů (elastázy a kolagenázy) uvolňovaných z bílých krvinek při zánětu. Vrozený defekt AAT způsobuje rozvoj plicního a jaterního postižení [36].
Sérová paraoxonáza 1 (PON1) je glykoprotein o molekulové hmotnosti 43 kDa, který je syntetizován v játrech a následně vyplavován do krevního řečiště, kde je asociován s lipoproteiny o vysoké hustotě (HDL) [37]. PON1 hraje významnou roli ve vzniku a rozvoji aterosklerotického postižení, diabetu mellitu II. typu a jeho komplikací [38]. Řada asociačních studií prokázala vztah mezi polymorfizmy PON1 s neurodegenerativními onemocněními, zejména s Parkinsonovou a Alzheimerovou nemocí [39].
Retinol-vazebný protein 4 je významný klinický biomarker pro detekci inzulinové rezistence a diabetu mellitu II. typu [40]. Fibrinogen je glykoprotein o molekulové hmotnosti 340 kDa, který se běžně vyskytuje v krevní plazmě a je nezbytný ke správnému zajištění hemokoagulace. Jeho plazmatické koncentrace se zvyšují např. při zánětu, poškození tkání a při nádorových onemocněních. Apolipoprotein E je významný plazmatický lipoprotein zodpovědný za transport a metabolizmus cholesterolu. Klinicky abnormální funkce ApoE je sdružena s mnoha onemocněními, jako je ateroskleróza a Alzheimerova nemoc [41].
Ačkoli použití metody ProteoMiner v kombinaci s 2-DE analýzou podstatně zvýšilo počet detekovaných proteinů ve srovnání s jinými komerčně dostupnými frakcionačními technikami, zvolený postup analýzy se ukázal jako nevhodný pro detailní studium mechanizmu lékové rezistence v krevní plazmě pacientů nemocných MM. Ke studiu minoritních složek bude nutné využít vhodné vícestupňové kombinace frakcionačních a separačních chromatografických technik s následnou citlivou hmotnostně spektrometrickou detekcí.
Poděkování
Děkuji Danuši Fridrichové za přípravu tryptických digestů.
Tato práce byla podpořena granty MŠMT ČR (LC06027 a MSM0021622434) a projektem IGA MZ (NS9683) a NT 12130) a grantem GAČR GAP304/10/1395..
Autoři deklarují, že v souvislosti s předmětem studie nemají žádné komerční zájmy.
Redakční rada potvrzuje, že rukopis práce splnil ICMJE kritéria pro publikace zasílané do bi omedicínských časopisů.
prof. MUDr. Hájek Roman, CSc.
Ústav patologické fyziologie
Lékařská fakulta MU
Kamenice 5
625 00 Brno
e-mail: r.hajek@fnbrno.cz
Obdrženo: 3. 6. 2011
Přijato: 8. 9. 2011
Zdroje
1. Ahn SM, Simpson RJ. Body fluid proteomics: Prospects for biomarker discovery. Proteomics Clin Appl 2007; 1(9): 1004–1015.
2. Apweiler R, Aslanidis C, Deufel T et al. Approaching clinical proteomics: current state and future fields of application in fluid proteomics. Clin Chem Lab Med 2009; 47(6): 724–744.
3. Anderson NL, Anderson NG. The human plasma proteome: history, character, and diagnostic prospects. Mol Cell Proteomics 2002; 1(11): 845–867.
4. Philips AV, Cooper TA. RNA processing and human disease. Cell Mol Life Sci 2000; 57(2): 235–249.
5. Mann M, Jensen ON. Proteomic analysis of post-translational modifications. Nat Biotechnol 2003; 21(3): 255–261.
6. O’Farrell PH. High resolution two-dimensional electrophoresis of proteins. J Biol Chem 1975; 25(10): 4007–4021.
7. Zelená J, Hájek R. Proteomické techniky a jejich aplikace u hematoonkologických onemocnění. Čas Lék Čes 2007; 146(7): 586–592.
8. Potáčová A, Čumová J, Hájek R. Proteomická analýza a její využití ve výzkumu mnohočetného myelomu. Klin Onkol 2008; 21 (Suppl 1): 243–246.
9. Kim HJ, Kim MR, So EJ et al. Comparison of proteomes in various human plasma preparations by two-dimensional gel electrophoresis. J Biochem Biophys Methods 2007; 70(4): 619–625.
10. Kim MR, Kim CW. Human blood plasma preparation for two-dimensional gel electrophoresis. J Chromatogr B Analyt Technol Biomed Life Sci 2007; 849(1–2): 203–210.
11. Cho SY, Lee EY, Lee JS et al. Efficient prefractionation of low-abundance proteins in human plasma and construction of a two-dimensional map. Proteomics 2005; 5(13): 3386–3396.
12. Vasudev NS, Ferguson RE, Cairns DA et al. Serum biomarker discovery in renal cancer using 2-DE and prefractionation by immunodepletion and isoelectric focusing; increasing coverage or more of the same? Proteomics 2008; 8(23–24): 5074–5085.
13. Polaskova V, Kapur A, Khan A et al. High-abundance protein depletion: comparison of methods for human plasma biomarker discovery. Electrophoresis 2010; 31(3): 471–482.
14. Roche S, Tiers L, Provansal M et al. Depletion of one, six, twelve or twenty major blood proteins before proteomic analysis: the more the better? J Proteomics 2009; 72(6): 945–951.
15. Björhall K, Miliotis T, Davidsson P. Comparison of different depletion strategies for improved resolution in proteomic analysis of human serum samples. Proteomics 2005; 5(1): 307–317.
16. Echan LA, Tang HY, Ali-Khan N et al. Depletion of multiple high-abundance proteins improves protein profiling capacities of human serum and plasma. Proteomics 2005; 5(13): 3292–3303.
17. Magagnotti C, Fermo I, Carletti RM et al. Comparison of different depletion strategies for improving resolution of the human urine proteome. Clin Chem Lab Med 2010; 48(4): 531–535.
18. Yocum AK, Yu K, Oe T et al. Effect of immunoaffinity depletion of human serum during proteomic investigations. J Proteome Res 2005; 4(5): 1722–1731.
19. Brand J, Haslberger T, Zolg W et al. Depletion efficiency and recovery of trace markers from a multiparameter immunodepletion column. Proteomics 2006; 6(11): 3236–3242.
20. Bellei E, Bergamini S, Monari E et al. High-abundance proteins depletion for serum proteomic analysis: concomitant removal of non-targeted proteins. Amino Acids 2011; 40(1): 145–156.
21. Tu C, Rudnick PA, Martinez MY et al. Depletion of abundant plasma proteins and limitations of plasma proteomics. J Proteome Res 2010; 9(10): 4982–4991.
22. Thulasiraman V, Lin SH, Gheorghiu L et al. Reduction of the concentration difference of proteins in biological liquids using a library of combinatorial ligands. Electrophoresis 2005; 26(18): 3561–3571.
23. Righetti PG, Boschetti E, Lomas L et al. Protein Equalizer Technology: the quest for a „democratic proteome“. Proteomics 2006; 6(14): 3980–3992.
24. Boschetti E, Righetti PG. The ProteoMiner in the proteomic arena: a non-depleting tool for discovering low-abundance species. J Proteomics 2008; 71(3): 255–264.
25. Keidel EM, Ribitsch D, Lottspeich F. Equalizer technology – Equal rights for disparate beads. Proteomics 2010; 10(11): 2089–2098.
26. Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 1976; 72: 248–254.
27. Havlis J, Thomas H, Sebela M et al. Fast-response proteomics by accelerated in-gel digestion of proteins. Anal Chem 2003; 75(6): 1300–1306.
28. Planeta J, Karasek P, Vejrosta J. Development of packed capillary columns using carbon dioxide slurries. J Sep Sci 2003; 26: 525–530.
29. Apweiler R, Martin MJ, O’Donovan C et al. The universal protein resource (UniProt) in 2010. Nucleic Acids Res 2010; 38: D142–D148.
30. Mi H, Lazareva-Ulitsky B, Loo R et al. The PANTHER database of protein families, subfamilies, functions and pathways. Nucleic Acids Res 2005; 33: D284–D288.
31. Tirumalai RS, Chan KC, Prieto DA et al. Characterization of the low molecular weight human serum proteome. Mol Cell Proteomics 2003; 2(10): 1096–1103.
32. Fang X, Zhang WW. Affinity separation and enrichment methods in proteomic analysis. J Proteomics 2008; 71(3): 284–303.
33. Sihlbom C, Kanmert I, Bahr H et al. Evaluation of the combination of bead technology with SELDI-TOF-MS and 2-D DIGE for detection of plasma proteins. J Proteome Res 2008; 7(9): 4191–4198.
34. Hartwig S, Czibere A, Kotzka J et al. Combinatorial hexapeptide ligand libraries (ProteoMiner™): an innovative fractionation tool for differential quantitative clinical proteomics. Arch Physiol Biochem 2009; 115(3): 155–160.
35. Porcel JM, Ordi J, Castro-Salomo A et al. The value of complement activation products in the assessment of systemic lupus erythematosus flares. Clin Immunol Immunopathol 1995; 74(3): 283–288.
36. American Thoracic Society, European Respiratory Society. American Thoracic Society/European Respiratory Society Statement: standards for the diagnosis and management of individuals with alpha-1 antitrypsin deficiency. Am J Respir Crit Care Med 2003; 168(7): 818–900.
37. Durrington PN, Mackness B, Mackness MI. Paraoxonase and atherosclerosis. Arterioscler Thromb Vasc Biol 2001; 21(4): 473–480.
38. Flekač M, Škrha J, Novotný Z. Faktory ovlivňující aktivitu a koncentraci antioxidačního enzymu paraoxonáza 1. Klin Biochem Metab 2006; 14(35): 33–39.
39. Goswami B, Tayal D, Gupta N et al. Paraoxonase: a multifaceted biomolecule. Clin Chim Acta 2009; 410(1–2): 1–12.
40. Graham TE, Yang Q, Blüher M et al. Retinol-binding protein 4 and insulin resistance in lean, obese, and diabetic subjects. N Engl J Med 2006; 354(24): 2552–2563.
41. Hofman A, Ott A, Breteler MM et al. Atherosclerosis, apolipoprotein E, and prevalence of dementia and Alzheimer’s disease in the Rotterdam Study. Lancet 1997; 349(9046): 151–154.
Štítky
Detská onkológia Chirurgia všeobecná OnkológiaČlánok vyšiel v časopise
Klinická onkologie

2012 Číslo 1
- Metamizol jako analgetikum první volby: kdy, pro koho, jak a proč?
- MUDr. Lenka Klimešová: Multiodborová vizita je kľúč k efektívnejšej perioperačnej liečbe chronickej bolesti
- Nejasný stín na plicích – kazuistika
Najčítanejšie v tomto čísle
- Žilní vstupy v onkologii
- Využitie elektroimpedančnej tomografie v diagnostike karcinómu prsníka
- Identifikácia molekulárnych markerov u detí s akútnou myeloblastovou leukémiou (AML)
- Šestileté sledování pacienta s mnohočetnou angiomatózou postihující skelet, břišní i hrudní dutinu a stěnu trávicí trubice