
Malaria-Induced NLRP12/NLRP3-Dependent Caspase-1 Activation Mediates Inflammation and Hypersensitivity to Bacterial Superinfection
Cyclic paroxysm and high fever are hallmarks of malaria and are associated with high levels of pyrogenic cytokines, including IL-1β. In this report, we describe a signature for the expression of inflammasome-related genes and caspase-1 activation in malaria. Indeed, when we infected mice, Plasmodium infection was sufficient to promote MyD88-mediated caspase-1 activation, dependent on IFN-γ-priming and the expression of inflammasome components ASC, P2X7R, NLRP3 and/or NLRP12. Pro-IL-1β expression required a second stimulation with LPS and was also dependent on IFN-γ-priming and functional TNFR1. As a consequence of Plasmodium-induced caspase-1 activation, mice produced extremely high levels of IL-1β upon a second microbial stimulus, and became hypersensitive to septic shock. Therapeutic intervention with IL-1 receptor antagonist prevented bacterial-induced lethality in rodents. Similar to mice, we observed a significantly increased frequency of circulating CD14+CD16−Caspase-1+ and CD14dimCD16+Caspase-1+ monocytes in peripheral blood mononuclear cells from febrile malaria patients. These cells readily produced large amounts of IL-1β after stimulation with LPS. Furthermore, we observed the presence of inflammasome complexes in monocytes from malaria patients containing either NLRP3 or NLRP12 pyroptosomes. We conclude that NLRP12/NLRP3-dependent activation of caspase-1 is likely to be a key event in mediating systemic production of IL-1β and hypersensitivity to secondary bacterial infection during malaria.
Published in the journal:
Malaria-Induced NLRP12/NLRP3-Dependent Caspase-1 Activation Mediates Inflammation and Hypersensitivity to Bacterial Superinfection. PLoS Pathog 10(1): e32767. doi:10.1371/journal.ppat.1003885
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1003885
Summary
Cyclic paroxysm and high fever are hallmarks of malaria and are associated with high levels of pyrogenic cytokines, including IL-1β. In this report, we describe a signature for the expression of inflammasome-related genes and caspase-1 activation in malaria. Indeed, when we infected mice, Plasmodium infection was sufficient to promote MyD88-mediated caspase-1 activation, dependent on IFN-γ-priming and the expression of inflammasome components ASC, P2X7R, NLRP3 and/or NLRP12. Pro-IL-1β expression required a second stimulation with LPS and was also dependent on IFN-γ-priming and functional TNFR1. As a consequence of Plasmodium-induced caspase-1 activation, mice produced extremely high levels of IL-1β upon a second microbial stimulus, and became hypersensitive to septic shock. Therapeutic intervention with IL-1 receptor antagonist prevented bacterial-induced lethality in rodents. Similar to mice, we observed a significantly increased frequency of circulating CD14+CD16−Caspase-1+ and CD14dimCD16+Caspase-1+ monocytes in peripheral blood mononuclear cells from febrile malaria patients. These cells readily produced large amounts of IL-1β after stimulation with LPS. Furthermore, we observed the presence of inflammasome complexes in monocytes from malaria patients containing either NLRP3 or NLRP12 pyroptosomes. We conclude that NLRP12/NLRP3-dependent activation of caspase-1 is likely to be a key event in mediating systemic production of IL-1β and hypersensitivity to secondary bacterial infection during malaria.
Introduction
Every year, approximately 250 million people are infected with Plasmodium, contributing to significant social and economic instability in the developing countries around the world [1]. One of the main physiological responses to Plasmodium infection is the paroxysm – characterized by cycles of sharp peaks of high fever accompanied by chills and rigors, which coincide with the release of parasites from synchronized infected red blood cells [2], [3]. Parasite components, such as DNA bound to hemozoin [4], [5] and glycosylphosphatidylinositol (GPI) anchors [6], trigger the production of proinflammatory cytokines, including interleukin-1 beta (IL-1β), via activation of Toll-Like receptors (TLRs) [7]. Furthermore, malaria sepsis [8] leads to an exquisite sensitivity to secondary bacterial infections, in particular non-typhoidal salmonellosis, that often associate with severe disease [9]–[12]. Hence, a better understanding of the mechanisms involved on this inflammatory stage during malaria is critical for the clinical management and prevention of severe disease.
TLRs are only one family of the receptors required for the release of active IL-1β, as cleavage of pro-IL-1β by caspase-1 also requires activation of Nod-Like Receptors (NLRs) [13], [14]. Upon stimulation, the respective NLRs oligomerize and recruit pro-caspase-1 directly via a N-terminal caspase recruitment domain (CARD) homotypic interaction (CARD-CARD) (e.g., CARD-containing NLRs such as NLRP1 or NLRC4) or indirectly via the adaptor molecule called apoptosis-associated speck-like protein containing a caspase recruitment domain (ASC), as is the case of NLRP3-inflammasome [13]. The inflammasome assembly culminates on activation of caspase-1, and consequent release of the active form of IL-1β. NLRP3 containing inflammasome is activated in response to a large range of insults, such as pathogens, bacterial RNA, and crystal structures [15]–[17]. The NLRP12 was the first NLR shown to associate with ASC and to form an active IL-1β-maturing inflammasome [18]. This receptor was initially placed as a negative regulator of inflammation [19], [20], but it was also shown to be involved in periodic fever of cryopyrinopathies [21], and to mediate host resistance to Yersinia pestis [22].
Here, we asked what are the molecular steps required for and the physiological role of inflammasome assembly during malaria sepsis. Our results indicate that symptomatic Plasmodium infection triggers inflammasome formation and caspase-1 activation via an intricate process that requires several inflammatory mediators as well as NLRP3 and NLRP12. Furthermore, we found that the malaria-primed monocytic cells produce deleterious amounts of IL-1β when exposed to a second microbial challenge, being an important component of the overwhelming inflammatory response observed during bacterial superinfection.
Results
Expression of inflammasome genes and ASC-dependent caspase-1 activation in rodent malaria
The Plasmodium chabaudi AS rodent model was used to evaluate the in vivo activation of inflammasome. The microarray analysis of splenocytes from C57BL/6 at 6 days post-infection demonstrates enhanced expression of various genes from the inflammasome pathway, including Casp1 and Il1b (Figure 1A). Consistently, the FLICA assay, which employs the fluorescent probe FAM-YVAD-FMK and Western blot, indicate that infection with P. chabaudi is sufficient to promote caspase-1 activation (Figures S1A and 1B). Immunoblots evidenced enhanced expression and cleavage of pro-caspase-1 in spleens from P. chabaudi infected mice (Figures S1B, S1C and 1C). The FLICA assay also revealed that macrophages (CD11b+F4/80+) and dendritic cells (DCs) (CD11c+MHC-II+) are the main source of active caspase-1 (Figure 1B) in the spleens from infected mice. We also observed a high frequency of macrophages and DCs undergoing inflammatory cell death (pyroptosis), as defined by damage of cell membranes evaluated by DNA-7AAD staining and augmented cell size associated with active caspase-1 (Figure 1B). Other splenic cell subsets did not express active caspase-1 or were undergoing pyroptosis during P. chabaudi infection (Figure S1D).
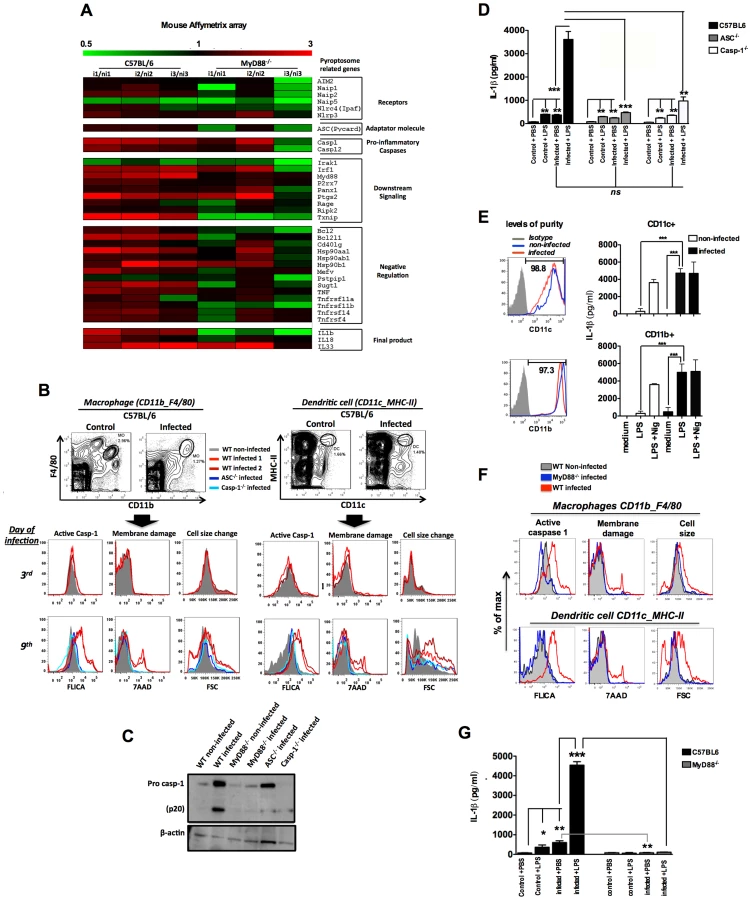
Importantly, macrophages and DCs from mice deficient for Asc (ASC−/−) and Casp1 (Casp-1−/−) were negative for both active caspase-1 and pyroptosis markers during P. chabaudi infection (Figures 1B). Similar results were obtained when we used splenocyte lysates in immunoblots to detect active caspase-1 (Figures 1C). We also evaluated the role of caspase-1 activation in host resistance to P. chabaudi. C57BL/6, ASC−/− and Casp-1−/− mice injected with 105 P. chabaudi infected erythrocytes displayed similar parasitemia beginning at day 5, peaking at days 7–8 days, and completely resolved at 25 days post-infection (Figure S2A). No lethality was observed until the end of the experiment.
Leuckocytes from mice and humans infected with Plasmodium are hyper-responsive to TLR agonists [23]. This hyperresponsiveness does not occur at all phases of infection. For example, as we show in Figure S2B, mice that are infected with P. chabaudi were hyperresponsive to small doses of TLR ligands, such as lipopolysaccharide (LPS), during the acute phase of the disease (day 7). This hyperresponsiveness was no longer observed when mice were challenged with low dose LPS 4 weeks after infection. Based on this and other studies [24], [25], we hypothesize that bacterial superinfection is a common co-factor for severe malaria. Thus, we used a challenge with low dose of bacterial LPS to mimic secondary bacterial infection and to evaluate the role of inflammasome and IL-1β in this process. C57BL/6 mice infected with P. chabaudi produced low, but significant levels of IL-1β, when compared to uninfected controls (Figures 1D and S2B). Surprisingly, these low levels of circulating IL-1β were produced in an ASC and Caspase-1-independent manner (Figure 1D). In contrast, production of IL-1β, which was extremely high in infected C57BL/6 mice challenged with LPS, was severely impaired in ASC−/− or Casp-1−/− mice (Figure S2B and Figure 1D). In order to check whether the active caspase-1+ cells were the main sources of active IL-1β, highly purified CD11b+ and CD11c+ cells from infected mice were stimulated with LPS and IL-1β measured thereafter. Both CD11b+ as well as CD11c+ cells produced high levels of IL-1β (Figure 1E).
MyD88-dependent expression of inflammasome genes and caspase-1 activation in rodent malaria
The in vivo proinflammatory priming promoted by Plasmodium infection is partially dependent on TLR9 activation by parasite DNA [4], [23], [26]. Consistently, expression of various inflammasome genes was no longer enhanced in Myd88-deficient mice (MyD88−/−) infected with P. chabaudi (Figure 1A and Table S1). Accordingly, we did not observe active caspase-1, and pyroptotic features in macrophages and DCs from MyD88−/− mice infected with P. chabaudi (Figure 1C and 1F). As expected, no systemic IL-1β was detected in sera of infected MyD88−/− mice challenged or not with LPS (Figure 1G). Furthermore, the data presented in Figure S3A show reduced expression of pro-caspase-1 in Tlr9-deficient mice (TLR9−/−). In view of this reduced level of basal expression of the pro enzyme, it is not surprising that expression levels of its active form were decreased in TLR9−/− mice infected with P. chabaudi. Likewise, the levels of circulating IL-1β were partially affected and survival rates increased in infected TLR9−/− mice challenged with the low LPS dose (Figures S3B and S3C). Finally, we found that priming with CpG immunostimulatory oligonucleotides mimics P. chabaudi infection leading to enhanced susceptibility to LPS challenge, but does not induce lethality when used to challenge P. chabaudi infected mice (Figure S3D).
IFN-γ is required for expression of pro-caspase-1 and pro-IL-1β in rodent malaria
The levels of circulating interferon gamma (IFN-γ), tumor necrosis factor-alpha (TNF-α) and IL-1β in sera of P. chabaudi infected mice, challenged or not with LPS, are presented in Figure S2B. Very low levels of IFN-γ, TNF-α, and IL-1β were detected in sera from uninfected mice challenged with low dose LPS. The hyperresponsiveness to LPS was observed in the first two weeks, but not at 28 days post-infection (Figure S2B). IFN-γ was shown to be a key mediator of inflammatory priming in febrile malaria patients and in mice infected with P. chabaudi [23]. Furthermore, TNF-α, a critical cytokine in malaria pathogenesis [27], [28] was shown to mediate expression of inflammasome components and pro-IL-1β [29], [30]. Indeed, even upon LPS challenge, active IL-1β was not produced in either Ifng or Tnfrsf1a-deficient mice (IFN-γ−/−) and (TNFR1−/−) respectively, when infected with P. chabaudi (Figure 2A). Normal activation of caspase-1 was observed in macrophages and DCs from TNFR1−/−, but not from IFN-γ−/− mice (Figures 2B, 2C and S4). In contrast, both IFN-γ and TNF-α were required for expression of pro-IL-1β by spleen cells of infected mice challenged with LPS (Figures 2D and S4).
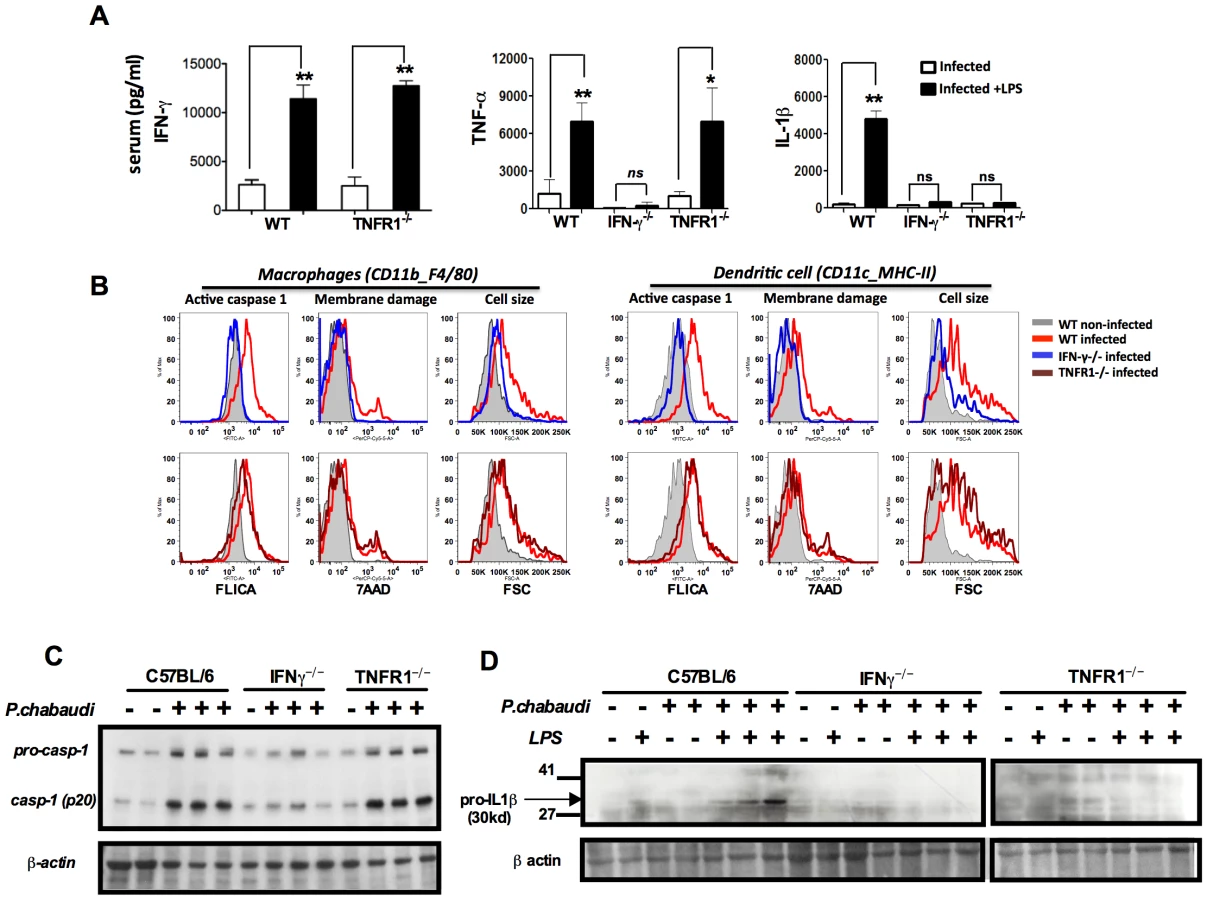
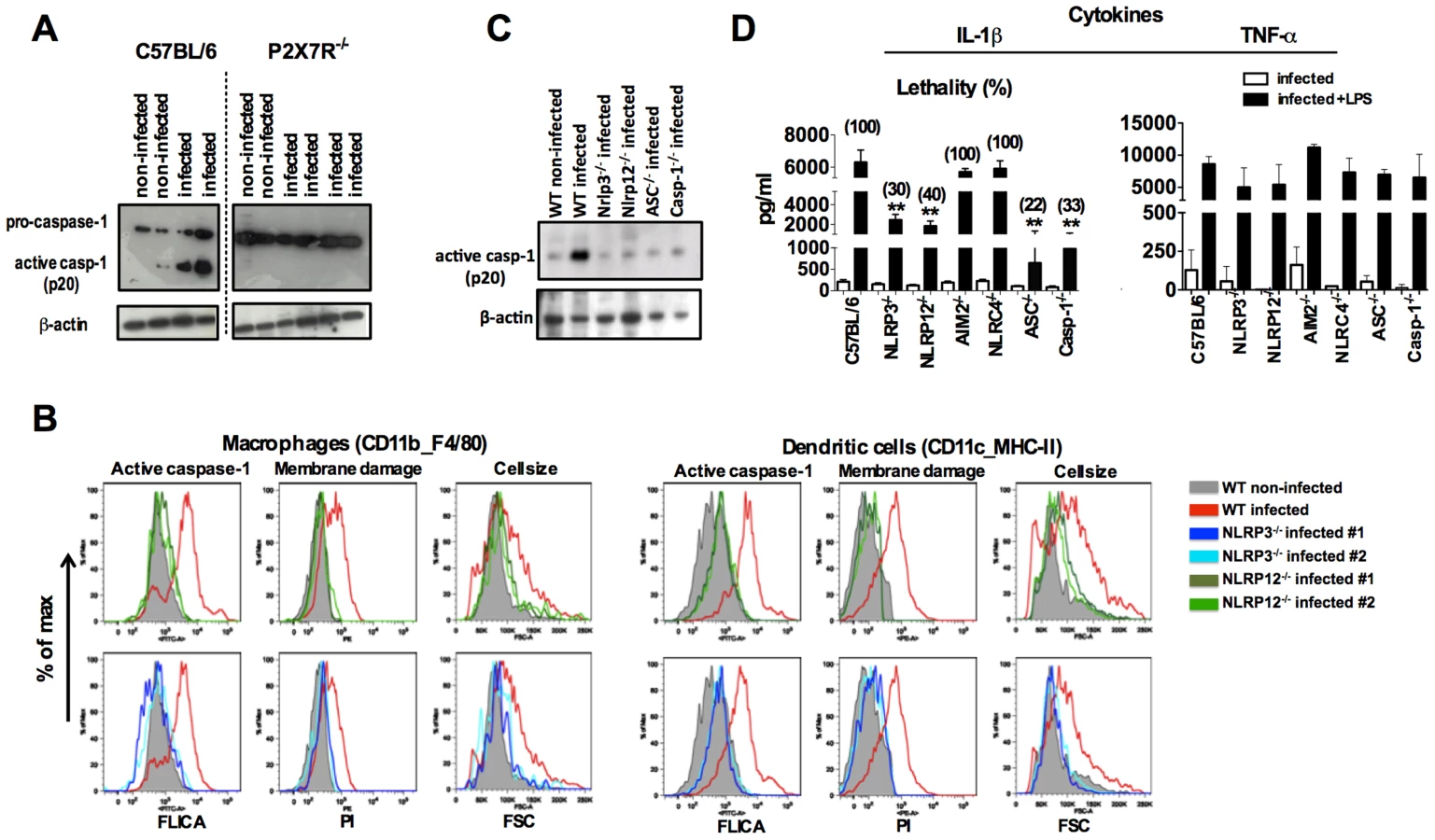
NLRP3 and NLRP12 are required for caspase-1 activation and optimum IL-1β production during rodent malaria
We next evaluated the requirement of specific NLRs for caspase-1 activation. The P2X7 receptor (P2X7R) which senses extracellular ATP, opens a cation-specific channel that alters the ionic environment of the cell [31] culminating on NLRP3-inflammasome assembly under certain conditions [32], [33]. Here we found that P2X7R is necessary for caspase-1 activation during in vivo infection with P. chabaudi (Figure 3A). Consistently, NLRP3 and NLRP12 were required for activation of caspase-1, systemic production of IL-1β and pyroptosis (Figures 3B, 3C and 3D). Other cytosolic receptors, i.e. NLRC4 and absent in melanoma 2 (AIM2) were not essential for IL-1β release (Figure 3D). The levels of circulating IL-1β in sera of C57BL6, NLRP3−/−, NLRP12−/−, ASC−/− as well as Casp-1−/− infected mice, but no challenged with LPS, were not statically different (Figure 3D).
Therapeutic effects of IL-1 receptor antagonist in rodent malaria
The infected Il1r-deficient mice (IL-1R−/−) mice are partially resistant to the LPS challenge, despite of the sustained levels of active caspase-1, IL-1β and TNF-α (Figures 4A and 4B). Importantly, in the different mouse lineages infected with P. chabaudi, we observed a striking correlation of high circulating IL-1β, but not necessarily active caspase-1, and lethality induced by LPS (Table 1 and Figure 3D). Consistently, treatment with IL-1 receptor antagonist (IL-1RA) prevented lethality of P. chabaudi infected mice challenged with LPS (Figure 4C).
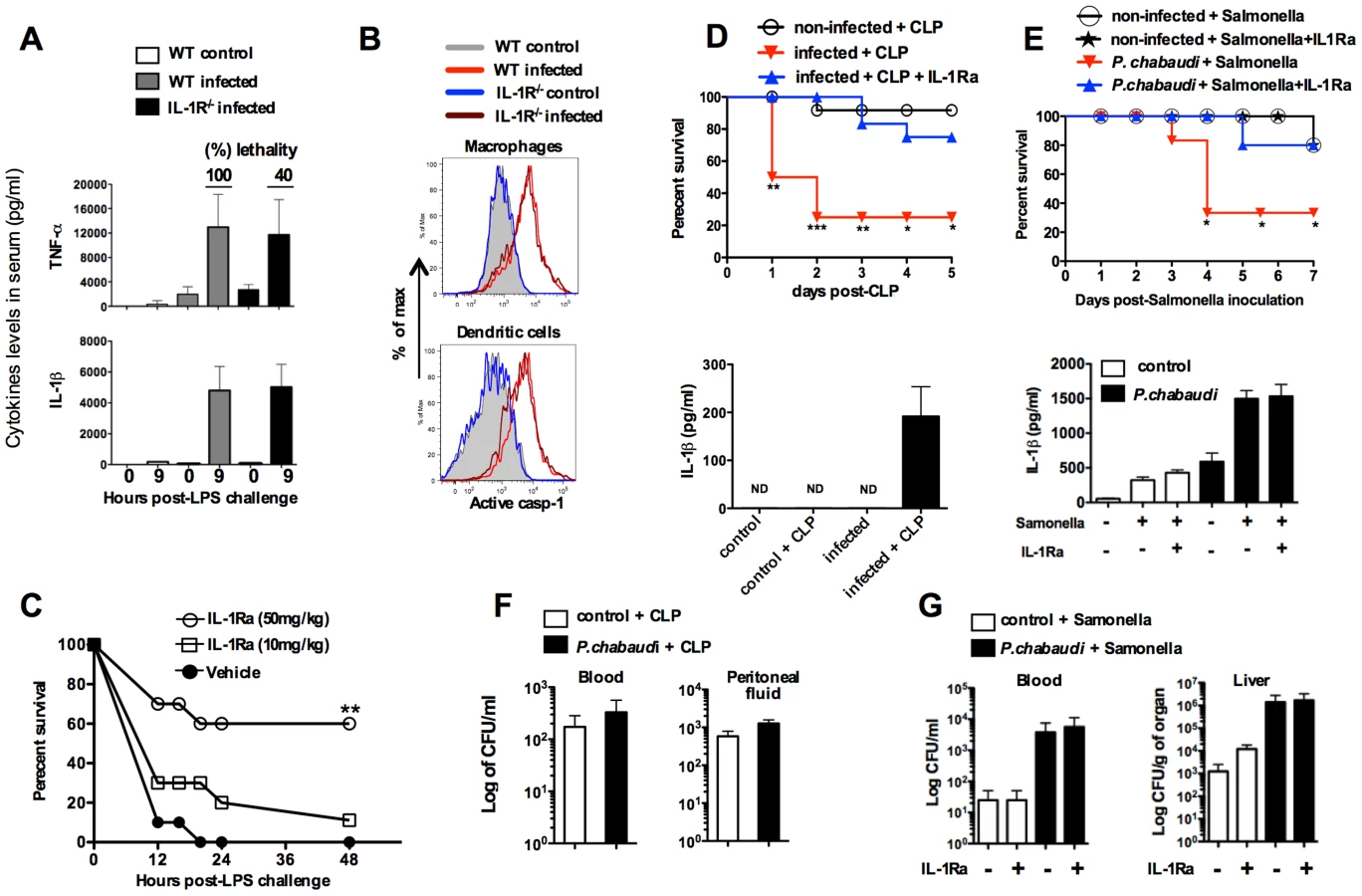
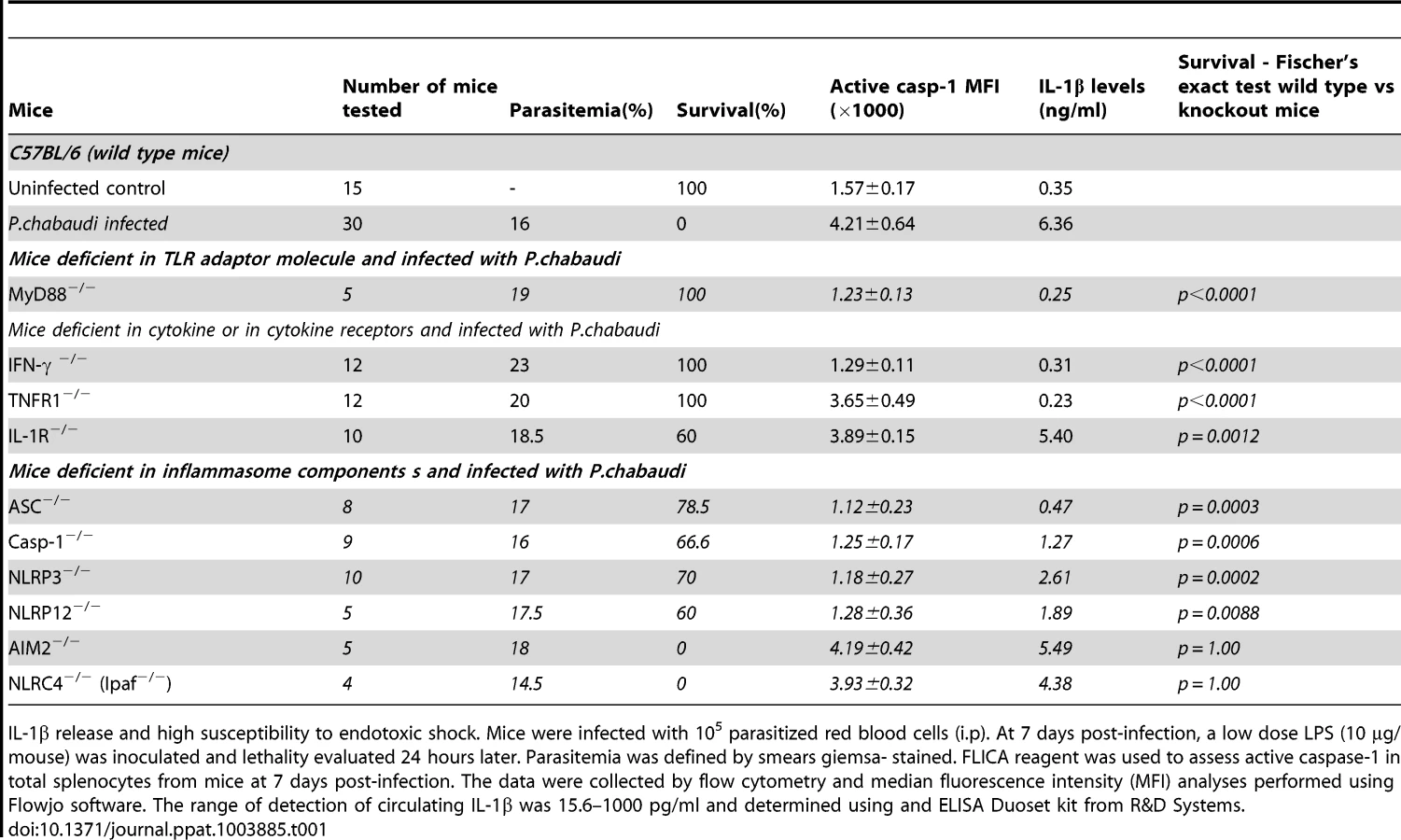
These results were validated in P. chabaudi infected mice challenged with sub-lethal cecal ligation puncture (CLP) (Figure 4D), a classic model for bacterial sepsis, as well as peroral infection with Salmonella typhimurium (Figure 4E). In both cases, bacterial superinfection leaded to rapid lethality that was associated with high circulating levels of IL-1β. Furthermore, mortality of co-infected mice was delayed or prevented by treatment with the IL-1RA. The loads of bacteria translocation were similar in co-infected mice treated or not with IL-1RA (Figures 4F and 4G).
Increased frequency of pyroptotic CD14dimCD16+Caspase-1+ monocytes in PBMCs from symptomatic malaria patients
We next studied caspase-1 activation and IL-1β release in peripheral blood mononuclear cells (PBMCs) from patients undergoing febrile malaria. Two different subsets of monocytes were closely examined: CD14+CD16− or CD14dimCD16+ monocytes (Figure 5A and S5A). Active caspase-1 was constitutively expressed in CD14+CD16− cells from healthy individuals, as previously described [34]. Nevertheless, the frequency of CD14+CD16− cells was augmented on average 3 fold in P. vivax malaria patients. The frequency and intensity of caspase-1 expression, indicated by median fluorescence intensity (MFI), in different monocyte subsets from healthy and P. vivax infected individuals before and after treatment are shown in Figure 5A. Expression of active caspase-1 in association with membrane damage and augmented cell size was also observed in CD14dimCD16+ monocytes (Figure S5A). The representative histograms presented in Figure S5A were obtained from counter plot gates shown in Figure 5A and illustrate the results for active caspase-1, membrane damage and cell size change from one malaria patient before and after treatment, and a healthy donor. Other cell populations found in PBMCs were negative for active caspase-1 and pyroptosis markers (Figure S5B).
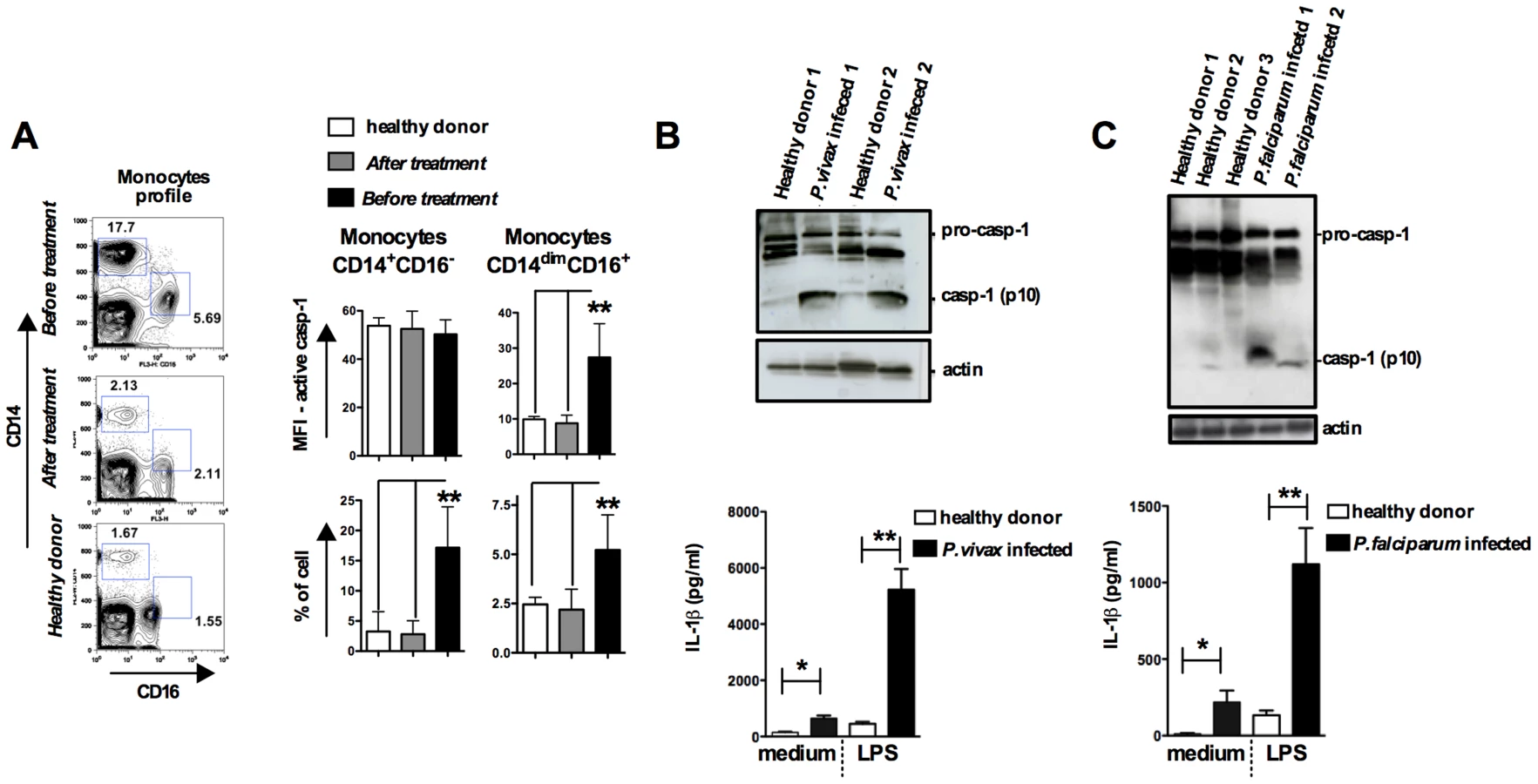
We also observed increased cleavage of caspase-1 (p10) in PBMCs from either P. vivax or P. falciparum malaria patients (Figures 5B, 5C and S5C). In addition, LPS-induced release of IL-1β is highly augmented in PBMCs from the same patients undergoing malaria sepsis (Figure 5B and 5C – bottom panels). Furthermore, we observed enhanced expression of inflammasome genes in PBMCs from P. falciparum malaria patients (Figure S5D and Table S2). As observed in the rodent malaria model (Figure 2), IFN-γ-priming of primary human monocytes mimics in vivo infection with Plasmodium and augments expression of pro-caspase-1, pro-IL-1β as well as IL-1β release induced by LPS stimulation (Figure S5E).
NLRP12/NLRP3 containing inflammasomes in febrile P. vivax infected patients
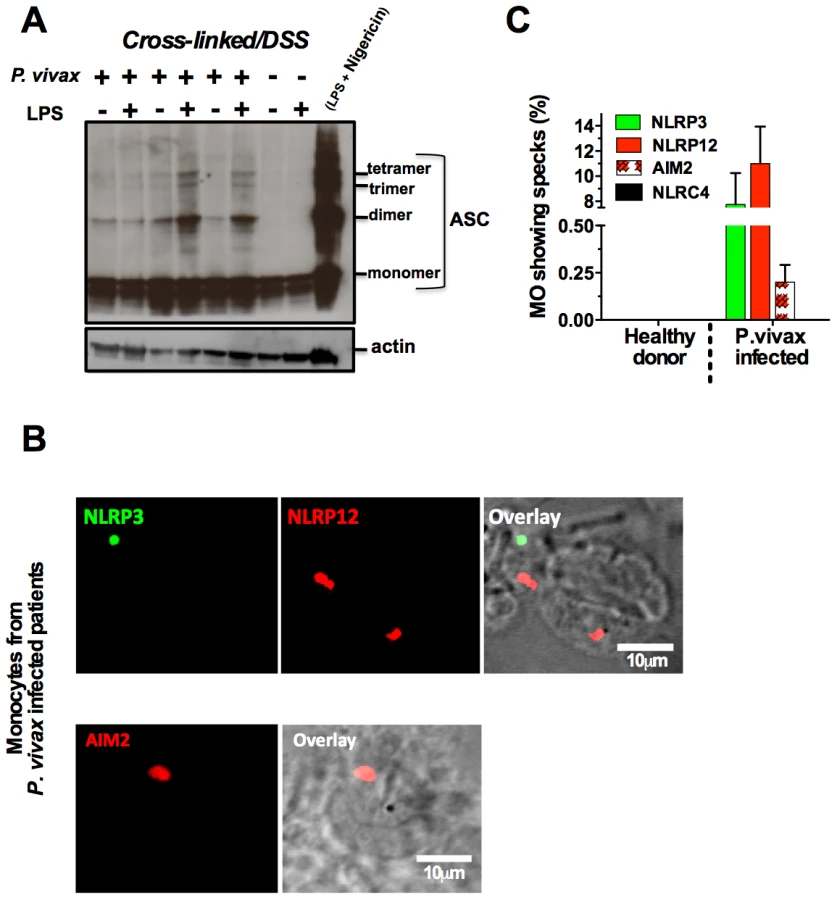
The nature of malaria-induced inflammasome was further explored by performing a crosslinking assay and confocal analyses. We observed an augmented multimerization of ASC (Figure 6A) in PBMCs from P. vivax infected individuals. Additionally, confocal microscopy indicated that inflammasome specks contained either NLRP3 (green) or NLRP12 (red), were present in 7 and 10% of monocytes from P. vivax malaria patients, respectively (Figure 6B and 6C). There was no co-localization of NLRP3 and NLRP12 specks. We did not detect monocytes containing NLRC4-inflammasome, and AIM2 specks appeared in a low frequency, ∼0.25% of monocytes from infected individuals. No specks were detected in monocytes from uninfected healthy donors (Figure 6C). Figure S6 provides controls of the confocal analysis.
Discussion
Paroxysm, an acute fever accompanied by chills and rigors, is a hallmark of Plasmodium infection [3], [35]. While the physiological role of fever is controversial, it can aid in host defense, delaying the growth of pathogens with strict temperature preferences [36]. In fact, prior to the discovery of antibiotics, deliberate infection with P. vivax was used to induce high fever and eliminate infection with Treponema pallidum in neurosyphilis patients [37]. However, fever is also associated with various pathological processes, such as respiratory distress, anemia, and neurological manifestations that cause morbidity and mortality in malaria [3], [35]. These clinical manifestations are related to the intensity of the systemic inflammatory response, yet the basic details of malaria-induced cytokinemia are not understood. Overall, our results indicate that Plasmodium infection primes innate immune cells leading to the oligomerization of inflammasomes containing, ASC, NLRP3 and NLRP12, resulting in activation of caspase-1 and the production of copious amounts of IL-1β upon a second TLR activation. An immediate consequence of this inflammatory priming is a drastic reduction in the threshold to septic shock-like syndrome caused by secondary bacterial infection.
Although there is a controversy about the role of different TLRs in the pathogenesis of malaria [38]–[41], various studies indicate that the initial cytokine storm during malaria is driven by TLR activation. Initial studies suggested that GPI anchors were the main P. falciparum molecules responsible for eliciting the production of proinflammatory cytokines during malaria [6]. However, recent studies indicate that parasite derived DNA containing both immunostimulatory CpG and AT-rich motifs, is instead the main force driving the cytokine storm during malaria sepsis [4], [5], [42]. Indeed, mice bearing non-functional TLR9 or MyD88, or treated with a TLR7/TLR9 antagonist display a less pronounced inflammatory response and attenuated pathology during experimental malaria [23], [26], [38], [41]. Moreover, mutations in TLR2, TLR9 and Mal/TIRAP appear to affect the outcome of human disease [24], [25], [43], [44].
Importantly, infection with Plasmodium in humans leads to a proinflammatory priming and hyperresponsiveness to microbial products [23], [45]–[47]. This enhanced ability to respond to microbes during immunosurveillance protects the host against infectious insult, but the innate immune response is the classic “double-edged sword”. Thus, the proinflammatory priming means that the innate immune system can overreact to secondary infection, leading to a septic shock syndrome with clinical manifestations. It is noteworthy that malaria is often associated with bacterial infections [12]. Furthermore, a recent study highlights that the chance to develop severe malaria is elevated 8.5 fold in children with bacteremia [9]. Similarly, bacterial and viral infections may also act as co-factors for severe P. vivax malaria [10], [35], [48]. We propose that inflammasome and IL-1β [35], [49], [50] are important components of the proinflammatory priming and the exquisite sensitivity to superinfection during malaria.
Here, we demonstrate that both in mouse and human malaria expression of inflammasome genes, caspase-1 activation and pyroptosis are induced in phagocytic cells. As a consequence, during Plasmodium infection the threshold for LPS sensitivity is decreased in at least 100 folds, as compared to non-infected control mice [51], [52]. Previous studies have shown that 1.0 mg of LPS induces the production of high IL-1β levels and lethality [52], [53]. In our model, challenge with 10 µg of LPS did not promoted or augmented caspase-1 activation in either uninfected or infected mice, respectively. Hence, Plasmodium infection is sufficient to induce inflammasome assembly and caspase-1 activation, but requires a challenge with a low dose of LPS to induce expression of pro-IL-1β and release of high levels of its active form.
Our studies in the P. chabaudi mouse model demonstrate that expression of inflammasome genes; pro-caspase-1 as well as pro-IL-1β are all dependent on intact Myd88 function. The MyD88 role on the process is independent of the IL-1 receptor signaling, as macrophages and DCs from P. chabaudi-infected IL-1R−/− mice show normal levels of caspase-1 activation and pyroptosis. In fact, we found that TLR9 explains, in part, the role of MyD88 on caspase-1 activation. As previously reported [54], treatment with CpG oligonucleotides mimics the P. chabaudi infection making the mice more susceptible to septic shock. Curiously, challenge of infected mice with CpG oligonucleotides did not result in lethality. As CpG DNA and LPS preferentially target DCs and monocyte/macrophages, respectively [55], our data suggest that the nature of the ligand and differential expression of TLRs are important determinants on priming and cytokine production by Plasmodium infected mice challenged with LPS.
We hypothesize that by targeting TLR9 on DCs, Plasmodium infection elicits the IL-12 production, and consequent IFN-γ-dependent inflammatory priming [23]. Priming with IFN-γ mediates caspase-1 activation through the induction of pro-caspase-1 expression, and the lack of either IFN-γ or TNFR1 results in impaired expression of pro- IL-1β. Hence, LPS, but not CpG, stimulates the production of high levels of TNF-α and pro-IL-1β culminating on the release of deleterious amounts of active IL-1β by IFN-γ-primed monocytes/macrophages. An alternative explanation is that by inducing Type I IFN production by DCs, Plasmodium infection inhibits the excessive production of IL-1β [56], [57], and prevents lethality induced by TLR9 activation.
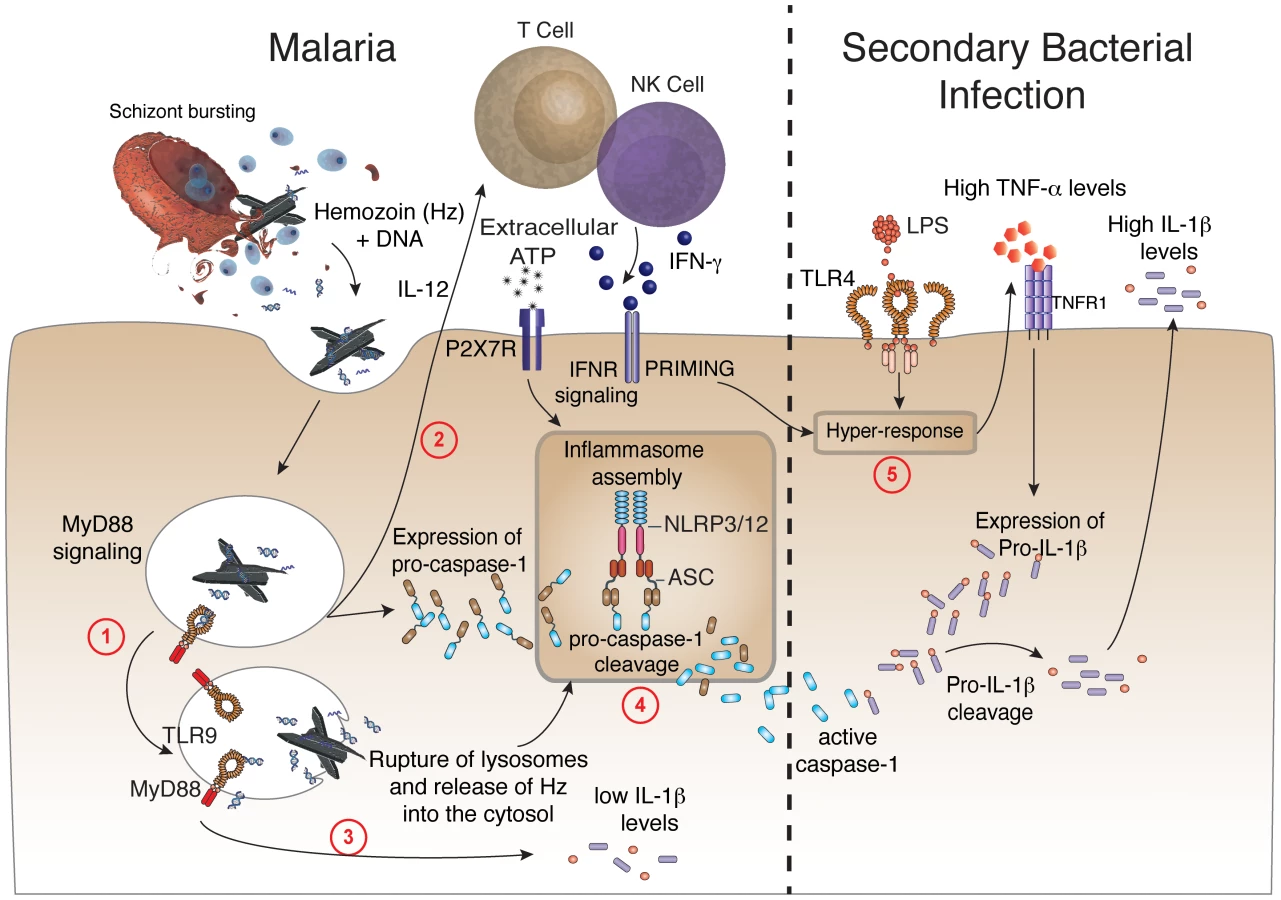
Another important requirement for caspase-1 activation in rodent malaria was the purinergic P2X7 receptor that under certain conditions mediates NLRP3-inflammasome activation [32], [58]. Indeed, in vitro experiments reported that synthetic hemozoin induces inflammasome formation, activation of caspase-1 and release of IL-1β by macrophages via NLRP3 [59]–[62]. Here, we demonstrated an in vivo requirement of both NLRP3 and NLRP12 for inflammasome formation and caspase-1 activation during in vivo infection with P. chabaudi. We also report that symptomatic infection with either P. falciparum or P. vivax leads to enhanced expression of inflammasome related genes and caspase-1 activation. Notably, we notice an in vivo assembly of NLRP3 and NLRP12 specks and ASC oligomerization in febrile malaria patients. A diagram detailing the different steps required for inflammasome assembly during malaria sepsis, and release of copious amounts of IL-1β during bacterial superinfection is presented in Figure 7.
It is noteworthy that periodic fever is a main symptom of cryopyrinopathies, a human inflammatory disorder that is associated with mutations in both NLRP3 and NLRP12 genes [21]. These patients have less severe symptoms when treated with IL-1R antagonist [63]. However, the relevance of this process during in vivo Plasmodium infection is controversial. While Dostert and colleagues [59] demonstrated a partial protection to experimental cerebral malaria in NLRP3−/− mice, other studies reported that this pathological process occurs independent of NLRP3, ASC, Caspase-1, IL-1R, IL-1β, and IL-18 [62], [64]. Furthermore, a study in the P. chabaudi model shows that caspase-12 (but not caspase-1), modulates cytokine responses and development of acquired immunity [65]. Thus, our results indicate that bacterial superinfection overcome the regulatory role of caspase 12.
Our results demonstrate that P. chabaudi infection triggers low levels of IL-1β release in an inflammasome-independent manner. Nevertheless, the parasite-induced NLRP3 and NLRP12 inflammasomes play a key role in the release of high IL-1β levels and hypersensitivity to LPS during malaria sepsis. Importantly, the lethality induced by low dose LPS, peroral infection with S. typhimurium, or sublethal CLP in P. chabaudi infected mice was prevented or delayed by treatment with IL-1R antagonist.
Relevant to our findings is the recently published study demonstrating that malaria impairs host resistance to Salmonella infection [11]. They propose that induction of heme oxygenase −1 (HO-1) by Plasmodium infection limits the generation of reactive oxygen species (ROS), an important mechanism of host resistance to Salmonella infection [66]. Another consequence of the decreased ROS generation during malaria would be the uncontrolled activation of caspase-1 and release copious amounts of active IL-1β by phagocytes, as previously reported in patients with chronic granulomatous disease (CGD) [67]–[69].
Similarly to murine malaria, extremely high levels of IL-1β are produced by PBMCs from P. vivax or P. falciparum malaria patients exposed to bacterial components [23], [47]. Importantly, the bacterial load in mice undergoing malaria sepsis was not different from mice treated with IL-1RA, suggesting that the acute lethality caused by bacterial superinfection is due to the deleterious inflammatory response. Overall, our data argue that the IFN-γ, TNF-α and MyD88 role on hypersensitivity to septic shock during malaria is, at least in part, mediated by inflammasome-dependent release of IL-1β.
In conclusion, years of research on malaria pathogenesis have funneled into the consensus that the clinical manifestations are often a result of the excessive activation of the innate immune cells. Recent reports have emphasized the important role of bacterial infections as co-factors for severe disease. Here we report that Plasmodium-induced NLRP3, NLRP12, ASC containing inflammasomes and caspase-1 activation, which are important events for the overwhelming IL-1β response and morbidity observed in bacterial superinfection during malaria sepsis.
Materials and Methods
Ethics statement
The study with Plasmodium infected patients and healthy controls was approved by the Ethical Committee of Research (CEP) from the Research Center of Tropical Medicine (CEP-CEPEM 096/09); the Brazilian National Committee of Research (CONEP/Ministry of Health – 15653); as well as the Institutional Research Board from the University of Massachusetts Medical School (UMMS) (IRB-ID11116_1). Informed written consent was obtained before enrollment of all subjects (Plasmodium infected patients and healthy control). All experiments involving animals were in accordance with guidelines set forth by the American Association for Laboratory Animal Science (AALAS) and with the recommendations in the Guide for the Care and Use of Laboratory Animals of the Brazilian National Council of Animal Experimentation (http://www.cobea.org.br/) and the Federal Law 11.794 (October 8, 2008). All protocols developed for this work were approved by the Institutional Animal Care and Use Committee (IACUC) at the UMMS (ID - 2371-12), (ID - 1369-11) and also were approved by the Council of Animal Experimentation of Oswaldo Cruz Foundation (CEUA protocol 38/10-3).
Reagents
LPS O55:B55 from E. coli, nigericin and RIPA buffer were obtained from SIGMA. All mAbs used in flow cytometry, as well as 7AAD and PI were purchased from Ebiosciences. Active caspase-1 detection kit was obtained from ImmunoChemistry Technologies, LLC (catalog no. 98). Human (SC-515) and mouse (SC-514) anti-caspase-1 (p10), ASC (SC-22514-R and SC-271054) primary antibodies were obtained from Santa Cruz Biotechnology and anti-actin (A2066) was purchased from SIGMA. Anti-caspase-1 (p20) was donated by Genentech. Anti-NLRP3 (ab4207 and ab17267), -NLRP12 (ab64928 and ab57906), -NLRC4 (ab99860), secondary Anti-rabbit IgG Texas Red (ab6800), secondary anti-goat IgG Texas Red (ab6883), secondary anti-goat IgG FITC (ab6881), and secondary anti-mouse IgG FITC (ab7057) were purchased from Abcam. The secondary antibodies used in western blots were purchased from KPL. Ficoll-Paque from GE Healthcare, and the crosslinker disuccinimidyl suberate [70] was obtained from Thermo Scientific. Protease inhibitor (EDTA free) was purchased from Roche and RPMI and DMEM from Gibco. Cytokine ELISA kits and Cytometric Bead Array were obtained from R&D Systems and BD Biosciences, respectively. Flow cytometry mAbs for mouse were from BD Biosciences: CD11c-PE (cat-557401), CD11b-PE (cat-557397), CD3-FITC (cat-553062), B220-APC (cat-561880), and from Ebioscience: MHC-II-PercpCy5 (cat-15-5321-82), and F4/80-APC (cat-17-4801-82), CD4-PE (cat-12-0041), CD8-PE (cat-12-008-81), and NKG2d-PE (cat-12-5872). Flow cytometry mAbs for human were from BD Biosciences: CD16-FITC (cat-555969), CD14-APC (cat-561708), CD19-FITC (cat-555412); and from Ebioscience: CD3-FITC (cat-55332), CD4-PE-Cy5 (cat-555348), CD8-PE-Cy5 (cat-555368), CD1c-APC (cat-17-0015), CD123-PercpCy5.5 (cat-45-1239), CD303-FITC (cat-11-9818-42), CD56-PE (cat-9012-0567).
Malaria patients
Patients with acute febrile malaria were seen in the outpatient malaria clinic in the Tropical Medicine Research Center in Porto Velho, Brazil. Patients infected with P. falciparum received a fixed dose of the artemeter (20 mg) and lumefetrine (120 mg) combination four times a day for three days. Patients infected with P. vivax were treated with chloroquine (150 mg) every 8 hours for three days and primaquine (15 mg) in a single dose per day for two weeks. Up to 100 cc of blood was obtained immediately after confirmation and differentiation of Plasmodium infection by a standard peripheral smear; and 30–40 days after therapy with confirmed parasitological cure by PCR. Non-infected subjects living in Porto Velho were also included in the study.
Mice and parasite
The knockout mice, ASC−/−, NLRP3−/−, and NLRP12−/− mice were generated by Millennium Pharmaceuticals and backcrossed 8–11 generations to C57BL/6 background. MyD88−/− and NLRC4−/− mice were provided by S. Akira and R. Flavel. AIM2−/− mice were provided by K. Fitzgerald. C57BL/6, IL-1R−/−, TNFR1−/−, P2X7R−/− and IFN-γ−/− mice were purchased from Jackson Laboratories. The caspase-1 knockout mice used in this work was originally provided by Dr. Flavell from Yale University School of Medicine. All mice used in experiments were 8–12 weeks of age and bred in isolated conditions in the animal house at CPqRR or at UMMS Animal Facility.
The Plasmodium chabaudi chabaudi AS strain was used for experimental infections. This strain was kept in our laboratory as described elsewhere [71]. Briefly, P. chabaudi strain was maintained in C57BL/6 mice by passages once a week. For experimental infection mice were injected i.p. with 105 infected red blood cells and parasitemia followed every three days. Although animals exhibit signs of disease, lethal infection is uncommon. The course of parasitemia in WT mice was similar to that reported in other studies [23], [72], .
Cecal ligation puncture (CLP) and S. typhimurium infection
For sub-lethal sepsis induced by CLP, mice were anesthetized, incision made on the anterior abdomen, cecal ligated and punctured two times with a 22-gauge needle. Bacterial load in exudates from the peritoneal cavity and blood 24 hours after CLP was evaluated on Mueller-Hinton agar dishes [74]. For Salmonella infections mice were inoculated intragastrically with Salmonella enterica serovar Typhimurium (ATCC 14028) (108 cfu). Three days after infection, mice were euthanized, liver aseptically collected, weighed, and homogenized in sterile PBS (1∶10, w/v). One hundred µl aliquots of serial decimal dilutions of liver homogenates and blood were plated onto MacConkey agar [75].
PBMC and monocytes
PBMCs were isolated from whole blood on Ficoll-paque Plus (GE Healthcare). Cells were then plated into 96-well cell culture plates at a density of 2×105 in DMEM containing 10% FCS and 10 µg/ml ciprofloxacin. Supernatants were collected 24 hours after stimulation and used to determine the levels IL-1β. Monocytes were purified from PBMCs of P. vivax infected patients and healthy donors by using a kit based on immunomagnetic negative selection from StemCell Technologies (catalog number 19058).
FACS analysis for caspase-1 activation, membrane damage, and change in cell size
PBMCs from acutely infected patients were stained with combinations of the following mAbs: monocytes (CD14/CD16), T lymphocytes (CD3+/CD4+ or CD3+/CD8+), B lymphocytes (CD19), myeloid DCs (CD1c+/CD19−), plasmacytoid DCs (CD123+/CD303+) and NK cells (CD3−/CD56+). Splenocytes from infected mice were stained with combinations of the following mAbs: macrophages (CD11b+/F4/80+), DCs (CD11c+/MHC-II+), T lymphocytes (CD3+/CD4+ or CD3+/CD8+), B lymphocytes (B220+) and NK cells (NKG2d+). To each sample, FLICA reagent and PI (or 7AAD) were added as indicated. The data were acquired using a LSRII cytometer.
Immunoblots for caspase-1 and pro-IL-1β
Ripa buffer (250 µl) plus protease inhibitor cocktail from Roche were added to a pellet containing 4×107 splenocytes or PBMCs. After 15 minutes on ice, lysates were centrifuged at 13,000 g for 20 min at 4°C. The supernatants were separated in a 15%-acrylamide SDS-PAGE, transferred onto nitrocellulose membranes. The membranes were incubated with caspase-1 or pro-IL-1β specific antibodies, and then revealed with HRP-conjugated antibody and the ECL system from Amersham (Bucks, UK).
Crosslinking ASC in PBMCs from malaria patients
PBMCs were resuspended in a hypotonic solution (10 mM Hepes - pH 7.9, 1.5 mM MgCl2, 10 mM KCl, 0.2 mM PMSF, 0.5 mM DTT, protease inhibitor cocktail Roche), incubated on ice for 15 minutes, homogenized (Kontes 22 mm) and centrifuged for 8 minutes at 10,000 g. The pellets were resuspended in 500 µl of CHAPs buffer (20 mM HEPES-KOH - pH 7.5, 5 mM MgCl2, 0.5 mM EGTA, 0.1% CHAPs, 0.1 mM PMSF, and protease inhibitor cocktail from Roche) and centrifuged for 8 minutes at 10,000 g. Finally, the pellets were resuspended in 200 µl of CHAPs buffer, 4 µl of a 100 mM DSS stock solution to a final concentration of 2 mM, and incubated for 30 min in the dark. The oligomers were resolved on a 12% SDS-PAGE and visualized by immunoblotting with an anti-ASC antibody (SC-22514-R).
Confocal analysis
Cells were fixed with paraformaldehyde 4%, permeabilized using Triton X-100 and stained with anti-NLRP3 (FITC), anti-NLRP12 (Texas Red), NLRC4 (Texas Red) and anti-AIM2 (Texas Red). Images were acquired using a Zeiss LSM510 Microscope and analyzed by ImageJ software. Dual color images were acquired by consecutive scanning with only one laser line active per scan to avoid cross-excitation.
Cytokine measurements
Measurements of mouse cytokines were performed using commercially available ELISA Duoset kits from R&D Systems. The ranges of detection are 15.6–1000 pg/ml for IL-1β; and 31.2–2000 pg/ml for TNF-α and IFN-γ. Human IL-1β was detected by ELISA kit from Ebioscience in a range of 4 to 500 pg/ml.
Microarray experiments and data analysis
For mouse experiments we used splenocytes from 3 C57BL/6 and 3 MyD88−/− mice at 6 days p.i. with P. chabaudi or uninfected. Gene expression was accessed by microarray analysis using a gene chip from Affymetrix (∼23,000 transcripts). Genes were clustered by Tiger Multi Experiment Viewer software using the fold increase value obtained by the reason of the signal intensity values from infected vs. non-infected mice. Differences in gene expression between the 2 conditions were considered significant if p<0.05 as defined by unpaired t test. Detailed methodological and analysis for human microarrays are presented in Table S1. Expression Omnibus (http://www.ncbi.nlm.nih.gov/gds/) accession numbers are GSE35083 for mouse and GSE15221 for human microarrays.
Statistical analysis
All data were analyzed using Graphpad Prism 5.0 Software. Cytokine measurements from human PBMCs were analyzed using two-tailed student's t test. Mann-Whitney testing was used for non-parametric analysis when data did not fit a Gaussian distribution. A p value≤0.05 was considered to be statistically significant.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. MurrayCJ, RosenfeldLC, LimSS, AndrewsKG, ForemanKJ, et al. (2012) Global malaria mortality between 1980 and 2010: a systematic analysis. Lancet 379: 413–431.
2. MuellerI, GalinskiMR, BairdJK, CarltonJM, KocharDK, et al. (2009) Key gaps in the knowledge of Plasmodium vivax, a neglected human malaria parasite. Lancet Infect Dis 9: 555–566.
3. MillerLH, AckermanHC, SuXZ, WellemsTE (2013) Malaria biology and disease pathogenesis: insights for new treatments. Nat Med 19: 156–167.
4. ParrocheP, LauwFN, GoutagnyN, LatzE, MonksBG, et al. (2007) Malaria hemozoin is immunologically inert but radically enhances innate responses by presenting malaria DNA to Toll-like receptor 9. Proc Natl Acad Sci U S A 104: 1919–1924.
5. SharmaS, DeOliveiraRB, KalantariP, ParrocheP, GoutagnyN, et al. (2011) Innate immune recognition of an AT-rich stem-loop DNA motif in the Plasmodium falciparum genome. Immunity 35: 194–207.
6. KrishnegowdaG, HajjarAM, ZhuJ, DouglassEJ, UematsuS, et al. (2005) Induction of proinflammatory responses in macrophages by the glycosylphosphatidylinositols of Plasmodium falciparum: cell signaling receptors, glycosylphosphatidylinositol (GPI) structural requirement, and regulation of GPI activity. J Biol Chem 280: 8606–8616.
7. O'NeillLA, GolenbockD, BowieAG (2013) The history of Toll-like receptors - redefining innate immunity. Nat Rev Immunol 13: 453–460.
8. VincentJL, OpalSM, MarshallJC, TraceyKJ (2013) Sepsis definitions: time for change. Lancet 381: 774–775.
9. WereT, DavenportGC, HittnerJB, OumaC, VululeJM, et al. (2011) Bacteremia in Kenyan children presenting with malaria. J Clin Microbiol 49: 671–676.
10. LacerdaMV, FragosoSC, AlecrimMG, AlexandreMA, MagalhaesBM, et al. (2012) Postmortem characterization of patients with clinical diagnosis of Plasmodium vivax malaria: to what extent does this parasite kill? Clin Infect Dis 55: e67–74.
11. CunningtonAJ, de SouzaJB, WaltherM, RileyEM (2012) Malaria impairs resistance to Salmonella through heme- and heme oxygenase-dependent dysfunctional granulocyte mobilization. Nat Med 18: 120–127.
12. ScottJA, BerkleyJA, MwangiI, OcholaL, UyogaS, et al. (2011) Relation between falciparum malaria and bacteraemia in Kenyan children: a population-based, case-control study and a longitudinal study. Lancet 378: 1316–1323.
13. SchroderK, TschoppJ (2010) The inflammasomes. Cell 140: 821–832.
14. DavisBK, WenH, TingJP (2011) The inflammasome NLRs in immunity, inflammation, and associated diseases. Annu Rev Immunol 29: 707–735.
15. HornungV, BauernfeindF, HalleA, SamstadEO, KonoH, et al. (2008) Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat Immunol 9: 847–856.
16. KannegantiTD, OzorenN, Body-MalapelM, AmerA, ParkJH, et al. (2006) Bacterial RNA and small antiviral compounds activate caspase-1 through cryopyrin/Nalp3. Nature 440: 233–236.
17. BrozP, NewtonK, LamkanfiM, MariathasanS, DixitVM, et al. (2010) Redundant roles for inflammasome receptors NLRP3 and NLRC4 in host defense against Salmonella. J Exp Med 207: 1745–1755.
18. WangL, ManjiGA, GrenierJM, Al-GarawiA, MerriamS, et al. (2002) PYPAF7, a novel PYRIN-containing Apaf1-like protein that regulates activation of NF-kappa B and caspase-1-dependent cytokine processing. J Biol Chem 277: 29874–29880.
19. WilliamsKL, LichJD, DuncanJA, ReedW, RallabhandiP, et al. (2005) The CATERPILLER protein monarch-1 is an antagonist of toll-like receptor-, tumor necrosis factor alpha-, and Mycobacterium tuberculosis-induced pro-inflammatory signals. J Biol Chem 280: 39914–39924.
20. LichJD, WilliamsKL, MooreCB, ArthurJC, DavisBK, et al. (2007) Monarch-1 suppresses non-canonical NF-kappaB activation and p52-dependent chemokine expression in monocytes. J Immunol 178: 1256–1260.
21. JeruI, Le BorgneG, CochetE, HayrapetyanH, DuquesnoyP, et al. (2011) Identification and functional consequences of a recurrent NLRP12 missense mutation in periodic fever syndromes. Arthritis Rheum 63: 1459–1464.
22. VladimerGI, WengD, PaquetteSW, VanajaSK, RathinamVA, et al. (2012) The NLRP12 inflammasome recognizes Yersinia pestis. Immunity 37: 96–107.
23. FranklinBS, ParrocheP, AtaideMA, LauwF, RopertC, et al. (2009) Malaria primes the innate immune response due to interferon-gamma induced enhancement of toll-like receptor expression and function. Proc Natl Acad Sci U S A 106: 5789–5794.
24. MockenhauptFP, CramerJP, HamannL, StegemannMS, EckertJ, et al. (2006) Toll-like receptor (TLR) polymorphisms in African children: Common TLR-4 variants predispose to severe malaria. Proc Natl Acad Sci U S A 103: 177–182.
25. KhorCC, ChapmanSJ, VannbergFO, DunneA, MurphyC, et al. (2007) A Mal functional variant is associated with protection against invasive pneumococcal disease, bacteremia, malaria and tuberculosis. Nat Genet 39: 523–528.
26. FranklinBS, IshizakaST, LamphierM, GusovskyF, HansenH, et al. (2011) Therapeutical targeting of nucleic acid-sensing Toll-like receptors prevents experimental cerebral malaria. Proc Natl Acad Sci U S A 108: 3689–3694.
27. GrauGE, FajardoLF, PiguetPF, AlletB, LambertPH, et al. (1987) Tumor necrosis factor (cachectin) as an essential mediator in murine cerebral malaria. Science 237: 1210–1212.
28. KarunaweeraND, CarterR, GrauGE, KwiatkowskiD, Del GiudiceG, et al. (1992) Tumour necrosis factor-dependent parasite-killing effects during paroxysms in non-immune Plasmodium vivax malaria patients. Clin Exp Immunol 88: 499–505.
29. BauernfeindFG, HorvathG, StutzA, AlnemriES, MacDonaldK, et al. (2009) Cutting edge: NF-kappaB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J Immunol 183: 787–791.
30. DinarelloCA, CannonJG, WolffSM, BernheimHA, BeutlerB, et al. (1986) Tumor necrosis factor (cachectin) is an endogenous pyrogen and induces production of interleukin 1. J Exp Med 163: 1433–1450.
31. JarvisMF, KhakhBS (2009) ATP-gated P2X cation-channels. Neuropharmacology 56: 208–215.
32. FranchiL, KannegantiTD, DubyakGR, NunezG (2007) Differential requirement of P2X7 receptor and intracellular K+ for caspase-1 activation induced by intracellular and extracellular bacteria. J Biol Chem 282: 18810–18818.
33. PetrilliV, PapinS, DostertC, MayorA, MartinonF, et al. (2007) Activation of the NALP3 inflammasome is triggered by low intracellular potassium concentration. Cell Death Differ 14: 1583–1589.
34. NeteaMG, Nold-PetryCA, NoldMF, JoostenLA, OpitzB, et al. (2009) Differential requirement for the activation of the inflammasome for processing and release of IL-1beta in monocytes and macrophages. Blood 113: 2324–2335.
35. AnsteyNM, DouglasNM, PoespoprodjoJR, PriceRN (2012) Plasmodium vivax: clinical spectrum, risk factors and pathogenesis. Adv Parasitol 80: 151–201.
36. DinarelloCA (2009) Immunological and inflammatory functions of the interleukin-1 family. Annu Rev Immunol 27: 519–550.
37. Wagner-JaureggJ, BruetschWL (1946) The history of the malaria treatment of general paralysis. Am J Psychiatry 102: 577–582.
38. AdachiK, TsutsuiH, KashiwamuraS, SekiE, NakanoH, et al. (2001) Plasmodium berghei infection in mice induces liver injury by an IL-12- and toll-like receptor/myeloid differentiation factor 88-dependent mechanism. J Immunol 167: 5928–5934.
39. TogbeD, SchofieldL, GrauGE, SchnyderB, BoissayV, et al. (2007) Murine cerebral malaria development is independent of toll-like receptor signaling. Am J Pathol 170: 1640–1648.
40. LepeniesB, CramerJP, BurchardGD, WagnerH, KirschningCJ, et al. (2007) Induction of experimental cerebral malaria is independent of TLR2/4/9. Med Microbiol Immunol
41. CobanC, IshiiKJ, UematsuS, ArisueN, SatoS, et al. (2007) Pathological role of Toll-like receptor signaling in cerebral malaria. Int Immunol 19: 67–79.
42. WuX, GowdaNM, KumarS, GowdaDC (2010) Protein-DNA complex is the exclusive malaria parasite component that activates dendritic cells and triggers innate immune responses. J Immunol 184: 4338–4348.
43. LeorattiFM, FariasL, AlvesFP, Suarez-MutisMC, CouraJR, et al. (2008) Variants in the toll-like receptor signaling pathway and clinical outcomes of malaria. J Infect Dis 198: 772–780.
44. Sam-AguduNA, GreeneJA, OpokaRO, KazuraJW, BoivinMJ, et al. (2010) TLR9 polymorphisms are associated with altered IFN-gamma levels in children with cerebral malaria. Am J Trop Med Hyg 82: 548–555.
45. McCallMB, NeteaMG, HermsenCC, JansenT, JacobsL, et al. (2007) Plasmodium falciparum infection causes proinflammatory priming of human TLR responses. J Immunol 179: 162–171.
46. HartgersFC, ObengBB, VoskampA, LarbiIA, AmoahAS, et al. (2008) Enhanced Toll-like receptor responsiveness associated with mitogen-activated protein kinase activation in Plasmodium falciparum-infected children. Infect Immun 76: 5149–5157.
47. LeorattiFM, TrevelinSC, CunhaFQ, RochaBC, CostaPA, et al. (2012) Neutrophil paralysis in Plasmodium vivax malaria. PLoS Negl Trop Dis 6: e1710.
48. MagalhaesBM, AlexandreMA, SiqueiraAM, MeloGC, GimaqueJB, et al. (2012) Clinical profile of concurrent dengue fever and Plasmodium vivax malaria in the Brazilian Amazon: case series of 11 hospitalized patients. Am J Trop Med Hyg 87: 1119–1124.
49. JakobsenPH, McKayV, Morris-JonesSD, McGuireW, van HensbroekMB, et al. (1994) Increased concentrations of interleukin-6 and interleukin-1 receptor antagonist and decreased concentrations of beta-2-glycoprotein I in Gambian children with cerebral malaria. Infect Immun 62: 4374–4379.
50. ArmahHB, WilsonNO, SarfoBY, PowellMD, BondVC, et al. (2007) Cerebrospinal fluid and serum biomarkers of cerebral malaria mortality in Ghanaian children. Malar J 6: 147.
51. KayagakiN, WongMT, StoweIB, RamaniSR, GonzalezLC, et al. (2013) Noncanonical Inflammasome Activation by Intracellular LPS Independent of TLR4. Science 341: 1246–1249 DOI: 10.1126/science.1240248
52. KayagakiN, WarmingS, LamkanfiM, Vande WalleL, LouieS, et al. (2011) Non-canonical inflammasome activation targets caspase-11. Nature 479: 117–121.
53. KawaiT, AdachiO, OgawaT, TakedaK, AkiraS (1999) Unresponsiveness of MyD88-deficient mice to endotoxin. Immunity 11: 115–122.
54. CowderyJS, ChaceJH, YiAK, KriegAM (1996) Bacterial DNA induces NK cells to produce IFN-gamma in vivo and increases the toxicity of lipopolysaccharides. J Immunol 156: 4570–4575.
55. KaishoT (2012) Pathogen sensors and chemokine receptors in dendritic cell subsets. Vaccine 30: 7652–7657.
56. GuermonprezP, HelftJ, ClaserC, DeroubaixS, KaranjeH, et al. (2013) Inflammatory Flt3l is essential to mobilize dendritic cells and for T cell responses during Plasmodium infection. Nat Med 19: 730–738.
57. GuardaG, BraunM, StaehliF, TardivelA, MattmannC, et al. (2011) Type I interferon inhibits interleukin-1 production and inflammasome activation. Immunity 34: 213–223.
58. MariathasanS, WeissDS, NewtonK, McBrideJ, O'RourkeK, et al. (2006) Cryopyrin activates the inflammasome in response to toxins and ATP. Nature 440: 228–232.
59. DostertC, GuardaG, RomeroJF, MenuP, GrossO, et al. (2009) Malarial hemozoin is a Nalp3 inflammasome activating danger signal. PLoS One 4: e6510.
60. GriffithJW, SunT, McIntoshMT, BucalaR (2009) Pure Hemozoin is inflammatory in vivo and activates the NALP3 inflammasome via release of uric acid. J Immunol 183: 5208–5220.
61. ShioMT, EisenbarthSC, SavariaM, VinetAF, BellemareMJ, et al. (2009) Malarial hemozoin activates the NLRP3 inflammasome through Lyn and Syk kinases. PLoS Pathog 5: e1000559.
62. ReimerT, ShawMH, FranchiL, CobanC, IshiiKJ, et al. (2010) Experimental cerebral malaria progresses independently of the Nlrp3 inflammasome. Eur J Immunol 40: 764–769.
63. JeruI, HentgenV, NormandS, DuquesnoyP, CochetE, et al. (2011) Role of interleukin-1beta in NLRP12-associated autoinflammatory disorders and resistance to anti-interleukin-1 therapy. Arthritis Rheum 63: 2142–2148.
64. KordesM, MatuschewskiK, HafallaJC (2011) Caspase-1 activation of interleukin-1beta (IL-1beta) and IL-18 is dispensable for induction of experimental cerebral malaria. Infect Immun 79: 3633–3641.
65. LabbeK, MiuJ, YeretssianG, SerghidesL, TamM, et al. (2010) Caspase-12 dampens the immune response to malaria independently of the inflammasome by targeting NF-kappaB signaling. J Immunol 185: 5495–5502.
66. MastroeniP, Vazquez-TorresA, FangFC, XuY, KhanS, et al. (2000) Antimicrobial actions of the NADPH phagocyte oxidase and inducible nitric oxide synthase in experimental salmonellosis. II. Effects on microbial proliferation and host survival in vivo. J Exp Med 192: 237–248.
67. MeissnerF, SegerRA, MoshousD, FischerA, ReichenbachJ, et al. (2010) Inflammasome activation in NADPH oxidase defective mononuclear phagocytes from patients with chronic granulomatous disease. Blood 116: 1570–1573.
68. van BruggenR, KokerMY, JansenM, van HoudtM, RoosD, et al. (2010) Human NLRP3 inflammasome activation is Nox1-4 independent. Blood 115: 5398–5400.
69. van de VeerdonkFL, SmeekensSP, JoostenLA, KullbergBJ, DinarelloCA, et al. (2010) Reactive oxygen species-independent activation of the IL-1beta inflammasome in cells from patients with chronic granulomatous disease. Proc Natl Acad Sci U S A 107: 3030–3033.
70. MouldsJM, BraiM, CohenJ, CortelazzoA, CucciaM, et al. (1998) Reference typing report for complement receptor 1 (CR1). Exp Clin Immunogenet 15: 291–294.
71. StevensonMM, TamMF, BelosevicM, van der MeidePH, PodobaJE (1990) Role of endogenous gamma interferon in host response to infection with blood-stage Plasmodium chabaudi AS. Infect Immun 58: 3225–3232.
72. CadmanET, AbdallahAY, VoisineC, SponaasAM, CorranP, et al. (2008) Alterations of splenic architecture in malaria are induced independently of Toll-like receptors 2, 4, and 9 or MyD88 and may affect antibody affinity. Infect Immun 76: 3924–3931.
73. OmerFM, RileyEM (1998) Transforming growth factor beta production is inversely correlated with severity of murine malaria infection. J Exp Med 188: 39–48.
74. RittirschD, Huber-LangMS, FlierlMA, WardPA (2009) Immunodesign of experimental sepsis by cecal ligation and puncture. Nat Protoc 4: 31–36.
75. MartinsFS, DalmassoG, ArantesRM, DoyeA, LemichezE, et al. (2010) Interaction of Saccharomyces boulardii with Salmonella enterica serovar Typhimurium protects mice and modifies T84 cell response to the infection. PLoS One 5: e8925.
Štítky
Hygiena a epidemiológia Infekčné lekárstvo LaboratóriumČlánok vyšiel v časopise
PLOS Pathogens
2014 Číslo 1
- Parazitičtí červi v terapii Crohnovy choroby a dalších zánětlivých autoimunitních onemocnění
- Očkování proti virové hemoragické horečce Ebola experimentální vakcínou rVSVDG-ZEBOV-GP
- Koronavirus hýbe světem: Víte jak se chránit a jak postupovat v případě podezření?
Najčítanejšie v tomto čísle
- Lyme Disease: Call for a “Manhattan Project” to Combat the Epidemic
- Origin, Migration Routes and Worldwide Population Genetic Structure of the Wheat Yellow Rust Pathogen f.sp.
- IFNγ/IL-10 Co-producing Cells Dominate the CD4 Response to Malaria in Highly Exposed Children
- Human and Plant Fungal Pathogens: The Role of Secondary Metabolites