
Současná léčba myelofibrózy na základě rizikové stratifikace pacientů
Current treatment of myelofibrosis based on risk stratification of patients
Primary myelofibrosis belongs to the group of Ph1-negative myeloproliferative neoplasms, along with essential thrombocythemia and polycythemia vera, which may also progress to a fibrotic stage. Risk stratification of patients is especially important for proper therapeutic decision making and selection of high risk patients suitable for allogeneic stem cell transplantation, the only curative option for myelofibrosis. Transplantation carries a significant risk of mortality and morbidity, therefore it is recommended in patients with a median survival of less than 5 years. The recently introduced Dynamic International Prognostic Scoring System-plus prognostic model uses eight independent predictors of inferior survival. Janus 2 kinase inhibitors present a new targeted therapy affecting pathogenetic signal pathways. The results of on-going studies will show their long-term efficacy, safety and impact on overall survival.
Key words:
myelofibrosis, splenomegaly, JAK2V617F mutation, JAK2 inhibitors
Autori:
M. Palová; K. Indrák; A. Hluší; T. Szotkowski
Pôsobisko autorov:
Hemato-onkologická klinika, LF UP a FN Olomouc
Vyšlo v časopise:
Transfuze Hematol. dnes,19, 2013, No. 3, p. 163-170.
Kategória:
Souhrnné práce, původní práce, kazuistiky
Súhrn
Primární myelofibróza je řazena do skupiny chronických Ph1-negativních myeloproliferativních chorob spolu s esenciální trombocytemií a pravou polycytemií, které ve svém průběhu taktéž mohou přecházet do fibrotického stadia. Riziková stratifikace těchto pacientů má velký význam pro správné terapeutické rozhodnutí a pomáhá selektovat vysoce rizikové nemocné vhodné k alogenní transplantaci krvetvorných buněk, která i nadále zůstává jedinou kurativní léčebnou modalitou myelofibrózy. Transplantační přístup je pro významnou mortalitu a morbiditu doporučován u pacientů s pravděpodobným mediánem přežití kratším než 5 let. Recentně užívané dynamické prognostické skóre The Dynamic International Prognostic Scoring System-plus hodnotí osm nezávislých rizikových faktorů zhoršujících prognózu. Do terapie myelofibrózy je nově zaváděna cílená léčba zasahující patogenetické signální dráhy, jejímiž představiteli jsou inhibitory Janus 2 kinázy. Jejich účinnost, bezpečnost a vliv na celkové přežití z dlouhodobějšího hlediska ukáží až výsledky probíhajících studií.
Klíčová slova:
myelofibróza, splenomegalie, mutace JAK2V617F, JAK2 inhibitory
ÚVOD
Primární myelofibróza (PMF) je chronickým klonálním onemocněním krvetvorby, vyznačujícím se abnormální cytokinovou expresí, fibrózou kostní dřeně, progresivní anémií, splenomegalií, extramedulární hematopoézou a konstitučními symptomy (slabost, teploty, noční poty, hmotnostní úbytek). Spolu s esenciální trombocytemií a pravou polycytemií patří do skupiny chronických Ph1 (Philadephia chromosome) – negativních myeloproliferativních nemocí (1). V patogenezi těchto chorob hraje zásadní roli hyperaktivace genu pro Janus 2 kinázu (JAK2) podílející se na autonomní aktivaci kinázy, jež vede k perzistentní fosforylaci STAT proteinů (Signal transducer and activator of transcription) a následně k aktivaci transkripce genů regulujících buněčnou proliferaci. Pro diagnózu myelofibrózy je nutné splnění dvou velkých kritérií (fibróza kostní dřeně, průkaz mutace JAK2V617F či jiného markeru klonality, nebo v případě jejich negativity vyloučení reaktivních příčin fibrózy kostní dřeně) a jakýchkoliv dvou z šesti malých diagnostických kritérií (leukoerytroblastóza, anémie, hmatná splenomegalie, dakryocyty v nátěrech periferní krve, konstituční symptomy, histologicky verifikovaná extramedulární hematopoeza) (2).
Nejčastějšími příčinami smrti je progrese myelofibrózy do akutní leukemie nebo rychle progredující myeloidní proliferace a komplikace cytopenie (3), dále pak přetížení železem, kachektizace při extrémní splenomegalii, ruptura sleziny, úmrtí po splenektomii nebo po alogenní transplantaci.
RIZIKOVÁ STRATIFIKACE PACIENTŮ
IPSS (the International Prognostic Scoring System) vyvinutý pracovní skupinou pro výzkum a léčbu myelofibrózy (IWG-MRT International Working Group for Myelofibrosis Research and Treatment) v roce 2009 byl jedním z prvních prognostických modelů užívaných u pacientů s myelofibrózou. Je aplikovatelný v době diagnózy a hodnotí pět nezávislých prognostických faktorů: věk nad 65 let, hemoglobin (Hb) pod 100 g/l, počet leukocytů nad 25 x 109/l, počet cirkulujících blastů v periferní krvi ≥ 1 % – tabulka 1 (4). Předpokládaná doba přežití se pohybuje v rozmezí od 135 měsíců u nízce rizikových pacientů (bez rizikových faktorů), přes 95 měsíců u kategorie intermediate-1 (1 rizikový faktor), 48 měsíců ve skupině intermediate-2 (2 rizikové faktory) a 27 měsíců u vysoce rizikových pacientů (3 a více rizikových faktorů) (5).
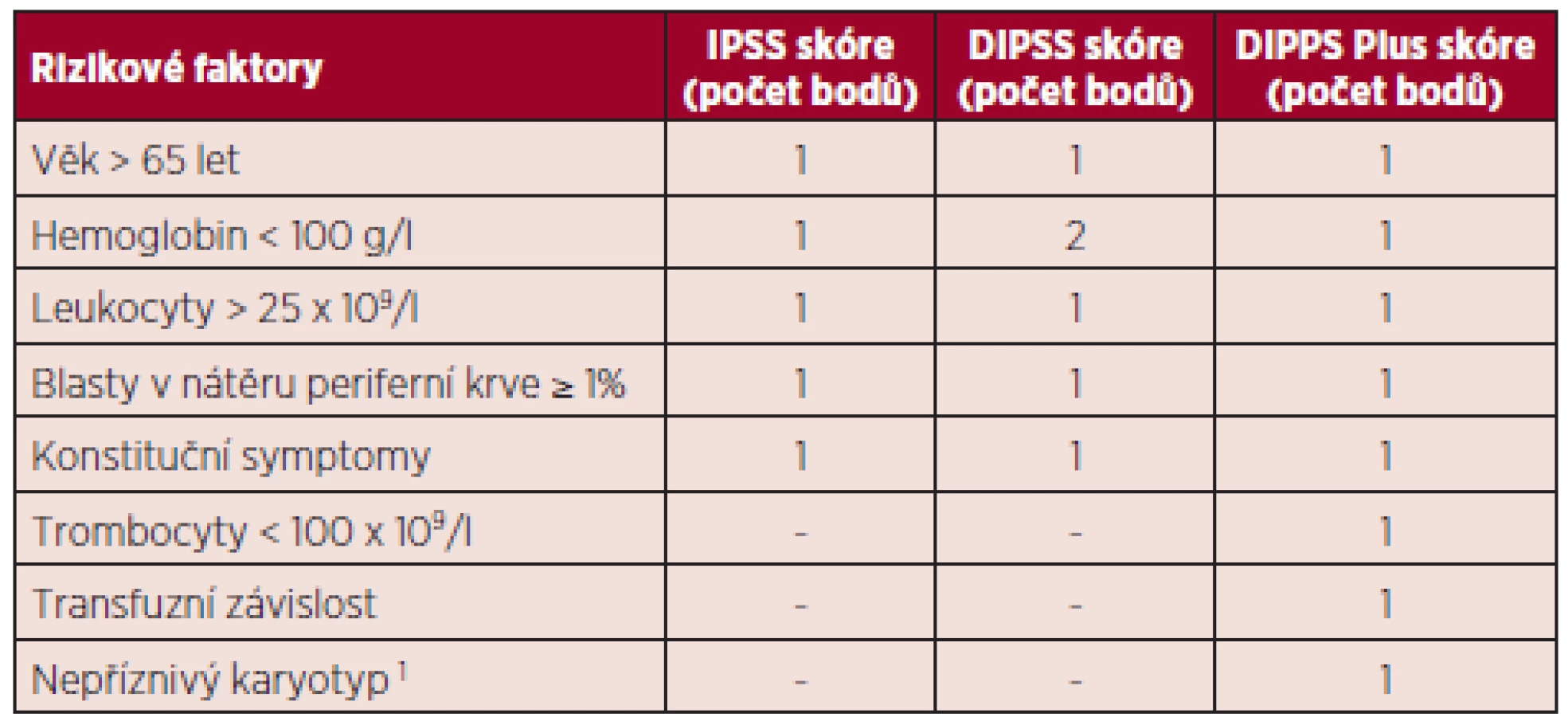
Následně, na základě výsledků studie s prodlouženým sledováním kohorty 525 pacientů s PMF, bylo vypracováno dynamické prognostické skóre DIPSS (the Dynamic International Prognostic Scoring System), jež hodnotí stejné faktory jako výše zmíněný model, ale reflektuje větší prognostický význam vzniklé anémie, která ovlivňuje přežití nemocných se zhruba dvojnásobným poměrným rizikem (hazard ratio) ve srovnání s dalšími parametry. Je navíc použitelný ke zhodnocení rizika choroby nejen v době diagnózy, ale kdykoliv v průběhu nemoci, proto dynamické prognostické skóre – viz tabulka 1 (6).
Recentně bylo DIPSS skóre modifikováno do DIPSS--plus skóre (the Dynamic International Prognostic Scoring System-plus) zahrnující osm nezávislých rizikových faktorů významně ovlivňujících horší přežití pacientů s primární myelofibrózou: věk nad 65 let, Hb pod 100 g/l, počet leukocytů nad 25 x 109/l, počet cirkulujících blastů v periferní krvi ≥ 1 %, transfuzní závislost, trombocytopenie pod 100 x 109/l a nepříznivý karyotyp (komplexní karyotyp, +8, -7/7q-, i(17q), inv(3), -5/5q-, 12p-, 11q23 přestavba) (7). Medián přežití je 185 měsíců u nízce rizikových pacientů (bez rizikového faktoru), 78 měsíců ve skupině intermediate-1 (1 rizikový faktor), 35 měsíců v kategorii intermediate-2 (2 až 3 rizikové faktory), 16 měsíců u pacientů s vysokým rizikem (4 a více rizikových faktorů) – tabulka 2 (5).
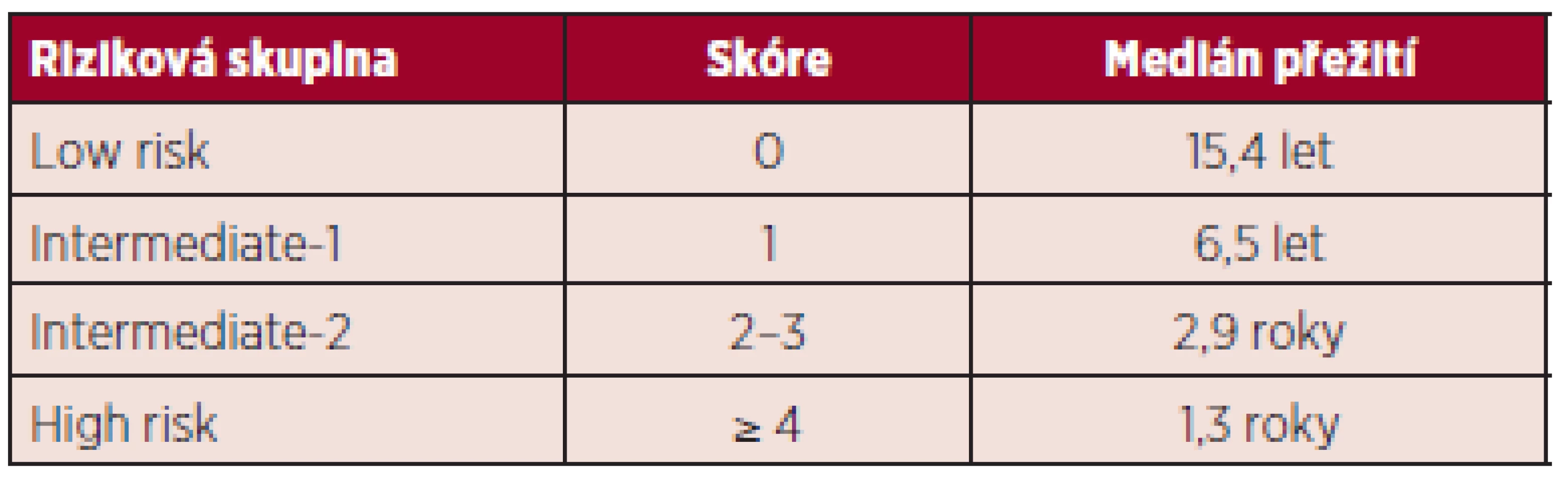
Mezi vysoce rizikovými pacienty dle DIPSS-plus skóre byla dále vyčleněna skupina nemocných „velmi vysokého“ rizika („very high risk“) se signifikantně kratším celkovým přežitím (p < 0,01) a s více než 80% mortalitou ve dvou letech predikovanou monosomálním karyotypem, inv(3)/i(17q) či přítomností dvou z následujících faktorů: počet cirkulujících blastů v periferní krvi nad 9 %, leukocytóza ≥ 40 x 109/l nebo jiný nepříznivý karyotyp. V těchto případech by mělo být rozhodnuto o eventuálním provedení alogenní transplantace krvetvorných buněk (ASCT) co nejčasněji (8). Nově byly identifikovány další přídatné faktory spojené s horším celkovým přežitím, a to mutace ASXL1, SRSF2, EZH2 a IDH1/2, zvýšená plazmatická hladina interleukinu 2 (IL-2), interleukinu 8 (IL-8) či volných lehkých řetězců séra, nicméně ještě bude nutná jejich další validace (1).
ALGORITMUS LÉČBY
V současné době máme stále omezené léčebné možnosti myelofibrózy. Jedinou potencionálně kurativní metodou zůstává alogenní transplantace kostní dřeně provázená poměrně vysokou mortalitou a morbiditou. Ostatní léčebné modality neprodlužují celkové přežití pacientů.
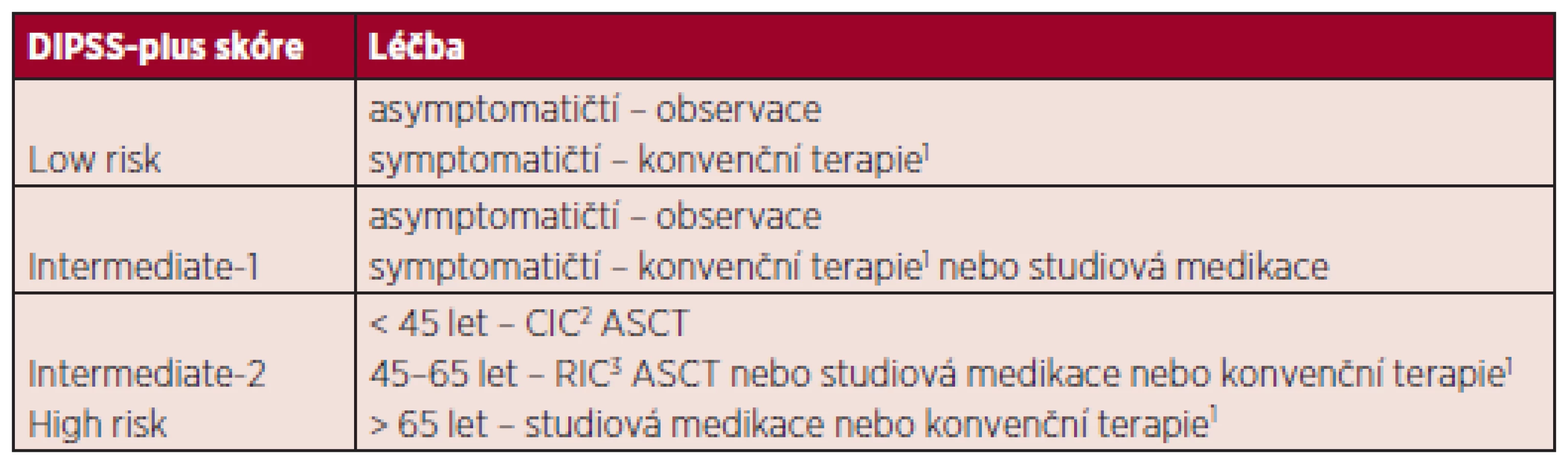
Léčba pacientů s nízkým rizikem a intermediate-1
Asymptomatičtí pacienti těchto rizikových skupin mohou být pouze sledováni bez nutnosti specifické terapeutické intervence. Jen symptomatičtí nemocní s anémií, splenomegalií, konstitučními symptomy nebo s extrémní leukocytózou či trombocytózou jsou indikováni ke konvenční terapii (viz dále) (9).
Léčba pacientů s vysokým rizikem a kategorie intermediate-2
U mladších pacientů (pod 65 let) spadajících do kategorie intermediate-2 či vysokého rizika dle DIPSS--plus skóre by měla být zvážena alogenní transplantace kostní dřeně či možnost zařazení do klinické studie. Zejména výše zmíněná skupina nemocných „velmi vysokého“ rizika s extrémně špatnou prognózou (monosomální karyotyp, inv(3), i(17q) nebo jakékoliv 2 faktory z následujících: cirkulující blasty v periferní krvi > 9 %, leukocytóza ≥ 40 x 109/l, další nepříznivý karyotyp, tj. komplexní karyotyp, +8, 12p-, 11q23 přestavba) je indikována k co nejčasnějšímu provedení alogenní transplantace kostní dřeně (1). Navzdory tomu, že alogenní transplantace představuje jedinou potencionálně kurativní metodu MF, je pro vysokou morbiditu a mortalitu nutný velmi pečlivý výběr pacientů. Publikované práce zahrnují značně heterogenní skupiny nemocných lišící se věkem, klinickým stavem, prognostickým rizikem, použitým přípravným režimem, typem dárce, imunosupresí po transplantaci. Analýzy výsledků největších publikovaných souborů naznačují, že peritransplantační mortalita se pohybuje od 10 % do více než 40 %, celkové přežití ve 3 letech je od 30 % do 50 % (5). Jedna z největších studií s 289 transplantovanými pacienty s primární myelofibrózou ze 118 center uvádí s transplantací spojenou mortalitu 18% u příbuzenských a 30% u nepříbuzenských transplantací, 5leté celkové přežití 37 % u transplantací od HLA-identického příbuzného a 30 % od HLA-identického nepříbuzného dárce, disease--free survival v 5 letech 33 % pro příbuzenské a 27 % pro nepříbuzenské transplantace. Jako pozitivní prediktory přežití byly v této práci identifikovány Karnofsky skóre nad 90 %, HLA-identický příbuzný dárce, chybění blastů v periferní krvi. U šedesáti pacientů, kteří dostali přípravný režim s redukovanou intenzitou (RIC), byla s transplantací spojená mortalita 15 %, tedy nižší než u myeloablativních režimů při srovnatelném celkovém přežití (10). Stewart et al. publikovali data 51 transplantovaných pacientů s myelofibrózou, z nichž 27 bylo léčeno myeloablativním režimem a 24 režimem s redukovanou intenzitou. Mezi oběma skupinami nebyl zjištěn signifikantní rozdíl v celkovém přežití či v přežití bez progrese (11). Užití přípravných režimů s redukovanou intenzitou snížilo vysokou mortalitu spojenou s transplantací a umožnilo indikovat i pacienty pokročilejšího věku. Nicméně bez randomizovaných studií srovnávajících myeloablativní režimy s RIC nelze jednoznačně určit rozdíl v celkovém přežití pacientů (12). Recentně publikované výsledky ASCT u myelofibrózy se zdají být optimističtější. Dosažení JAK2V617F negativity po transplantaci je asociováno s nižší incidencí relapsů (p = 0,03) (13). V poslední době Bacigalupo et al. formulovali prediktivní faktory pro přežití pacientů s PMF po alogenní transplantaci kostní dřeně. V multivariantní analýze byly nezávislými faktory pro horší přežití: aplikace více než 20 transfuzních jednotek erytrocytů, velikost sleziny nad 22 cm a HLA--neshodný dárce. V přítomnosti dvou a více z výše zmíněných rizikových faktorů bylo 5leté přežití pacientů po transplantaci 8% (14). Zvláště u těchto nemocných, kteří nemusí profitovat z alogenní transplantace kostní dřeně, by proto měla být zvážená studiová medikace či jiné léčebné možnosti. Vliv masivní splenomegalie na výsledek přežití po transplantaci je stále diskutován, a proto není splenektomie rutinně doporučována v přípravě před ASCT (5). U pacientů nevhodných pro transplantační léčbu volíme konvenční symptomatickou terapii.
Léčba anémie asociované s myelofibrózou
K léčbě anémie asociované s myelofibrózou jsou nejčastěji užívány kortikoidy (prednizon 0,5 mg/kg/den), na některých pracovištích androgeny (např. fluoxymesterone 10 mg p. o. 3x denně nebo testosteron 400–600 mg i. m. týdně) či danazol (600 mg/den) (1). Nízce dávkovaný thalidomid (50 mg/den ± prednizon) bývá pacienty často špatně tolerován a výskyt vedlejších účinků (periferní neuropatie, konstipace, somnolence) vede ve většině případů k přerušení terapie (15–17). Četnost léčebných odpovědí u výše zmíněných preparátů se pohybuje mezi 15–25 %, s průměrnou délkou trvání jeden až dva roky.
U nemocných s prokázanou delecí 5q (del5q; 5q-) je v první linii léčby doporučován lenalidomid (± prednizon), přičemž úpravy anémie dosáhne 30 % pacientů a regrese splenomegalie 42 % pacientů. Jsou publikovány i případy, kdy při léčbě lenalidomidem došlo ke snížení alelické nálože JAK2V617F či dosažení molekulární remise (18–19).
Kontroverzní zůstává otázka užití erytropoezu stimulujících proteinů (ESA). Mohou být aplikovány u anemických pacientů bez transfuzní závislosti s hodnotou erytropoetinu (EPO) v séru pod 125 U/l a bez splenomegalie. V různých studiích je uváděna léčebná odpověď u 20–60 % pacientů, s délkou trvání kratší než 1 rok (20). Vzhledem k zaznamenaným případům rychlého zvětšení sleziny u pacientů, jímž byly podávány ESA, a k jejich neefektivnosti u transfuzně závislých pacientů není jejich použití některými autory doporučováno (5).
Léčba splenomegalie asociované s myelofibrózou
Objemná splenomegalie vzniklá v důsledku extramedulární krvetvorby významně snižuje kvalitu života nemocných. Bývá spojená s abdominální bolestí, pocitem plnosti břicha, kachektizací, zhoršením fyzické aktivity, rozvojem portální hypertenze a zhoršením cytopenie díky sekvestraci ve zvětšené slezině (21). U 10 % pacientů se vyskytuje symptomatická splenomegalie již v době diagnózy a u dalších 50 % pacientů se vyvine v průběhu 4 let (22).
Navzdory omezené efektivitě v redukci velikosti sleziny je hydroxyurea (HU) stále nejčastějším lékem první volby splenomegalie asociované s myelofibrózou. Ve studii hodnotící 69 nemocných s PMF byla odpověď na léčbu hydroxyureou dokumentována u 28 % pa-cientů, z toho 16 pacientů dosáhlo klinického zlepšení a 3 parciální odpovědi dle kritérií IWG-MRT. Dále byla prokázána signifikantní asociace mezi pozitivním mutačním stavem JAK2V617F a léčebnou odpovědí na HU (23). Celkové zlepšení klinického stavu při medikaci HU (ústup konstitučních symptomů, bolestí kostí, pruritu, splenomegalie) je pozorováno u zhruba 40 % pacientů, s mediánem délky trvání léčebné odpovědi 13,2 měsíců (22). U většiny pacientů dochází ke ztrátě efektivity léčby díky rozvoji rezistence, intoleranci, či nutnosti redukce dávky pro cytopenii (24).
Dříve používaný melfalan nebo busulfan vedl u části nemocných k redukci masivní splenomegalie, avšak jejich dlouhodobé podávání bylo spojeno se zvýšenou incidencí leukemické transformace. Ve studii s nízce dávkovaným melfalanem (2,5 mg tři krát týdně) je udáváno zmenšení velikosti sleziny u 66 % pacientů, přičemž u 26 % léčených byla potvrzena transformace do akutní leukemie (25). Tyto alkylační látky by proto měly být považovány za terapeutickou alternativu jen pro starší symptomatické nemocné, kteří netolerují nebo jsou rezistentní k ostatním léčebným možnostem.
Interferon alfa (INFα) byl vzhledem ke své prokázané efektivitě v cytoredukční léčbě pravé polycytemie (26) zkoušen také v terapii PMF. V kontrastu s účinností INFα u pravé polycytemie a esenciální trombocytemie byly výsledky klinického testování standardních i pegylovaných preparátů v léčbě PMF neuspokojivé. Dosud nebyla jednoznačně prokázána jejich efektivita a ve více než 50 % případů byly špatně tolerovány (27). Ve studii s pegylovaným INFα-2b (PEG-IFN-α-2b) nebyla zaznamenána léčebná odpověď u žádného pacienta s PMF (28). Ianotto et al. uvádějí slibnější data s PEG-INF-α-2a naznačující jeho možné terapeutické využití v hypercelulární fázi choroby s akceptovatelným stupněm toxicity. Nicméně závěry této retrospektivní studie vyžadují potvrzení na větším souboru pacientů (29). Dále byla publikována i kazuistická sdělení pacientů s myeloproliferativní neoplazií, u nichž byl pro nechutenství a hmotnostní úbytek nasazen megestrol-acetát. Jako „vedlejší účinek“ bylo pozorováno významné zmenšení splenomegalie a snížení transfuzní závislosti (30).
Radioterapie sleziny je užívána u vybraných refrakterních symptomatických pacientů s dostatečným počtem trombocytů, kteří nejsou vhodnými kandidáty pro splenektomii. Efekt radioterapie je různými autory uváděn u 63–95 % pacientů, s mediánem délky trvání odpovědi 3,5–6 měsíců (31). Mezi závažné až život ohrožující nežádoucí účinky patří komplikace spojené s rozvojem těžké cytopenie.
Splenektomie prováděná u pacientů se symptomatickou splenomegalií zůstává výkonem se signifikantní peri- a postoperační morbiditou a mortalitou. Retrospektivní studie Mayo Clinic hodnotící 314 pa-cientů s MF, kteří podstoupili splenektomii, uvádí perioperační komplikace u 27 % pacientů a 6% mortalitu. Předoperační trombocytopenie ≤ 50 x 109/l byla spojena se signifikantně horším celkovým přežitím a anamnéza splenického infarktu s vyšší incidencí postoperačního krvácení (21). Mezi nejčastější komplikace splenektomie patří krvácení, trombózy, infekce a „myeloproliferativní“ reakce s trombocytózou a leukocytózou, která bývá obvykle dobře kontrolována HU. Někteří autoři pro snížení rizika trombózy doporučují udržovat počet trombocytů kolem 200 x 109/l v pre- a postoperačním období s profylaktickým podáváním LMWH po dobu alespoň 4–6 týdnů. Také jsou popsány případy progrese velikosti jater po splenektomii v důsledku „migrace“ extramedulární hematopoézy, výrazná hepatomegalie by proto měla být považována za kontraindikaci výkonu.
Pro snížení rizika peri- a postoperačních komplikací je nutný velmi pečlivý výběr pacientů. Splenektomie je alternativou pro nemocné s výrazně symptomatickou splenomegalií neodpovídající na medikamentózní léčbu, kteří mají relativně dobrý performance status a předpokládanou délku života více než 1 rok (22).
Imunomodulační léčba – thalidomid s konkomitantní medikací prednizonem má omezený vliv na splenomegalii, ve studiích je popisována redukce velikosti sleziny u 8–19 % pacientů (17, 32). Lepších výsledků bylo dosaženo při léčbě lenalidomidem v kombinaci s prednizonem.
I přes zatím omezené zkušenosti byly u pacientů s delecí 5q léčených lenalidomidem zaznamenány i kompletní hematologické a molekulární remise se zlepšením dřeňové fibrózy (19).
Extramedulární hematopoéza
Extramedulární hematopoéza může mimo sleziny a jater postihnout také peritoneum, plíce, lymfatické uzliny, torakolumbální páteř, kosti horních a dolních končetin. Klinicky se v pokročilé fázi manifestuje rozvojem ascitu, plicní hypertenzí, adenomegalií nebo refrakterní bolestí kostí. Léčbou volby pro symptomatické pacienty je nízce dávkovaná radioterapie.
Trombotické komplikace
PMF je spojena s vyšším rizikem trombózy. Ve velkém souboru 707 pacientů byly popsány trombotické komplikace u 7,2 % nemocných (1,75 % paciento-roků), což je srovnatelný výskyt jako u esenciální trombocytemie. V multivariantní analýze byly jako nezávislé faktory signifikantně zvyšující riziko trombózy identifikovány: věk nad 60 let (p = 0,01) a pozitivní mutační status JAK2V617F (p = 0,02). Zatímco samotná leukocytóza nad 15 x 109/l měla hraniční statistickou významnost (p = 0,06), největší incidence trombóz byla pozorována při současné přítomnosti leukocytózy a mutace JAK2V617F. Na druhé straně nebyla v této práci prokázána korelace mezi počtem trombocytů a incidencí trombózy (33). U nemocných se zvýšeným rizikem trombózy, zejména tedy u starších pacientů nebo pacientů s již prodělanou trombózou, je doporučována HU s nízce dávkovanou kyselinou acetylsalicylovou (ASA).
NOVÉ LÉČEBNÉ MOŽNOSTI MYELOFIBRÓZY
Inhibitory Janusovy kinázy 2 (JAK2). Do rodiny Janusových kináz patří čtyři proteiny s tyrozinkinázovou aktivitou: JAK1, JAK2, JAK3 a Tyk2. Růstové faktory jako erytropoetin (EPO), trombopoetin (TPO) a granulocytární kolonie stimulující faktor (G-CSF) mají své transmembránové receptory asociované s JAK2. Po vazbě ligandu na svůj receptor dochází k aktivaci receptoru s následnou fosforylací JAK2 kinázy a přenosu signálu do jádra cestou dráhy JAK-STAT (34). V roce 2005 byla popsána specifická mutace JAK2 genu způsobující substituci valinu fenylalaninem v pozici 617 pseudokinázové domény JAK2 proteinu (35). Mutace JAK2V617F je příčinou autofosforylace JAK2 kinázy následně vedoucí k perzistentní aktivaci dalších signálních drah zahrnujících STAT3, STAT5, MAPK, ERK, PI3K-AKT. Inhibitory Janusovy kinázy 2 (JAK2 inhibitory) jsou malé molekuly kompetitivně se vážící na vazebné místo pro ATP v kinázové doméně JAK2 proteinu. Ruxolitinib (Jakavi) je prvním ze skupiny těchto inhibitorů schválený FDA (Food and Drug Administration) a EMA (European Medicines Agency) pro léčbu myelofibrózy. Registrován byl na základě předběžných výsledků dvou randomizovaných studií. V první studii COMFORT I, jež hodnotí efekt ruxolitinibu oproti placebu v souboru 309 pacientů s MF, byla redukce velikosti sleziny o ≥ 35 % potvrzena u 41,9 % pacientů (36). Prvotní data druhé studie COMFORT II srovnávající ruxolitinib s „nejlepší dostupnou péčí“ u 219 nemocných prokázala zmenšení velikosti sleziny u 28,5 % pacientů (p < 0,0001) (37). V obou souborech došlo u většiny pacientů ke zmírnění konstitučních symptomů. Ruxolitinib je potentním inhibitorem nejen JAK2, ale také JAK1 a Tyk2. Právě inhibice JAK1 je spojována s dramatickým snížením hladin prozánětlivých cytokinů jako IL-6 a TNFα a zlepšením konstitučních symptomů. Vzhledem k tomu, že vazebné místo ruxolitinibu se nachází v kinázové doméně JAK2 proteinu, tzn. mimo mutovanou oblast, je léčebná odpověď nezávislá na mutačním stavu JAK2V617F. Dle výsledků prodlouženého sledování 51 nemocných z Mayo Clinic iniciálně zařazených do klinického testování ruxolitinibu fáze I/II byla dosud léčba ukončena u 92 % pacientů, nejčastěji z důvodu ztráty léčebné odpovědi, toxicity a progrese choroby. Po přerušení léčby se u 11 % pacientů objevil závažný rebound fenomén s nutností hospitalizace (rychlý, bolestivý nárůst splenomegalie, ojediněle šokový stav) (38). Nevýhodou ruxolitinibu je také jeho poměrně vysoká cena a možnost podání jen u nemocných s trombocyty nad > 50 x 109/l. Řada dalších JAK2 inhibitorů prochází klinickým testováním, ale zdá se, že žádný z nich, včetně ruxolitinibu, nevede k eradikaci nádorového klonu. Jejich klinický efekt je dán zejména silnou anticytokinovou aktivitou (tab. 3).
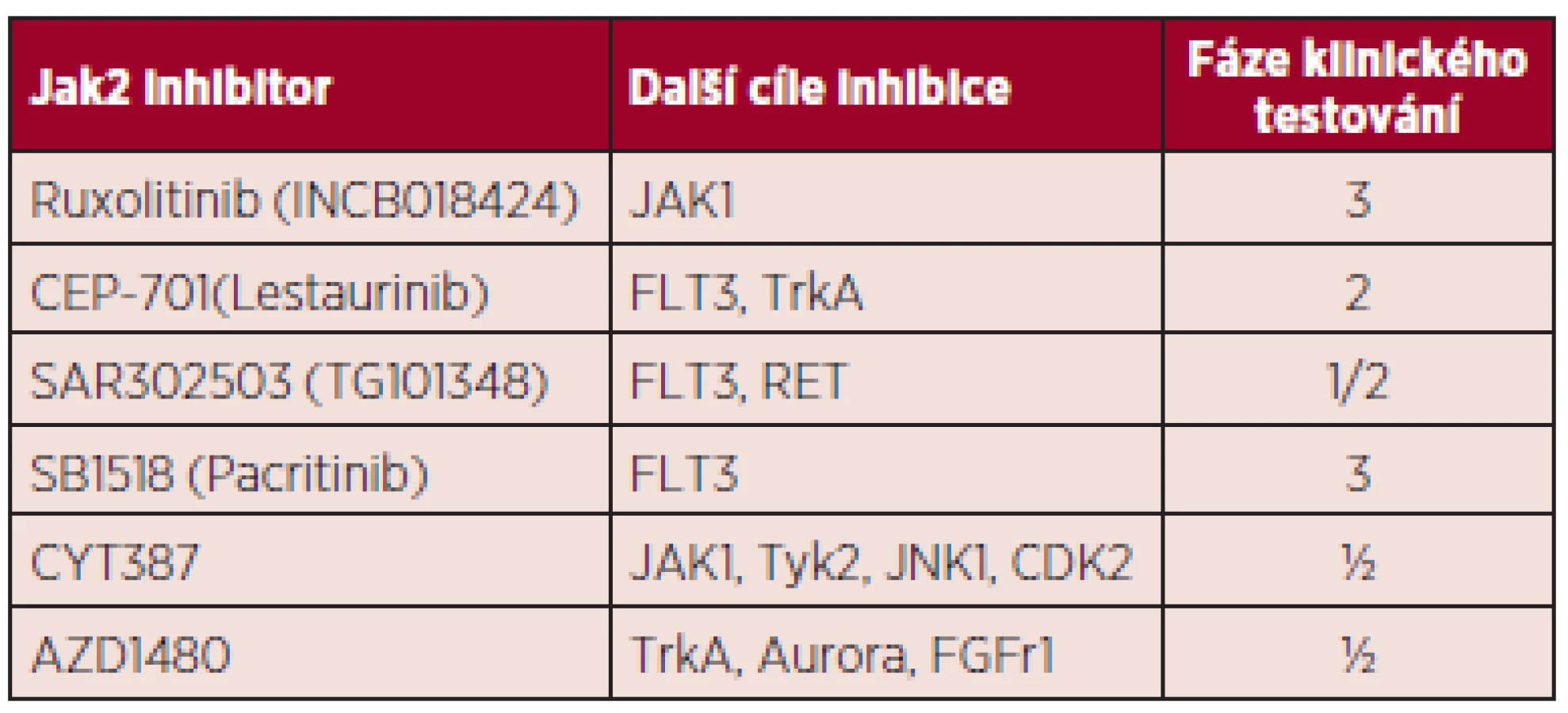
Pacritinib, orální selektivní JAK2/FLT3 inhibitor, právě prochází III. fází klinického testování a na rozdíl od ruxolitinibu nezhoršuje trombocytopenii.
Pomalidomid patřící mezi 2. generaci imunomodulačních látek má nižší toxicitu než thalidomid či lenalidomid. Prochází klinickým testováním a zlepšení anémie je uváděno u 25 % pacientů (1).
Everolimus. Skutečnost, že cestou JAK-STAT je zvýšeně aktivována také signální dráha Akt/mTOR, vyústila v testování účinnosti mTOR inhibitorů u pacientů s myelofibrózou. Celková léčebná odpověď hodnocená dle kritérií IGW-MRT byla u 23 % pacientů, u 69 % nemocných došlo k ústupu konstitučních symptomů (39).
ZÁVĚR
Prognóza myelofibrózy je u středně a vysoce rizikových pacientů ve srovnání s ostatními Ph1 negativní myeloproliferativními onemocněními značně nepříznivá, jejich celkové přežití je 3-5 let. V současné době je stále jedinou potencionálně kurativní léčebnou modalitou alogenní transplantace krvetvorných buněk, provázená vysokou morbiditou a mortalitou a vhodná pouze pro vybranou skupinu mladších nemocných. Transplantace je doporučována u pacientů s předpokládaným mediánem přežití kratším než 5 let, tzn. u rizikových skupin intermediate-2 a vysokého rizika dle DIPSS-plus skóre. Dle některých autorů by však měli být vyloučeni nemocní s přítomností dvou a více z následujících faktorů: více než 20 aplikovaných transfuzních jednotek erytrocytů, velikost sleziny nad 22 cm a HLA-neshodný dárce. Tato skupina nemocných s udávaným 5letým přežitím po transplantaci menším než 10 % z alogenní transplantace krvetvorných buněk neprofituje. Naopak pacienti velmi vysokého rizika s více než 80% mortalitou ve dvou letech predikovanou monosomálním karyotypem, inv(3)/i(17q) či přítomností dvou z následujících faktorů: počet cirkulujících blastů v periferní krvi nad 9 %, leukocytóza ≥ 40 x 109/l nebo jiný nepříznivý karyotyp, by měli být k alogenní transplantaci indikováni co nejčasněji. U pacientů s masivní splenomegalií indikovaných k transplantaci by měla být před výkonem zvážena nízce dávkovaná radioterapie sleziny. Pacienti nízkého rizika s mediánem přežití 15 let a nemocní rizikové skupiny intermediate-1 dle DIPSS-plus skóre s mediánem přežití 6,5 let nejsou vzhledem k uvedeným rizikům vhodnými kandidáty transplantační léčby.
Poznání mutace JAK2V617F a signální dráhy JAK-STAT vedlo k rychlému vývoji JAK2 inhibitorů. Ruxolitinib, selektivní ATP-kompetitivní JAK1/JAK2 inhibitor, je prvním ze skupiny testovaných inhibitorů schválený k léčbě vysoce a středně rizikových pacientů s myelofibrózou. Nicméně velká očekávání vkládána do JAK2 inhibitorů se ne zcela naplnila. Jak se ukazuje, dosud žádný z JAK2 inhibitorů nevede k eradikaci nádorového klonu a významně neovlivňuje fibrózu kostní dřeně. Na druhou stranu výrazně zlepšují kvalitu života nemocných a je otázkou blízké budoucnosti, než naleznou své místo v léčbě myelofibrózy, zejména u pacientů se symptomatickou splenomegalií a konstitučními symptomy. Léčba myelofibrózy se vyvíjí a pokrok je možno očekávat s hlubším poznáním etiopatogeneze a rizikových faktorů pro přežití.
Seznam použitých zkratek
PMF – primární myelofibróza
JAK 2 – Janusova 2 kináza
STAT – Signal transducer and activator of transcription
IPSS – International Prognostic Scoring System
DIPSS – Dynamic International Prognostic Scoring System
IWG-MRT – International Working Group for Myelofibrosis Research and Treatment
ASCT – Allogeneic stem cell transplantation
RIC – Reduced intensity conditioning
ESA – erythropoiesis stimulating agents
EPO – erytropoetin
HU – hydroxyurea
INFα – interferon alfa
PEG-INFα – pegylovaný interferon alfa
LMWH – Low molecular weight heparin
TPO – trombopoetin
G-CSF – Granulocyte colony-stimulating factor
FDA – Food and Drug Administration
EMA – European Medicines Agency
IL 6 – interleukin 6
TNFα – Tumor necrosis factor alpha
mTOR – Mammalian target of rapamycine
Podíl autorů na rukopisu
M. Palová – příprava rukopisu
K. Indrák – revize rukopisu
A. Hluší – revize rukopisu
T. Szotkowski – kontrola rukopisu
Poděkování: Práce byla podporována grantem IGA UP LF-2013-004.
Doručeno do redakce: 16. 7. 2013
Přijato po recenzi: 22. 8. 2013
MUDr. Miroslava Palová
Hemato-onkologická klinika LF UP a FN Olomouc
I. P. Pavlova 6
775 20 Olomouc
e-mail: miroslava.palova@fnol.cz
Zdroje
1. Tefferi A. Primary myelofibrosis: 2013 update on diagnosis, risk-stratification, and management. Am J Hematol 2013; 88: 142-150.
2. Reilly JT, McMullin MF, Beer PA, et al. Guideline for the diagnosis and management of myelofibrosis. Br J Haematol 2012; 158: 453-471.
3. Mesa RA, Li CY, Ketterling RP, et al. Leukemic transformation in myelofibrosis with myeloid metaplasia: A single-institution experience with 91 cases. Blood 2005; 105: 973-977.
4. Cervantes F, Dupriez B, Pereira A, et al. New prognostic scoring system for primary myelofibrosis based on a study of the International working group for myelofibrosis research and treatment. Blood 2009; 113: 2895-2901.
5. Vannucchi AM. Management of myelofibrosis. Hematology Am Soc Hematol Educ Program 2011; 2011: 222-230.
6. Passamonti F, Cervantes F, Vannucchi AM, et al. A dynamic prognostic model to predict survival in primary myelofibrosis: a study by the IWG-MRT (International working group for myelofibrosis research and treatment). Blood 2010; 115: 1703-1708.
7. Gangat N, Caramazza D, Vaidya R, et al. DIPSS plus: a refined Dynamic international prognostic scoring system for primary myelofibrosis that incorporates prognostic information from karyotype, platelet count, and transfusion status. J Clin Oncol 2011; 29: 392-397.
8. Tefferi A, Jimma T, Gangat N, et al. Predictors of greater than 80% 2-year mortality in primary myelofibrosis: a Mayo Clinic study of 884 karyotypically annotated patients. Blood 2011; 118(17): 4595-4598.
9. Barosi G, Rosti V, Vannucchi AM. Therapeutic approaches in myelofibrosis. Expert Opin Pharmacother 2011; 12(10): 1597-1611.
10. Ballen K, Shrestha S, Sobocinski KA, et al. Outcome of transplantation for myelofibrosis. Biol Blood Marrow Transplant 2010; 16: 358-367.
11. Stewart WA, Pearce R, Kirkland KE, et al. The role of allogeneic SCT in primary myelofibrosis: a British society for blood and marrow transplantation study. Bone Marrow Transplant 2010; 45: 1587-1593.
12. Ballen K. How to manage the transplant question in myelofibrosis. Blood Cancer J; publikováno elektronicky 2. března 2012. DOI 10.1038/bcj-2012-3
13. Steckel NK, Koldehoff M, Ditschkowski M, et al. Use of the activating gene station of the tyrosine kinase JAK2 as a minimal disease marker in patients with myelofibrosis and myeloid metaplasia after allogeneic stem cell transplantation. Transplantation 2007; 83: 1518-1520.
14. Bacigalupo A, Soraru M, Dominietto A, et al. Allogeneic hematopoietic SCT for patients with primary myelofibrosis: a predictive transplant score based on transfusion requirement, spleen size, and donor type. Bone Marrow Transplant 2010; 45: 458-463.
15. Elliott MA, Mesa RA, Li CY, et al. Thalidomide treatment in myelofibrosis with myeloid metaplasia. Br J Haematol 2002; 117: 288-296.
16. Mesa RA, Steensma DP, Pardanani A, et al. A phase 2 trial of combination low-dose thalidomide and prednisone for the treatment of myelofibrosis with myeloid metaplasia. Blood 2003; 101: 2534-2541.
17. Thapaliya P, Tefferi A, Pardanani A, et al. International working group for myelofibrosis research and treatment response assessment and long-term follow-up of 50 myelofibrosis patients treated with thalidomide-prednison based regimens. Am J Hematol. 2011; 86(1): 96-98.
18. Quintás-Cardama A, Kantarjian H, Manshouri T, et al. Lenalidomide plus prednisone results in durable clinical, histopathologic, and molecular responses in patients with myelofibrosis. J Clin Oncol 2009; 27: 4760-4766.
19. Tefferi A, Lasho TL, Mesa RA, et al. Lenalidomide therapy in del(5q)(q31)-associated myelofibrosis: cytogenetic and JAK2V617F molecular remissions. Leukemia 2007; 21(8): 1827-1828.
20. Tefferi A. How I treat myelofibrosis. Blood 2011; 117(13): 3949-3504.
21. Mesa RA, Nagomey DS, Schwager S, et al. Palliative goals, patient selection, and perioperative platelet management: outcomes and lessons from 3 decades of splenectomy for myelofibrosis with myeloid metaplasia at the Mayo Clinic. Cancer 2006; 107: 361-370.
22. Randhawa J, Ostojic A, Vrhovac R, Atallah E, Verstovsek S. Splenomegaly in myelofibrosis – new options for therapy and the therapeutic potential of Janus kinase 2 inhibitors. J Hematol Oncol; publikováno elektronicky 1. srpna 2012. DOI: 10.1186/1756-8722-5-43
23. Sirhan S, Lasho TL, Hanson CA, et al. The presence of JAK2V617F in primary myelofibrosis or its allele burden on polycythemia vera predicts chemosensitivity to hydroxyurea. Am J Hematol 2008; 83: 363-365.
24. Mesa RA. How I treat symptomatic splenomegaly in patients with myelofibrosis. Blood 2009; 113: 5394-5400.
25. Petti MC, Latagliata R, Spadea T, et al. Melphalan treatment in patients with myelofibrosis with myeloid metaplasia. Br J Haematol 2002; 116: 576-581.
26. Kiladjian JJ, Cassinat B, Chevret S, et al. Pegylated interferon-alfa-2a induces complete hematologic and molecular responses with low toxicity in polycythemia vera. Blood 2008; 112: 3065-3072.
27. Kiladjian JJ, Mesa RA, Hoffman R. The renaissance of interferon therapy for the treatment of myeloid melignancies. Blood 2011; 117: 4706-4715.
28. Verstovsek S, Lawhorn K, Giles F, et al. PEG-intron for myeloproliferative diseases: an update of ongoing phase II study. Blood 2004; 104: Abstract 633.
29. Ianotto JC, Kiladjian JJ, Demory JL, et al. PEG-INF-alpha-2a therapy in patients with myelofibrosis: a study of the french groupe d etudes des myelofibroses (GEM) and France intergroupe des syndromes myéloprolifératifs (FIM). Br J Haematol 2009; 146: 223-225.
30. Hubáček J, Faber E, Indrák K. Podpůrná terapie megestrol acetátem u myeloproliferativních neoplazií. Transfuze Hematol dnes 2012; 4: 174-176.
31. Elliott MA, Chen MG, Silverstein MN, et al. Splenic iradiation for symptomatic splenomegaly associated with myelofibrosis with myeloid metaplasia. Br J Haematol 1998; 103: 505-511.
32. Mesa RA, Steensma DP, Pardanani A, et al. A phase 2 trial of combination low-dose thalidomide and prednison for the treatment of myelofibrosis with myeloid metaplasia. Blood 2003; 101: 2534-2541.
33. Barbui T, Carrobio A, Cervantes F, et al. Thrombosis in primary myelofibrosis: incidence and risk factors. Blood 2010; 115(4): 778-782.
34. Cross NC. Genetic and epigenetic komplexity in myeloproliferative neoplasms. Hematology Am Soc Hematol Educ Program 2011; 2011: 208-214.
35. Kralovics R, Passamonti F, Buser AS, et al. A gain-of-function station of JAK2 in myeloproliferative disorders. N Engl J Med 2005; 352: 1779-1790.
36. Verstovsek S, Mesa RA, Gotlib J, et al. A double-blind, placebo-controlled trial of ruxolitinib for myelofibrosis. N Engl J Med 2012; 366: 799-807.
37. Harrison C, Kiladjian JJ, Al-Ali HK, et al. JAK inhibition with ruxolitinib versus best available therapy for myelofibrosis. N Engl J Med 2012; 366: 787-789.
38. Tefferi A, Litzow MR, Pardanani A. Long-term outcome of treatment with ruxolitinib in myelofibrosis. N Engl J Med 2011; 365: 1455-1457.
39. Guglielmelli P, Barosi G, Rambaldi A, et al. Safety and efficacy of everolimus, a mTOR inhibitor, as single agent in a phase 1/2 study in patients with myelofibrosis. Blood 2011; 118(8): 2069-2076.
Štítky
Hematológia Interné lekárstvo OnkológiaČlánok vyšiel v časopise
Transfuze a hematologie dnes

2013 Číslo 3
- Nejasný stín na plicích – kazuistika
- Realita liečby bolesti v paliatívnej starostlivosti v Nemecku
- Těžké menstruační krvácení může značit poruchu krevní srážlivosti. Jaký management vyšetření a léčby je v takovém případě vhodný?
- MUDr. Lenka Klimešová: Multiodborová vizita je kľúč k efektívnejšej perioperačnej liečbe chronickej bolesti
Najčítanejšie v tomto čísle
- Současná léčba myelofibrózy na základě rizikové stratifikace pacientů
- Lymfomy gastrointestinálního traktu – klinicko-patologický přehled
- Význam stanovení sérových hladin volných lehkých řetězců imunoglobulinu u AL amyloidózy
- Plazmocelulární leukemie